

Abstract
Glucocorticoid stress hormones (GCs) are well known for being anti-inflammatory, but some reports suggest that GCs can also augment aspects of inflammation during acute brain injury. Because the GC receptor (GR) is ubiquitously expressed throughout the brain, it is difficult to know which cell types might mediate these unusual “proinflammatory” GC actions. We examined this with cell type-specific deletion or overexpression of GR in mice experiencing seizure or ischemia. Counter to their classical anti-inflammatory actions, GR signaling in myeloid cells increased Iba-1 and CD68 staining as well as nuclear p65 levels in the injured tissue. GCs also reduced levels of occludin, claudin 5, and caveolin 1, proteins central to blood–brain-barrier integrity; these effects required GR in endothelial cells. Finally, GCs compromised neuron survival, an effect mediated by GR in myeloid and endothelial cells to a greater extent than by neuronal GR.
Introduction
Acute CNS injury activates the stress response in mammals. A major component of this response is the secretion of adrenal glucocorticoid (GC) hormones into the blood. These lipophilic hormones cross the blood–brain-barrier (BBB) and bind to GC receptors (GRs) in neurons, astrocytes, oligodendrocytes, and microglia (Vielkind et al., 1990; Sierra et al., 2008). Despite the central role of GCs in the stress response and their abundant use in medicine, the cell-specific effects of GCs during acute CNS injury are still poorly understood.
GCs have context-dependent effects where the timing, duration, and magnitude of GC exposure can lead to different, even opposite outcomes. A singular example of this is the effects of GCs on inflammation. Their well described anti-inflammatory properties (Coutinho and Chapman, 2011) might be expected to have therapeutic benefit for the injured CNS, where excessive inflammation can be detrimental (Block et al., 2007; Perry et al., 2010); however, recent evidence suggests that GCs do not always decrease inflammation and, depending on the context of their exposure, may even increase inflammatory responses (for review, see Dhabhar, 2009; Sorrells et al., 2009). For example, acute GC exposure stimulates lymphocyte recruitment to injury sites instead of inducing apoptosis as was previously assumed (Dhabhar et al., 1996; Viswanathan and Dhabhar, 2005). GC exposure due to moderate amounts of stress for 1–2 weeks augments the CNS inflammatory response to challenge with lipopolysaccharide (LPS) (Munhoz et al., 2006, 2010; de Pablos et al., 2006) and excitotoxins (Dinkel et al., 2003; MacPherson et al., 2005). One emerging hypothesis is that both short- and long-term GC exposure before an injury exacerbate the subsequent inflammatory response to the injury (Frank et al., 2010; Munhoz et al., 2010).
Neuroinflammation is a composite of responses from cells of many different types, and, while it is an important part of the repair process, it is also frequently a source of secondary injury to neurons (Perry et al., 2010). Both CNS resident and activated peripheral immune cells respond quickly to necrotic cell death, activating transcription factors like nuclear factor-κ B (NF-kB) and releasing proinflammatory cytokines. During ischemic injury, the BBB is also disrupted, worsening edema and neuron death. It is likely that GCs have divergent cell-specific effects on each of these processes. To determine which cells GCs are acting on to increase inflammation, we conditionally deleted or overexpressed the GR in myeloid cells, neurons, or endothelial cells in mouse models of excitotoxicity and stroke.
Materials and Methods
Animals.
All experiments were conducted following protocols approved by the Stanford Institutional Animal Care and Use Committee. Mice were housed in a 12 h light/dark cycle with ad libitum food and water. Great care was taken to reduce uncontrolled stressors in their environment at all times throughout their lives. C57 BL/6J, CaMKIIα-cre, and Tie2/TEK-cre mice were obtained from Jackson Laboratories. LysM-CRE and floxed-GR mice were a gift from Luis Muglia (Cincinnati Children's Hospital Medical Center in Cincinnati, Ohio). TRE-rGR mice were a gift from Sam Okret (Karolinska Institutet, Stockholm, Sweden) and were mated to ROSA-LSL-rtTA-GFP mice from Jackson Laboratories. All mice were male littermates used between 2–4 months of age, except CaMKIIα-cre mice that were used between 4–6 months of age. No mice used for middle cerebral artery occlusion (MCAO) were <3 months of age.
GC manipulations.
Mice were given a subcutaneous corticosterone (Sigma) pellet weighing 10 mg that was implanted between the scapulae using a 12 ga trochar (Innovative Research of America). Vehicle animals were subjected to the same trochar injection but no pellet was implanted. For overexpression induction, doxycycline hyclate (Sigma) was given in 5% sucrose drinking water at 2 mg/ml for 7 d, replaced every third day.
Excitotoxic injury model.
Mice were given 5% isoflurane (air mixture) to induce anesthesia, which was then maintained with 1.5% isoflurane. Kainic acid (KA) was injected via stereotactic surgery into the dentate gyrus of the hippocampus using the following coordinates: 2.0 mm posterior and 2.1 mm lateral to bregma; 1.9 mm ventral to dura. A total volume of 200 nl was infused over the course of 4 min for a final dose of 50 ng of KA. Buprenorphine was administered as a postoperative analgesic.
Histology.
Mice were transcardially perfused at 72 h after KA with 0.9% saline followed by 4% PFA. Brains were postfixed in 4% PFA for 1 h and transferred to 30% sucrose for 24 h before cutting several representative sets of 20 μm frozen cryostat sections across the hippocampus. Nissl stain and hippocampus lesion quantification were performed as previously described (Dinkel et al., 2003). Briefly, tissue sections were dehydrated and stained with cresyl violet, and 40× images of the hippocampus were acquired on an Olympus IX70 microscope using a Hamamatsu digital camera. Total hippocampal damage was calculated as the damaged cell area within the hippocampal pyramidal layer divided by the total pyramidal area. The next serial set of representative hippocampus tissue sections was used for immunofluorescent staining. Sections were air dried for 30 min, blocked in 5% NGS in PBS + 0.3% Triton X-100 (PBST) for 1 h at room temperature, and stained overnight at 4°C in primary antibody. Unless specified otherwise, antibodies were diluted (1:500) and the GR antibody used for all applications was clone M-20 (Santa Cruz Biotechnology). The C-terminal GR antibody used was clone PA-516 (Affinity Bioreagents). FITC-conjugated tomato lectin was L0401 (Sigma 1:1000), NeuN (Millipore Bioscience Research Reagents, 1:250), Iba-1 (Dako), CD68 (Abcam), and anti-GFP (Invitrogen). Sections were washed with PBST 3 times for 10 min and incubated 1 h at room temperature with 488 or 594 nm Alexa secondary antibodies (1:200, Invitrogen) in PBST, washed again, and mounted in polyvinyl alcohol (Sigma). MetaMorph (Universal Imaging) was used to quantify the amount of immunoreactive area adhering to guidelines for quantifying fluorescent signal (Waters, 2009). All staining and imaging to be compared was counterbalanced by treatment group and performed and imaged on the same day. Slides were background corrected, the hippocampus was traced, and artifacts were excluded while blind to experimental condition. Signal was counted if it was between 8 and 40 μm in diameter and was >10 pixel intensity units above background.
Focal cerebral ischemia.
Mice were anesthetized as for stereotactic surgery. The middle cerebral artery (MCA) was exposed and permanently occluded by electrocoagulation as previously described (Lambertsen et al., 2005). Briefly, an incision perpendicular to the line connecting the lateral canthus of the left eye and the external auditory canal was made to expose and retract the temporalis muscle. A burr hole was drilled to expose the MCA for elevation and cauterization by cutting and retracting the dura. Body temperature was maintained at 37°C with a thermal pad and postoperative care was administered as for stereotactic surgery.
Flow cytometry.
Mononuclear cells were extracted from adult brains as described previously (Cardona et al., 2006a). Briefly, brains were homogenized and mononuclear cells were collected from the 70/30% Percoll interface, washed, and stained with antibodies for flow cytometry. Cells were stained with directly conjugated antibodies in FACS buffer for CD11b-APC, CD45-FITC, and Ly6G-PE or their isotype controls (Abcam) and analyzed on an LSR 1 analyzer (BD Biosciences). Gating and analysis were performed in FlowJo (Treestar). Cells were first gated for forward and side scatter to restrict events to live cells. This gate (G1) was then further gated to only include CD45+ cells and CD11b+ cells. This gate (G2) was then broken into three populations for analysis; granulocytes were Ly6G+CD45low (P1) and activated mononuclear cells were Ly6G-CD45high (P2). The largest isolated population (P3) was Ly6G-CD45low microglia. The abundance of these populations in the infarcted and contralateral hemispheres was expressed as a percentage of the parent gate (G2). The same gating strategy (excluding P3) was used for peripheral blood mononuclear cell isolations, which were performed on blood collected from a terminal cardiac bleed, collected in 1 mm EDTA. Before staining for flow cytometry, peripheral blood was incubated in 1% Dextran (T-500) (Sigma) for 30 min at 37°C. The supernatant was then centrifuged at 300–400 × g at 4°C and the pellet was resuspended in red blood cell lysis buffer (155 mm NH4Cl, 12 mm NaHCO3, 0.1 mm EDTA). After a 10 min incubation at 20°C, lysis was stopped by adding a large volume of PBS. Cells were washed and stained for flow cytometry as described above for CNS mononuclear cells.
