

Abstract
Rickettsiae are arthropod-borne intracellular bacterial pathogens that primarily infect the microvascular endothelium leading to systemic spread of the organisms and the major pathophysiological effect, increased microvascular permeability, and edema in vital organs such as the lung and brain. Much work has been done on mechanisms of immunity to rickettsiae, as well as the responses of endothelial cells to rickettsial invasion. However, to date, no one has described the mechanisms of increased microvascular permeability during acute rickettsiosis. We sought to establish an in vitro model of human endothelial-target rickettsial infection using the etiological agent of Rocky Mountain spotted fever, Rickettsia rickettsii, and human cerebral microvascular endothelial cells. Endothelial cells infected with R. rickettsii exhibited a dose-dependent decrease in trans-endothelial electrical resistance, which translates into increased monolayer permeability. Additionally, we showed that the addition of pro-inflammatory stimuli essential to rickettsial immunity dramatically enhanced this effect. This increase in permeability correlates with dissociation of adherens junctions between endothelial cells and is not dependent on the presence of nitric oxide. Taken together, these results demonstrate for the first time that increased microvascular permeability associated with rickettsial infection is partly attributable to intracellular rickettsiae and partly attributable to the immune defenses that have evolved to protect the host from rickettsial spread.
Keywords: Bacterial infection, cytokines, nitric oxide, microvascular permeability, rickettsiae
Introduction