Western blots.
Antibodies for p65, actin (Santa Cruz Biotechnology), GAPDH (Sigma), occludin, caveolin-1, claudin-5, glut-1, lamin B1, and CX3CL1 (Abcam) were used at a concentration of 1:1000. The immunoreactive density of equally loaded lanes was quantified using ImageJ, and all samples were normalized to internal β-actin load controls. To quantify nuclear p65 levels, the same procedure was used on total tissue nuclear extracts using lamin B1 as a nuclear load control.
ELISAs.
Hippocampus was microdissected 12 or 72 h after KA and supernatant and nuclear extracts were prepared as previously described (Munhoz et al., 2006). Total protein was quantified using the BCA method (Pierce) and ELISAs for IL-1β, IL-6, and brain-derived neurotrophic factor (BDNF; R&D) were run on the supernatants (cytosolic fraction) as per manufacturer instructions and quantified with a SoftMax 2000 plate reader (Molecular Devices). To measure cortisol levels, blood was allowed to clot at room temperature and serum was collected following a 5 m spin at 3000 rpm. This serum was diluted 1:20 before using a competitive enzyme immunoassay (Assay Designs) to measure serum corticosterone levels following the manufacturer's instructions.
Infarct size quantification.
Brains were removed 24 h after MCAO and cut into seven 1 mm coronal brain slices using a brain matrix (Braintree Scientific), which were stained in 1% 2,3,5-triphenyltetrazolium chloride (TTC, Merck) in 0.1 mol/L phosphate buffer. Infarct volumes were measured by imaging stained sections with a digital camera. With the observer masked to the experimental conditions, the contralateral hemisphere perimeter was overlapped onto the ipsilateral hemisphere to exclude edema, and infarct borders were delineated with an operator-controlled cursor in ImageJ. The unstained area of infarct was determined by counting the pixels contained within the outlined regions of interest and expressed in square millimeters. Infarct volumes (in mm3) were integrated from the infarct areas over the extent of the infarct calculated as an orthogonal projection. All animals displayed infarcts after the occlusion procedure, which included the cortex, subcortex, and striatum, depending on the intensity of the lesion.
Blood vessel isolation.
Whole-brain blood microvessels were extracted from the brains of Tie2-GRKO, Tie2-GROV, and WT littermates, and GR levels were measured for each animal by Western blot. Mice were transcardially perfused with 0.9% saline, the meninges were removed, and brains were rinsed with cold PBS. Each brain was diced into small pieces and homogenized in a 1 ml Dounce tissue grinder, with eight strokes. Homogenates were diluted to 5 ml with cold PBS and then resuspended in 5 ml of a cold 30% (w/v) dextran solution (molecular weight 35,000–45,000, Sigma). Samples were spun at 4000 rpm at 4°C without using the brake on the centrifuge and the myelin layer was aspirated. The supernatant and pellet were split, washed in cold PBS, and spun again. Pellets in each tube were resuspended in 1 ml PBS and pooled. This suspension was passed through a 70 μm cell strainer (BD Biosciences) and then collected on a 40 μm filter. After washing the 40 μm filter with 2 ml PBS, the filter was flipped over, blood vessels were eluted from the underside, washed in PBS, and centrifuged at 2000 rpm for 10 min at 4°C. These pellets were kept frozen at −80°C before immunoblotting.
Statistics.
Significance levels were established by two-way ANOVA and Bonferroni post hoc tests, whereas main treatment effects are noted in the figure legends. Significance of the endothelial GR data was established with unpaired, two-tailed t tests. All data are expressed as means ± SEM. Statistical tests were performed in Prism (GraphPad), vector art was assembled in Adobe Illustrator, and graphic art was assembled in Adobe Photoshop.
Results
The two acute neurological injury models we examined were KA-induced excitotoxic injury in the hippocampus, and the MCAO model of stroke. Stereotactic injections of KA produce localized, reproducible damage and inflammation that progresses through a well studied cascade of necrotic events. GCs have been reported to increase neuron death (Sapolsky and Pulsinelli, 1985; Kaufer et al., 2004) and cellular inflammation during KA-injury in rats (Dinkel et al., 2003; MacPherson et al., 2005). In contrast to the more localized microglial inflammatory response to KA-injury, MCAO injury involves broader necrosis and secondary injury coupled with greater peripheral inflammation and vascular disruption.
To study the effects of GCs on immune responses during these injuries, we tested two different patterns of exposure to corticosterone (the endogenous GC in mice): (1) GC treatment of mice before injury, where mice received subcutaneous GC pellets 3 d before injury to raise circulating GC levels into the high stress range (Fig. 1A). Equivalent GC levels increase inflammation during inflammatory injury in rats (Munhoz et al., 2006; Munhoz et al., 2010). (2) Control mice, where endogenous GC secretion was raised into the stress range only by the stress of the necrotic insult. This exposure pattern was expected to suppress inflammation in the classic GC manner (Munck et al., 1984; Sapolsky et al., 2000) and has been characterized both for KA injury (Stein and Sapolsky, 1988) and MCAO injury (Smith-Swintosky et al., 1996).
Figure 1.

Study design and animal models. A, Male WT and MGRKO littermates were given vehicle or a 10 mg, s.c. GC pellet 72 h before either KA or MCAO injury. For excitotoxic injury, KA was stereotactically delivered into the dentate gyrus of the hippocampus and the injury was assessed 12 and 72 h later. For MCAO injury, the MCA was permanently occluded via electrocoagulation and injury was monitored 24 h later. Serum corticosterone levels in WT mice at different times after vehicle or GC pellet implantation (n = 3–5 different mice per day). B, Mouse brain map indicating regions affected by KA and MCAO injury where immunostaining was assessed in the transgenic mouse models. Images in C–J are from uninjured mice. C, Antibodies against both the N and C terminus of the GR were used to stain the CA3 region of the hippocampus in WT (top) and MGRKO mice (bottom). In MGRKO mice, many CD68+ cells lack GR colocalization (purple only, arrowheads) but some CD68+ cells can be found that retain GR immunoreactivity (white, arrows). N-terminal GR antibodies were used for all other applications unless otherwise indicated. Scale bar, 35 μm. D, In MGRKO mice, some CD68+ cells in the uninjured cortex were GR+ (arrows, bottom), and others lacked GR-immunoreactivity (arrowheads, top). Two different fields from the same animal are shown. Scale bar, 10 μm. E, The CA3 pyramidal neurons in FBGRKO mice (bottom) also lacked GR signal compared with WT littermates (top). Scale bar, 100 μm. F, After crossing the CaMKIIa-cre driver to a ROSA-LSL-rtTA-IRES-GFP mouse, GFP expression could be observed in CA3 pyramidal neurons after 3 d of DOX treatment (top) and was more extensive after 10 d (bottom). Scale bar, 100 μm. G, Tie2-cre caused similar GFP expression in the blood vessels in the cortex of Tie2-GROV mice compared with no expression in ROSA-LSL-rtTA-IRES-GFP littermates lacking the cre (inset). Scale bar, 30 μm. In the cortex (H), CA1 (I), and CA3 (J) of FBGROV mice, GR immunostaining intensity in neurons labeled with NeuN was increased the longer the mice received DOX in their drinking water. The biggest increase in protein levels occurred in the cortex by 3 d of DOX treatment, but took closer to 5 d of DOX treatment to be visible in the CA1 and CA3. Scale bars: in H–J, 100 μm.
To determine which GC effects directly require myeloid GR, we used homozygous floxed-GR mice (Brewer et al., 2003) carrying the LysM-cre driver to generate a myeloid GR-deficient animal (MGRKO) (Bhattacharyya et al., 2007). MGRKO mice are reported to have efficient GR deletion in peripheral myeloid cells (Bhattacharyya et al., 2007) and microglia (Ros-Bernal et al., 2011). To further verify this, we isolated mononuclear cells from the uninjured CNS on a Percoll gradient (Cardona et al., 2006a) and GR protein levels were analyzed by Western blot. This fraction contained significantly lower GR levels in MGRKO mice, at 33.6% (±11.1% SEM n = 4 pools of 5 mice per genotype) of the GR protein levels in their WT littermates (p = 0.007, unpaired, one-tailed t test). Because only exons 1C and 2 of the GR are floxed in these mice, it is possible that C-terminal fragments of the GR are still produced, so we compared N- and C-terminal antibody staining in WT and MGRKO mice (Fig. 1C). Many but not all CD68+ cells were missing the GR in the MGRKO brain, regardless of the GR fragment against which the antibody was raised. Examples of CD68+ cells that were GR+ and GR− could be found in the uninjured cortex via confocal microscopy, indicating that not all myeloid cells had the GR deleted (Fig. 1D).
Secondary GC effects on inflammation may result from GR signaling in neurons, so in some experiments we also examined forebrain neuron GR-KO mice (FBGRKO) after injury. In agreement with prior studies, we also found that neuronal GR deletion in these mice was most efficient if animals were >4 months old (Boyle et al., 2005); however, many but not all pyramidal neurons in the CA3 lacked GR immunostaining in FBGRKO mice even by 2 months of age (Fig. 1E).
Particularly for MCAO injury, additional secondary injury may develop from disruption of the BBB. To investigate the effects of GR signaling in endothelial cells we used Tie2-cre mice crossed to the same floxed-GR allele (Tie2-GRKO). The Tie2-cre promoter expresses in endothelial cells throughout the body, so to verify GR deletion in brain endothelial cells, we conducted Western blots on blood microvessel extracts from the brains of Tie2-GRKO and WT mice. GR levels in the extracts were significantly lower at 67.12% (±7.217% SEM n = 5) in Tie2-GRKO mice relative to 100% expression in WT mice (p = 0.033, one-tailed t test with Welch's correction).
To measure GC effects on neuron death during these injuries, we also wanted to know whether increasing GR signaling in myeloid cells, neurons, or endothelial cells might further worsen neuron death. We developed an overexpression system for the GR using the same cre drivers and ROSA-loxP-stop-loxP-rtTA-eGFP (ROSA-LSL-rtTA-IRES-GFP) mice also carrying an inducible TetO-GR allele. The resulting triple-transgenic mouse overexpresses the GR in myeloid cells (MGROV), forebrain neurons (FBGROV), or endothelial cells (Tie2-GROV) following 7 d of doxycycline (DOX) administration. To verify overexpression, MGROV and WT mice were given DOX in their drinking water for 7 d and CNS monocytes were isolated on a Percoll gradient as for MGRKO mice. Anti-GR immunoblotting determined that GR overexpression was significantly higher at 176.5% (±14.13%, SEM n = 5 pools of 5 mice per genotype) in monocytes isolated from MGROV brains relative to 100% in monocytes from WT brains (p = 0.001, unpaired, one-tailed t test). GR overexpression in blood microvessels extracted from Tie2-GROV mice was significantly higher at 156.0% (±18.42% SEM n = 7) relative to 100% in WT littermates (p = 0.017, unpaired, one-tailed t test). For FBGROV mice, we examined IRES-GFP expression by immunostaining (Fig. 1F), which agreed with the blood vessel localization of the IRES-GFP expression in Tie2-GROV mice (Fig. 1G). Immunostaining of neurons in the cortex, CA1, and CA3 in FBGROV mice revealed visible increases in GR immunofluorescence after 5 or 7 d of DOX treatment (Fig. 1H–J). These increases were less evident before 5 d of DOX treatment, and were absent in mice that received vehicle.
With these tools we sought to determine how GR signaling in myeloid cells and neurons affects inflammation and neuron survival following these acute CNS injuries.
Prior GCs activate and endogenous GCs suppress immune responses to KA
To measure myeloid cell activation after KA, brain sections were collected 72 h after injury and stained for CD68 (Fig. 2A,C,D), a protein upregulated upon myeloid cell activation. Prior GC treatment significantly increased both CD68 area and intensity, but only the increased intensity required myeloid GR signaling to occur (Fig. 2D, compare GC-treated WT and GC-treated MGRKO). Despite typically reducing inflammation (Nadeau and Rivest, 2003), the endogenous GCs secreted after injury failed to reduce CD68 area or intensity via direct effects on myeloid GR (Fig. 1C,D, compare vehicle-treated WT and vehicle-treated MGRKO).
Figure 2.
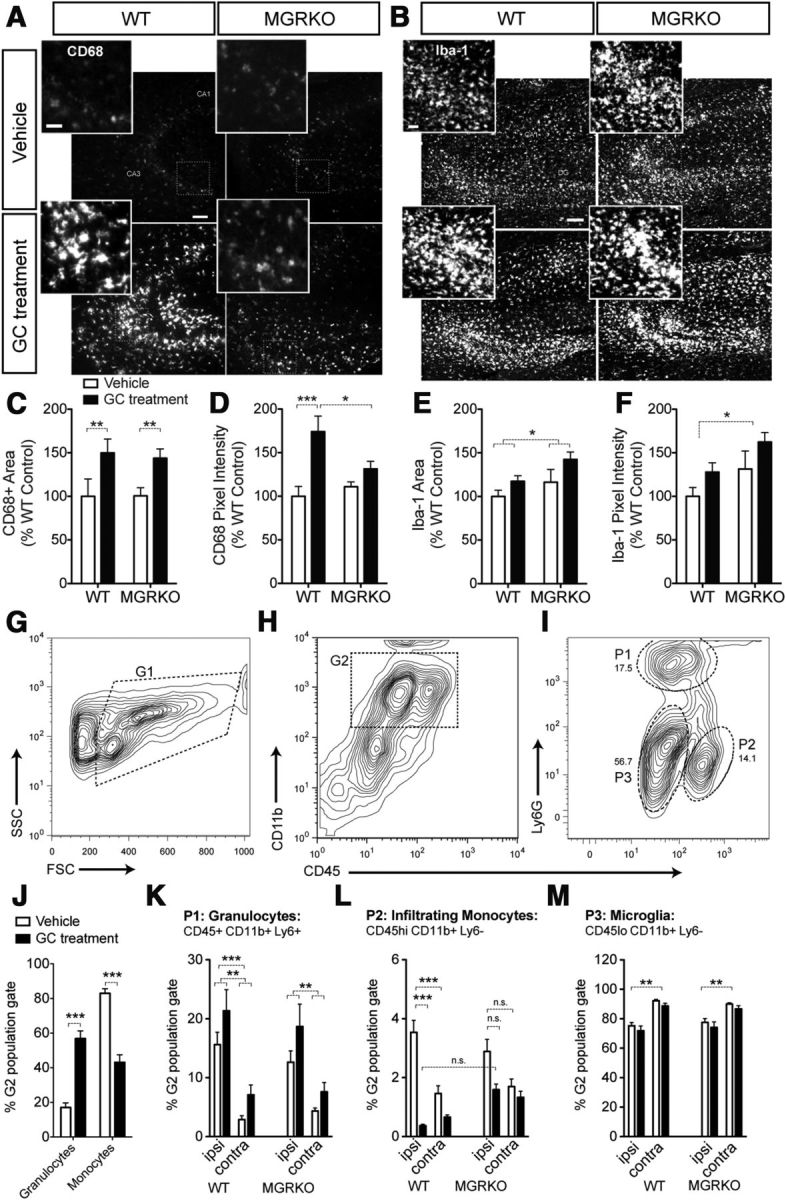
GCs increase some aspects of the cellular response to KA and MCAO injury and suppress others. CD68 and Iba-1 staining was measured in stereologically sampled coronal sections across the hippocampus following published guidelines (Waters, 2009). Stained area of specific size and intensity relative to the local background was quantified in eight sections per mouse, n = 6–11 mice/group. All staining was performed and imaged on the same day for all groups. A, Representative images of CD68 staining in the CA3 region of the hippocampus 72 h after KA. Scale bar, 100 μm. Morphology of activated cells (inset). Scale bar, 40 μm. B, Iba-1 staining in the next subsequent section. Scale bar, 100 μm. Morphology of activated cells (inset). Scale bar, 40 μm. C, CD68 staining area was increased by prior GC treatment in both WT and MGRKO mice. **p < 0.01, two-way ANOVA, Bonferroni post hoc test (D) CD68 pixel intensity was increased by GC treatment in WT but not MGRKO mice. *p = 0.03, significant interaction between treatment and genotype, two-way ANOVA. *p < 0.05, ***p < 0.01, two-way ANOVA, Bonferroni post hoc tests. E, Iba-1 staining area was also increased by prior GC treatment and was nearly significantly increased in MGRKO mice relative to WT.*p < 0.05, two-way ANOVA main effect, genotype main effect p = 0.058. F, Iba-1 pixel intensity was increased in MGRKO mice relative to WT, and was nearly significantly increased by prior GC treatment.*p < 0.05, two-way ANOVA main effect, GC main effect p = 0.0515. (C–F, n = 4–11 per group.) G–M, CNS myeloid cells from WT and MGRKO mice were isolated 24 h after MCAO on a Percoll gradient and analyzed via flow cytometry for expression of CD45, LY6G, and CD11b. Peripheral blood mononuclear cells in J were analyzed using the same staining panel and a similar gating strategy. G, Events were first gated based on forward and side scatter (G1) to eliminate debris. H, Cells from G1 were gated for CD11b+ and CD45+ signal (G2). I, The G2 gate was split into three populations for CNS isolations using the granulocyte marker Ly6G and relative expression of CD45 (high or low). Granulocytes (P1) were defined as Ly6G+ and CD45 low. Monocytes isolated from the CNS were Ly6G− and were split into a CD45 high population (P2), which is infiltrating mononuclear cells and a CD45 low population (P3) that is largely microglia The abundance of these populations was calculated as a percentage of the G2 gate (CD11b+ CD45+). J, To analyze peripheral blood mononuclear cells, a similar gating strategy was performed to define CD11b+CD45+ cells as gate G2, with the primary difference being the absence of the P3 population. Within the CD11b+CD45+ G2 population, Ly6G+ granulocytes were significantly elevated by GC treatments alone, whereas Ly6G− monocytes were significantly reduced by prior GC treatments. ***p < 0.001, two-tailed t tests. n = 4–5 mice per group. K, In the injured CNS, prior GC treatments also increased granulocytes (P1) in both the infarcted and contralateral hemispheres in both WT and MGRKO mice. **p < 0.01, two-way ANOVA, Bonferroni post hoc tests. This population was also increased in the infarcted hemisphere over the contralateral hemisphere ***p < 0.001, two-way ANOVA main effect. L, Prior GCs also reduced the infiltrating monocyte cell population (P2) in the infarcted hemisphere in WT, but not MGRKO mice. ***p < 0.01, two-way ANOVA, Bonferroni post hoc test. *p = 0.0264, significant interaction between prior GC treatment and genotype in the ipsilateral hemisphere. This population was also higher in the infarcted hemisphere than the contralateral ***p < 0.001, two-way ANOVA main effect. M, Microglia (P3) were significantly lower in the infarcted hemisphere compared with the uninjured contralateral hemisphere **p < 0.01 two-way ANOVA main effect. GCs did not significantly affect this population. (K–M, n = 8–16 per group). Data shown as mean ± SEM.
The next serial section was stained for Iba-1, to reveal all myeloid cells, not just activated ones (Fig. 2B,E,F). As for CD68 area, prior GC treatment significantly increased Iba-1 area and nearly its intensity. Unlike CD68, these increases did not require myeloid GR to occur (compare GC-treated WT and MGRKO). In contrast, the endogenous GC activation of myeloid GR suppressed Iba-1 intensity and nearly its area (compare vehicle-treated WT and MGRKO).
GCs have opposing effects on granulocytes and activated monocytes after MCAO
To measure myeloid cell responses after MCAO, mononuclear cells were isolated from the infarcted and contralateral hemispheres 24 h after injury on a Percoll gradient and labeled with CD11b, CD45, and Ly6G antibodies for analysis by flow cytometry (Fig. 2 G–M). These antibodies were used to differentiate the mononuclear fraction into granulocytes, infiltrating monocytes, and microglia (Fig. 2G–I), all of which express the LysM-cre driver and are lacking GR in MGRKO mice (Bhattacharyya et al., 2007).
To first measure the effects of our GC treatments on peripheral leukocyte populations we examined the peripheral blood composition in vehicle and GC-treated WT mice at the time when the neurological injury would have been given. The CD11b+CD45+ peripheral blood mononuclear cell population was subdivided into granulocytes (Ly6G+) and monocytes (Ly6G−). Prior GC treatments increased the abundance of granulocytes (CD11b+ CD45+ Ly6G+) and decreased monocyte (CD11b+ CD45+ Ly6G−) numbers in the peripheral blood (Fig. 2J), so we next investigated whether these changes were also reflected in the injured CNS.
Prior GC treatments also significantly increased granulocyte (P1) abundance in both the infarcted and contralateral hemispheres in WT and MGRKO mice alike (Fig. 2K). Endogenous GR signaling in granulocytes was not required for their normal response (compare vehicle-treated WT and MGRKO). In contrast, prior GC treatments reduced the number of infiltrating monocytes (P2) in the infarcted hemisphere (Fig. 2L), an effect that was not present in MGRKO mice, suggesting that it requires myeloid GR to occur. Finally, GCs did not significantly affect the CD45lo microglia (P3) (Fig. 2M), but this population was reduced in the infarcted hemisphere most likely reflecting the influx of activated cells, and the activation of endogenous CNS myeloid cells.
Thus, prior GCs increased the abundance of granulocytes, but suppressed the numbers of monocytes both in peripheral blood and in the CNS after MCAO. Only the suppressive effect of GCs required myeloid GR signaling to occur; the GC-stimulated granulocyte numbers were myeloid cell GR-independent.
GR signaling in myeloid cells increases NFkB nuclear localization after KA and MCAO
To investigate the cellular mechanisms behind how GCs were stimulating and suppressing myeloid cells after injury, we next measured nuclear p65 levels in the injured hippocampus and infarcted hemisphere. Prior GC treatments increased nuclear p65 levels following both injuries (Fig. 3A,B,E,F), and myeloid GR was required for this to occur either 72 h after KA or 24 h after MCAO, but not 12 h after KA (Fig. 3A,B, compare vehicle and GC-treated MGRKO). Nuclear p65 levels were also increased by endogenous GR signaling in myeloid cells following both injuries (compare vehicle-treated WT and MGRKO). Cytosolic p65 levels were significantly reduced by prior GC treatments 12 h after KA, supporting the conclusion that the increased nuclear p65 levels at this time were due to nuclear translocation (Fig. 3C,D). We detected only traces of the cytosolic protein GAPDH in the nuclear fraction, indicating successful enrichment of the nuclear fraction (Fig. 3E). To see how these p65-activating GC effects altered cytokine production, we next measured IL-1β and IL-6 levels in the injury site.
Figure 3.

Both prior GC treatments and endogenous GCs increase p65 nuclear localization. A, Two representative bands per treatment from Western blots for the p65 subunit of NF-kB in nuclear extracts from the injured hippocampus at 12 or 72 h after KA. Lamin B1 was used as a load control for the nuclear fraction. n = 3–6 mice per treatment group. B, Quantification of nuclear p65 band density at different times after KA normalized to lamin B1 and expressed as a percentage of 12 h WT levels. At 12 h after KA, prior GC treatment increased p65 nuclear localization in both WT and MGRKO mice; however, this did not happen in MGRKO mice at 72 h. Nuclear p65 levels were also significantly higher in WT mice than MGRKO mice. The interaction was nonsignificant at both times. C, Representative Western blots for p65 from the cytosolic fraction using β-tubulin as a load control. D, Quantification of cytosolic p65 band density at 12 and 72 h after KA, normalized to β-tubulin, and expressed as a percentage of 12 h WT levels. Prior GC treatment decreased p65 levels in the cytosol in both WT and MGRKO mice at 12 h, but this reduction was not significant at 72 h. **p = 0.004, two-way ANOVA main effect. Cytosolic p65 levels were not significantly higher in MGRKO mice than WT mice. n = 3–4 mice per group. E, Western blotting for the p65 subunit of NF-kB in the nuclear fraction isolated from the infarcted hemisphere 24 h after MCAO. Two representative bands per treatment are shown and lamin B1 is shown as a load control for the nuclear fraction. n = 5–6 mice per group. F, Nuclear p65 band density 24 h after MCAO normalized to lamin B1. GC treatment increased p65 nuclear localization in WT but not MGRKO mice. Nuclear p65 levels were significantly lower in MGRKO mice than WT mice. The interaction was nonsignificant (p = 0.089). G, Representative Western blots demonstrating successful nuclear and cytosolic fractionation in the different treatment groups. The cytosolic protein GAPDH is present only at low levels in the enriched nuclear fraction. *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA, Bonferroni post hoc tests.
GCs have bidirectional effects on cytokine levels after injury
We next examined production of the cytokines IL-1β and IL-6 after injury to see which stimulatory and suppressive GC effects required myeloid GR (Fig. 4). Prior GC exposure can increase levels of IL-1β following KA (Dinkel et al., 2003; MacPherson et al., 2005), and we observed the same outcome after KA (Fig. 4A), but not MCAO (Fig. 4B). This outcome did not require myeloid GR to occur (compare vehicle and GC-treatments in MGRKO mice). In contrast, endogenous GCs did suppress IL-1β via myeloid GR as evidenced by MGRKO mice having higher IL-1β levels after both injuries. Prior GC treatments increased IL-6 levels after KA, but only in MGRKO mice, suggesting a suppressive effect of myeloid GR on this cytokine (Fig. 4C). In contrast, prior GCs suppressed IL-6 levels after MCAO, and this effect required myeloid GR to occur (Fig. 4D).
Figure 4.

GCs have mixed stimulatory and suppressive effects on IL-1β and IL-6 after KA and MCAO. A, IL-1β levels in the injured hippocampus at 12 h after KA were increased by prior GC treatments. IL-1β levels were also significantly higher in MGRKO mice than WT, *p = 0.018 for GC treatments and *p = 0.026 for genotype, two-way ANOVA main effects. By 72 h after KA, IL-1β levels were negligible and not significantly higher in MGRKO mice. B, IL-1β levels in the infarcted hemisphere 24 h after MCAO were also higher in MGRKO mice than WT, *p = 0.039, two-way ANOVA main effect. Prior GC treatments had opposite effects on IL-1β levels in WT and MGRKO mice. *p < 0.05, two-way ANOVA, Bonferroni post hoc test. Interaction was nonsignificant. C, IL-6 levels in the injured hippocampus at 12 h after KA were elevated only in GC-treated MGRKO mice. **p < 0.01, two-way ANOVA, Bonferroni post hoc test. By 72 h after injury, IL-6 levels were lower and there were no significant differences between experimental groups. Interaction was nonsignificant. (p = 0.09 at 12 h). D, GCs reduced IL-6 in WT mice 24 h after MCAO, but not in MGRKO mice. Vehicle-treated MGRKO mice also had lower levels of IL-6 in the infarcted hemisphere relative to WT. *p < 0.05, **p < 0.01, two-way ANOVA, Bonferroni post hoc tests, significant interaction, p = 0.005. n = 5–8 at 12 h after KA, 3–7 at 72 h after KA, and n = 6 after MCAO.
Together, these results indicate that GCs can have both stimulatory and suppressive net effects on the levels of cytokines produced in the injury site, only some of which are due to their direct effects on myeloid cells. This led us to next investigate how the cell-specific effects of GCs on other aspects of the injury response might influence GC effects on inflammation. For example, reduced production of neurotrophins like BDNF could result in reduced neuron survival leading to increased inflammation.
GCs suppress BDNF release in a cell-specific manner
In contrast to the mixed effects of GCs on cytokine signaling in these injuries, their effects on levels of BDNF were more consistent. Following KA-injury, GC treatments suppressed BDNF levels in WT and MGRKO mice alike (Fig. 5A), but FBGRKO mice had reduced BDNF levels that could not be further reduced by GC treatments (Fig. 5B). The same GC suppressive effects on BDNF levels were also observed following MCAO; however, in this injury model, myeloid GR was required for GCs to suppress BDNF levels and neuron GR was not required (Fig. 5C,D). Endogenous GC signaling in neurons had opposite effects on BDNF levels in these two injury models, increasing BDNF after KA and reducing it after MCAO (Fig. 5B,D, compare vehicle-treated WT and FBGRKO). Prior GCs most consistently reduced BDNF, and could therefore be acting on myeloid cells in both direct and indirect ways to increase different inflammatory mediators.
Figure 5.

GCs have cell-specific suppressive effects on BDNF levels after KA and MCAO. A, BDNF levels in the injured hippocampus at 12 h after KA were suppressed by GC treatments in both WT and MGRKO mice, **p < 0.005, two-way ANOVA main effect. At 72 h after KA, BDNF levels were nearly significantly reduced by GC treatments, p = 0.056, two-way ANOVA main effect. B, Vehicle-treated FBGRKO mice had significantly lower BDNF levels 12 h after KA. GCs suppressed BDNF levels in WT, but not FBGRKO mice. *p < 0.05, two-way ANOVA, Bonferroni post hoc tests. C, BDNF levels after MCAO were reduced by GC treatment in WT but not MGRKO mice. *p < 0.05, two-way ANOVA, Bonferroni post hoc test. Interaction was nonsignificant (p = 0.08). D, BDNF levels after MCAO were elevated in FBGRKO mice relative to WT, and were suppressed by GCs in both WT and FBGRKO mice. *p < 0.05, **p < 0.01, two-way ANOVA, Bonferroni post hoc tests. n = 5–8 at 12 h after KA, 3–7 at 72 h after KA, and n = 6 after MCAO.
GCs exert opposing, injury-specific effects on CX3CL1
We next considered the possibility that the myeloid cell-activating effects of GCs that we observed were further compounded by GC effects on anti-inflammatory homeostatic mechanisms within the CNS. One example of a system like this is the soluble chemokine CX3CL1, a cellular “off” signal from neurons to microglia that maintains a less neurotoxic, reduced inflammatory response to injury (Biber et al., 2007; Lucin and Wyss-Coray, 2009; Ransohoff and Cardona, 2010). GCs can suppress CX3CL1 production in peripheral cells (Bhavsar et al., 2008), so if this occurs in the CNS, it could partially explain why GC exposure before the injury might lead to an increased inflammatory response. Additionally, prior GC exposure has previously been found to suppress other CNS proteins with similar immunosuppressive function (Frank et al., 2010).
Prior GC treatments suppressed hippocampal CX3CL1 protein levels at 12 h after KA and these effects required both myeloid and neuron GR (Fig. 6A–C). In addition, endogenous myeloid GR activation after injury suppressed CX3CL1 levels at 72 h after injury, indicated by WT mice having lower levels than MGRKO mice (Fig. 6B).
Figure 6.

GCs suppress CX3CL1 after KA but increase it after MCAO. A, Two representative bands per treatment from Western blots for CX3CL1 from the injured hippocampus at 12 and 72 h after KA. Beta-actin was used as a load control. n = 5–6 mice per group. B, Quantification of CX3CL1 band density at different times after KA normalized to β-actin and expressed as a percentage of 12 h WT levels. GC treatment reduced CX3CL1 levels in WT mice but not MGRKO mice. *p < 0.05, two-way ANOVA, Bonferroni post hoc test. After 72 h, MGRKO mice had significantly higher CX3CL1 levels than WT mice and GC treatment further increased levels in MGRKO mice. *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA, Bonferroni post hoc tests. *p = 0.015, significant interaction at 72 h, nonsignificant at 12 h, two-way ANOVA. C, GC treatments reduced CX3CL1 levels in the hippocampus 12 h after KA in WT mice but not FBGRKO mice. *p < 0.05, two-way ANOVA, Bonferroni post hoc test. Interaction was nonsignificant. D, Western blotting for CX3CL1 in the infarcted hemisphere 24 h after MCAO. Two representative bands per treatment are shown and β-actin is shown as a load control. n = 5–6 mice per group. E, CX3CL1 levels after MCAO were increased by GC treatment in both WT and MGRKO mice. CX3CL1 levels were also significantly higher in WT mice than MGRKO mice. **p < 0.01, ***p < 0.001, two-way ANOVA, Bonferroni post hoc tests. F, CX3CL1 levels after MCAO were increased by GC treatment in both WT and FBGRKO mice and were significantly higher in WT than FBGRKO mice. *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA, Bonferroni post hoc tests.
In contrast to its CNS role as a suppressive inflammatory mediator released by neurons, soluble CX3CL1 can promote the recruitment of peripheral immune cells to injury, increasing stroke infarct volume (Fong et al., 1998; Soriano et al., 2002). GC exposure before MCAO resulted in increased CX3CL1 levels after injury, an opposite result to that observed for KA (Fig. 6D–F). Endogenous GC signaling in both myeloid cells and neurons was also required for higher CX3CL1 levels, and prior GC treatments increased CX3CL1 levels independently of myeloid or neuron GR signaling (Fig. 6D–F).
These injury-specific GC effects suggested that the divergent effects of GCs on neurons and myeloid cells might lead to differences in neuron survival. To address this we next quantified neuron death following each injury.
GR signaling in myeloid cells endangers neurons
Quantification of neuron death revealed that timing of GC-exposure was again critical. Exposure to only the GCs secreted after injury was not damaging to neurons, as neither MGRKO nor FBGRKO mice were protected from excitotoxic cell death relative to WT littermates (Fig. 7A–F, compare vehicle-treated WT and KOs). In contrast, prior GC exposure increased lesion size in WT mice by nearly two-fold (Fig. 7A–F, compare vehicle and GC-treated WT mice). MGRKO mice were completely protected from this GC endangerment (Fig. 7A,B), but FBGRKO mice were not (Fig. 7D,E).
Figure 7.

GC-augmented neuron death after KA and after MCAO requires myeloid cell GR, but not neuronal GR. A, Representative Nissl-stained hippocampal images from WT and MGRKO mice with or without GC treatment at 72 h after injury. Scale bar, 200 μm. The CA3 region of the hippocampus is magnified on the bottom. Scale bar, 50 μm. Arrows indicate damaged regions. B, Quantification of the damaged area in the hippocampal pyramidal layer. GC treatment (black bars) worsened excitotoxic damage in WT mice but not in MGRKO littermates. *p < 0.05, **p < 0.01, two-way ANOVA, Bonferroni post hoc tests. n = 6–11 mice per group. Treatment and genotype interaction, p = 0.06, two-way ANOVA. C, GC treatment increased neuron death in mice overexpressing the GR in myeloid cells (MGROV) as well as WT controls. ***p = 0.0006, two-way ANOVA main effect. n = 8–11 mice per group. D, Representative Nissl-stained hippocampal images from WT and FBGRKO mice with or without GC treatment. Scale bar, 200 μm. The CA3 subfield of the hippocampus is magnified on the bottom. Scale bar, 50 μm. Arrows indicate damaged regions. E, Quantification of damaged area in the hippocampal pyramidal layer. GC treatment worsened excitotoxic damage in both WT and FBGRKO mice. *p = 0.033, two-way ANOVA main effect. n = 5–11 mice per group. F, GC treatment did not increase neuron death in mice overexpressing the GR in neurons (FBGROV), but did in their WT littermates. *p < 0.05, two-way ANOVA, Bonferroni post hoc test. n = 6–11 mice per group. G, Representative TTC stained tissue sections 24 h after MCAO from WT and MGRKO mice with or without GC pellets. The white infarcted region is indicated by a dotted line. H, Quantification of the infarct volume 24 h after MCAO. GC treatment significantly increased infarct volume in WT but not MGRKO littermates. **p < 0.01, ***p < 0.001, two-way ANOVA Bonferroni post hoc tests. n = 5–12 mice per group. Treatment and genotype interaction p = 0.08, two-way ANOVA. I, GC treatment increased infarct volume in both WT and MGROV mice. **p = 0.006, two-way ANOVA main effect. n = 6–9 mice per group. J, Representative TTC sections from WT or FBGRKO mice with or without GC pellets. K, Quantification of infarct volume 24 h after MCAO. GC treatment increased MCAO infarct in both WT and FBGRKO mice. *p < 0.05, ***p < 0.001, two-way ANOVA Bonferroni post hoc tests. n = 6–9 mice per group. L, GC treatment also increased MCAO infarct in both WT and FBGROV mice. **p < 0.01, ***p < 0.001, two-way ANOVA Bonferroni post hoc tests. n = 5–15 mice per group.
To investigate whether increased GR signaling was cell-autonomously sufficient to worsen neuron death after KA, we next measured neuron death in MGROV and FBGROV mice. Prior GC exposure again worsened neuron death in WT mice (Fig. 7C,F), but myeloid GR overexpression did not alter this (Fig. 7C). In contrast, neuronal GR overexpression caused a near-significant increase in damage in vehicle-treated animals (Fig. 7F).
To assess stroke damage, brains were stained with TTC 24 h after MCAO and total infarct volume was quantified (Fig. 7G–L). All mice had significant cortical damage but some GC-treated mice had damage extending into the striatum (Fig. 7G,J). As was the case with KA, prior GCs worsened MCAO damage in WT mice (Fig. 7H,I,K,L). Also similar to KA, this was dependent on myeloid GR, but not neuronal GR (Fig. 7H,K, compare vehicle and GC-treated KOs). Neither neuron nor myeloid GR overexpression had a significant effect on infarct volume (Fig. 7I,L).
Because MCAO injury involves significant vascular pathology and GCs are frequently given for their effects on the BBB, we next tested how GC signaling in endothelial cells influenced neuron death after MCAO.
GR signaling in endothelial cells reduces BBB protein levels
Necrotic insults such as MCAO compromise the integrity of the BBB, with adverse consequences for neurons. GCs classically reduce BBB permeability by increasing the production of tight-junction proteins (Förster et al., 2005). We next used our cell-specific GR manipulations to investigate whether GR signaling in endothelial cells after ischemia actually increases several different components of the BBB. The parents of Tie2-GRKO mice had low fecundity, so we used Tie2-GROV mice as an alternative to prior GC treatment to measure the effects of increased endothelial-GR signaling.
We subjected Tie2-GRKO and Tie2-GROV mice to MCAO and examined levels of (1) occludin and claudin 5, the two primary proteins in BBB tight junctions, (2) caveolin 1, an integral part of the transcytotic pathway, and (2) the glucose transporter glut-1 (Fig. 8A–E). Contrary to the expectation that GCs increase production of proteins involved in BBB integrity, Tie2-GRKO mice had increased levels of occludin (Fig. 8B), claudin 5 (Fig. 8C), and caveolin 1 (Fig. 8D), suggesting that endogenous GCs released after injury signal through endothelial cell GR to reduce levels of these proteins. In addition, occludin and caveolin 1 were further suppressed in Tie2-GROV mice relative to WT, indicating that GC signaling via endothelial GR is both necessary and sufficient to suppress the production of these proteins after MCAO. We observed only a trend toward GC suppression of endothelial glut-1 (Fig. 8E) despite GC suppression of this protein in peripheral endothelial cells (Olgemöller et al., 1985). These GC effects on endothelial cell proteins were counter to their classic anti-inflammatory effects on the BBB and suggest an additional mechanism by which GCs might contribute to worsened ischemic infarct.
Figure 8.

Endothelial cell GR signaling decreases BBB protein levels after MCAO. A, Two representative bands from Western blots for the endothelial cell proteins occludin, claudin-5, caveolin-1, and glut-1 in the infarcted hemisphere 24 h after MCAO. Protein levels were quantified in WT (Tie2-cre+/−::GRflox/WT, left gray bars) and Tie2-GRKO (Tie2-cre+/−::GRflox/null, open bars) mice, and overexpression WT (ROSA-LSL-rtTA::TetO-rGR, right gray bars) and Tie2-GROV (Tie2-cre+/−::ROSA-LSL-rtTA::TetO-rGR, black bars) mice normalized to β-actin as a loading control and expressed as a percentage of the corresponding WT. n = 5 mice per group. B, Tie2-GRKO mice had higher occludin levels than WT mice, which in turn had higher occludin levels than Tie2-GROV mice. *p = 0.012, ***p = 0.0002 unpaired, two-tailed t test. C, Claudin 5 levels were higher in Tie2-GRKO mice than WT littermates and GR overexpression had no effect. **p = 0.0014, unpaired, two-tailed t test. D, Caveolin protein levels were significantly increased in Tie2-GRKO mice, (**p = 0.008, unpaired, two-tailed t test) and reduced in Tie2-GROV mice. (**p = 0.0017, unpaired, two-tailed t test). E, Glut-1 protein levels were not significantly altered by these GR manipulations in endothelial cells. (p = 0.059, unpaired, two-tailed t test). F, Representative Western blots for occludin and claudin-5 in WT and MGRKO mice 24 h after MCAO. There were no observable differences in the levels of these two BBB proteins between WT and MGRKO mice. G, Representative TTC stained tissue sections 24 h after MCAO from Tie2-GRKO and Tie2-GROV mice and WT controls. The white infarcted region is indicated by a dotted line. H, Quantification of the infarct volume. Tie2-GRKO mice had smaller infarcts than WT control mice. *p = 0.037, unpaired, two-tailed t test. n = 7 mice per group. I, GC treatment worsened infarct volume in Tie2-GROV mice and WT controls alike. ***p < 0.0001, two-way ANOVA main effect. Tie2-GROV mice had a nearly significant increase in infarct relative to WT. p = 0.057, two-way ANOVA main effect.
One potential problem with the Tie2-cre GR mice is that the Tie2 promoter can also express in the myeloid lineage (Koni et al., 2001). To determine whether GR signaling in myeloid cells might affect these BBB proteins we examined occludin and claudin-5 levels in WT and MGRKO mice 24 h after MCAO (Fig. 8F). The levels of these proteins were the same in WT and MGRKO mice, suggesting that their increased expression was not due to myeloid GR signaling. We next measured the infarct volume in these mice after MCAO to see how these changes affected neuron survival.
Stroke infarct is worsened by GR signaling in myeloid and endothelial cells
Infarct volume was quantified in Tie2-GRKO and Tie2-GROV mice (Fig. 8G–I). Deletion of endothelial cell GR decreased infarct volume (Fig. 8G,H), and overexpression of endothelial cell GR produced a strong trend toward increased infarct volume (Fig. 8I, compare open bars). As in MGROV and FBGROV mice, prior GCs worsened infarct in WT and Tie2-GROV mice alike (Fig. 8G,I). From this we inferred that GR signaling in endothelial cells, whether triggered by endogenous GC secretion after ischemia or prior increases in GR levels, is detrimental to neuron survival.
Discussion
GCs are some of the most potent and widely used anti-inflammatory compounds known, making it important to characterize the situations where they are not effective or even worse, have opposite effects (Dinkel et al., 2003; Frank et al., 2007, 2010; de Pablos et al., 2006; Munhoz et al., 2010). This study used clinically relevant injury models to identify which of these unusual proinflammatory GC effects on inflammation are mediated directly via GR signaling in myeloid cells themselves. Together, our findings support the hypothesis that GCs can act directly on myeloid cells to increase some indicators of their activation, an unexpected result given the classical anti-inflammatory actions of this receptor in immune cells. Furthermore, the cellular and temporal context of GR signaling is crucial in determining how GCs will affect the immune response. GCs also had secondary effects on nonmyeloid cells to indirectly increase inflammation, including reduced neurotrophin levels and effects on endothelial cells to decrease levels of proteins that control BBB patency. Finally, GCs worsened neuron death during these injuries and this was mediated more by their signaling in myeloid and endothelial cells than by their signaling in injured neurons themselves.
Following any acute injury, GCs are released into the blood stream where they activate GR signaling in cells throughout the body. Following excitotoxic brain injury, circulating GC levels are in the high stress range within an hour, fall into the moderate-stress range by 3 h, and remain there for three more hours before returning to baseline (Stein and Sapolsky, 1988). The resulting widespread transcriptional changes suppress immune responses as a part of the recovery from injury-induced immune activation (Yeager et al., 2004). Despite the classical expectation that endogenous secretion of GCs should suppress immune cell activation, we identified several notable instances of the opposite following both injury models (summarized in Fig. 9). Endogenous GC-stimulated myeloid GR signaling after KA decreased the anti-inflammatory signaling of CX3CL1 and increased nuclear p65 levels in the injury site. Following MCAO, endogenous GC-stimulated myeloid GR signaling was required for myeloid cells to migrate to the injury site and produce more IL-6. In neither injury model, however, did these endogenous GC effects substantially affect neuron death. This result may reflect the balance between the need for immune recruitment to protect neurons and the detrimental effects of excessive or prolonged immune activation (El Khoury et al., 2007). Finally, this study did not address the role of the GC-binding mineralocorticoid receptor (MR), but others have shown that endogenous GC activation of MR signaling in myeloid cells also significantly worsens ischemic injury and increases the production of proinflammatory cytokines (Frieler et al., 2011).
Figure 9.

Summary of GC effects on immune responses to excitotoxicity and ischemia. Summary of how GCs affected each inflammatory mediator, broken into injury type (KA or MCAO), timing of GC exposure (before or after injury), and cellular context (neuron, myeloid, or endothelial cell). Up and down arrows indicate the direction of significant changes determined in this study.
In contrast, GC exposure before an injury has previously been shown to augment aspects of the subsequent inflammatory response to several types of CNS injury (Dinkel et al., 2003; MacPherson et al., 2005; Frank et al., 2007, 2010; Munhoz et al., 2006, 2010; de Pablos et al., 2006). Our results determined which of these proinflammatory effects are mediated directly by myeloid GR signaling and which are indirect effects of GCs on other cells. Of these myeloid GR-dependent effects, we found GR signaling to be necessary for both pro-inflammatory and anti-inflammatory changes depending on the immune activation marker measured. For example, despite the fact that myeloid GR signaling suppressed many proinflammatory cytokines in a classical manner, it increased other indicators of immune activation like p65 and CD68, particularly in the later phase of the response to injury. Unlike the GCs released by the injury, this prior exposure to GCs for as little as 3 d promoted an altered myeloid cell activation state that was detrimental to neuron survival.
We found that prior GC exposure also affected neuronal regulation of myeloid cell responses by altering the production of the protein CX3CL1. Myeloid GR signaling regulated the production of this chemokine oppositely between the two injuries we studied. During excitotoxic injury, prior GC exposure suppressed CX3CL1 production in a myeloid GR-dependent manner. This decrease could explain the increased myeloid cell activation measured by CD68 and nuclear p65 levels. The fact that myeloid GR was necessary for a decrease in the release of a neuronal protein suggests that myeloid cells may communicate to neurons in a feedback loop, a common theme with cytokine signaling (Ransohoff et al., 2007). During hypoxic-ischemic injury, however, prior GC exposure increased CX3CL1 production and this increase did not require myeloid GR. These myeloid GR-independent effects may instead be mediated by GR in neurons directly. During ischemia, such increases in CX3CL1 are thought to lead to greater recruitment of damaging peripheral cells, increasing infarct volume (Fong et al., 1998; Soriano et al., 2002). Our results suggest that in addition, the cells that arrive are more activated in certain ways.
The type of immune activation that we found to result from prior-GC exposure was not observed across all inflammatory mediators, implicating influences that could come from GC effects on other cell types. Increased NF-kB activation (Cho et al., 2008) and lower CX3CL1 levels (Cardona et al., 2006b) would both be expected to increase proinflammatory cytokine levels, but these effects were inconsistent between injury models. It is possible that GCs can suppress proinflammatory cytokine production independently of their activating effects on other inflammatory mediators in ways that could depend on the type of injury. Elevated nuclear p65 levels are predictive of GC-augmented p65 DNA-binding activity in vivo (Munhoz et al., 2006), but GR also exerts effects via protein–protein interactions with transcription factors like p65. It is unclear whether GC-augmented inflammation results from a disruption of inhibitory protein–protein interactions or increased DNA binding by p65.
Prior exposure to GCs caused approximately twice the amount of neuron death in both injury models and this required myeloid GR signaling, but not neuron GR signaling. Increased myeloid cell NF-kB signaling is responsible for the majority of neuron death in both excitotoxicity and ischemia (Cho et al., 2008); however, this is thought to occur via increased release of neurotoxic proinflammatory cytokines (Cardona et al., 2006b; Cho et al., 2008). Instead, GC-augmented inflammation is characterized by a state of activated cellular responses in the absence of consistently elevated proinflammatory cytokines. It is known that GCs decrease neuron survival by reducing glutamate re-uptake, calcium sequestration, and reactive oxygen species scavenging (Elliott and Sapolsky, 1993; Roy and Sapolsky, 2003). Perhaps then, it is the effects of GCs on myeloid cells that are required to worsen these energetic catastrophes in neurons. Indeed, activated microglia produce oxygen radicals, which can reduce glutamate re-uptake (Trotti et al., 1996).
Whether GCs increase or decrease inflammatory cell activation may also differ between brain regions. GCs induce classical anti-inflammatory effects after injury in subcortical regions like the substantia nigra (Ros-Bernal et al., 2011) and hypothalamus (Munhoz et al., 2006), whereas their immune-activating properties have been primarily reported in forebrain regions like the hippocampus and frontal cortex (de Pablos et al., 2006; Munhoz et al., 2010). In these brain regions, even chronic exposure to GCs (1–2 weeks) can be immune activating, particularly if it occurs before the injury (Munhoz et al., 2006).
GCs ameliorate vascular edema in brain tumors (Gomes et al., 2005) by directly inducing transcription of the BBB tight junction protein occludin (Förster et al., 2005) in the endothelial cells of the BBB, thereby decreasing BBB permeability (Förster et al., 2006; Silwedel and Förster, 2006). GCs are less frequently helpful in reducing poststroke edema (Gomes et al., 2005), however, so we measured the effects of endothelial GR signaling on several BBB proteins after MCAO. In contrast to the picture with brain tumors, the tight junction proteins occludin and claudin 5 were reduced by endothelial GR signaling after ischemia, as was the transcytotic protein caveolin 1. Such effects would be expected to contribute to reduced BBB integrity, increased permeability, and the worsened neuron death we observed. Because the Tie2/TEK promoter also expresses in myeloid cells, the possibility cannot be excluded that some GC suppression of BBB proteins might be mediated via myeloid cells rather than exclusively through endothelial GR. At least for occludin and caveolin-1, however, GR overexpression in these cell types was also sufficient to suppress their levels. GC effects on myeloid cells and endothelial cells may have similar consequences, because the neuron death outcomes we observed in LysM-cre mice were only further exacerbated in Tie2-cre mice. These results indicate that endothelial GR signaling reduces the production of proteins involved in BBB patency following ischemic brain injury, likely contributing to increased neuron death.
Together, these results demonstrate that GCs have both myeloid-dependent and myeloid-independent effects to increase or decrease different aspects of the inflammatory response during two common acute CNS injuries. Moreover, GC effects on myeloid cells have detrimental consequences for neuron survival. The notion that GCs are only anti-inflammatory is becoming outdated as new approaches permit a more comprehensive picture of what happens during acute CNS injury. If these observations are also true in humans, then there may be critical implications for the widespread clinical use of GCs.
Footnotes
This work was funded by NIH Grants 1F31NS063491 to S.F.S. and 1R01NS059918 to R.M.S. J.R.C. was a recipient of a MICINN postdoctoral fellowship. We thank Louis Muglia for the generous gift of floxed-GR mice and Sam Okret for the generous gift of TRE-rGR mice. Nathan Manley, Trevor Sorrells, and Joanna Lee provided helpful suggestions for this manuscript. This work is dedicated to the memory of Angela Riepel who made significant contributions to this work and passed away on August 29th, 2011.
The authors declare no competing financial interests.
References
- Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. 2007;109:4313–4319. doi: 10.1182/blood-2006-10-048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar PK, Sukkar MB, Khorasani N, Lee KY, Chung KF. Glucocorticoid suppression of CX3CL1 (fractalkine) by reduced gene promoter recruitment of NF-kappaB. FASEB J. 2008;22:1807–1816. doi: 10.1096/fj.07-094235. [DOI] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A. 2005;102:473–478. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JA, Khor B, Vogt SK, Muglia LM, Fujiwara H, Haegele KE, Sleckman BP, Muglia LJ. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. 2003;9:1318–1322. doi: 10.1038/nm895. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat Protoc. 2006a;1:1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006b;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Cho IH, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim CH, Kim DW, Jo EK, Lee KE, Karin M, Lee SJ. Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain. 2008;131:3019–3033. doi: 10.1093/brain/awn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2–13. doi: 10.1016/j.mce.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pablos RM, Villáran RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006;26:5709–5719. doi: 10.1523/JNEUROSCI.0802-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16:300–317. doi: 10.1159/000216188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Stress-induced changes in blood leukocyte distribution. Role of adrenal steroid hormones. J Immunol. 1996;157:1638–1644. [PubMed] [Google Scholar]
- Dinkel K, MacPherson A, Sapolsky RM. Novel glucocorticoid effects on acute inflammation in the CNS. J Neurochem. 2003;84:705–716. doi: 10.1046/j.1471-4159.2003.01604.x. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Elliott EM, Sapolsky RM. Corticosterone impairs hippocampal neuronal calcium regulation-possible mediating mechanisms. Brain Res. 1993;602:84–90. doi: 10.1016/0006-8993(93)90245-I. [DOI] [PubMed] [Google Scholar]
- Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster C, Silwedel C, Golenhofen N, Burek M, Kietz S, Mankertz J, Drenckhahn D. Occludin as direct target for glucocorticoid-induced improvement of blood–brainbarrier properties in a murine in vitro system. J Physiol. 2005;565:475–486. doi: 10.1113/jphysiol.2005.084038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster C, Waschke J, Burek M, Leers J, Drenckhahn D. Glucocorticoid effects on mouse microvascular endothelial barrier permeability are brain specific. J Physiol. 2006;573:413–425. doi: 10.1113/jphysiol.2006.106385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. 2010;24:19–30. doi: 10.1016/j.bbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Frieler RA, Meng H, Duan SZ, Berger S, Schütz G, He Y, Xi G, Wang MM, Mortensen RM. Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke. 2011;42:179–185. doi: 10.1161/STROKEAHA.110.598441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes JA, Stevens RD, Lewin JJ, 3rd, Mirski MA, Bhardwaj A. Glucocorticoid therapy in neurologic critical care. Crit Care Med. 2005;33:1214–1224. doi: 10.1097/01.CCM.0000166389.85273.38. [DOI] [PubMed] [Google Scholar]
- Kaufer D, Ogle WO, Pincus ZS, Clark KL, Nicholas AC, Dinkel KM, Dumas TC, Ferguson D, Lee AL, Winters MA, Sapolsky RM. Restructuring the neuronal stress response with anti-glucocorticoid gene delivery. Nat Neurosci. 2004;7:947–953. doi: 10.1038/nn1296. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Meldgaard M, Ladeby R, Finsen B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:119–135. doi: 10.1038/sj.jcbfm.9600014. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson A, Dinkel K, Sapolsky R. Glucocorticoids worsen excitotoxin-induced expression of pro-inflammatory cytokines in hippocampal cultures. Exp Neurol. 2005;194:376–383. doi: 10.1016/j.expneurol.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, Lima Lde S, Avellar MC, Sapolsky RM, Scavone C. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. 2006;26:3813–3820. doi: 10.1523/JNEUROSCI.4398-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munhoz CD, Sorrells SF, Caso JR, Scavone C, Sapolsky RM. Glucocorticoids exacerbate lipopolysaccharide-induced signaling in the frontal cortex and hippocampus in a dose-dependent manner. J Neurosci. 2010;30:13690–13698. doi: 10.1523/JNEUROSCI.0303-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23:5536–5544. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgemöller B, Schön J, Wieland OH. Endothelial plasma membrane is a glucocorticoid-regulated barrier for the uptake of glucose into the cell. Mol Cell Endocrinol. 1985;43:165–171. doi: 10.1016/0303-7207(85)90080-2. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, Alvarez-Fischer D, Carrillo-de Sauvage MA, Saurini F, Coussieu C, Kinugawa K, Prigent A, Höglinger G, Hamon M, Tronche F, Hirsch EC, Vyas S. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A. 2011;108:6632–6637. doi: 10.1073/pnas.1017820108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Sapolsky RM. The exacerbation of hippocampal excitotoxicity by glucocorticoids is not mediated by apoptosis. Neuroendocrinology. 2003;77:24–31. doi: 10.1159/000068337. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Pulsinelli WA. Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications. Science. 1985;229:1397–1400. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/er.21.1.55. [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia. 2008;56:659–674. doi: 10.1002/glia.20644. [DOI] [PubMed] [Google Scholar]
- Silwedel C, Förster C. Differential susceptibility of cerebral and cerebellar murine brain microvascular endothelial cells to loss of barrier properties in response to inflammatory stimuli. J Neuroimmunol. 2006;179:37–45. doi: 10.1016/j.jneuroim.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Smith-Swintosky VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab. 1996;16:585–598. doi: 10.1097/00004647-199607000-00008. [DOI] [PubMed] [Google Scholar]
- Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/S0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64:33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein BA, Sapolsky RM. Chemical adrenalectomy reduces hippocampal damage induced by kainic acid. Brain Res. 1988;473:175–180. doi: 10.1016/0006-8993(88)90332-0. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem. 1996;271:5976–5979. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- Vielkind U, Walencewicz A, Levine JM, Bohn MC. Type II glucocorticoid receptors are expressed in oligodendrocytes and astrocytes. J Neurosci Res. 1990;27:360–373. doi: 10.1002/jnr.490270315. [DOI] [PubMed] [Google Scholar]
- Viswanathan K, Dhabhar FS. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc Natl Acad Sci U S A. 2005;102:5808–5813. doi: 10.1073/pnas.0501650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC. Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol. 2009;185:1135–1148. doi: 10.1083/jcb.200903097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand. 2004;48:799–813. doi: 10.1111/j.1399-6576.2004.00434.x. [DOI] [PubMed] [Google Scholar]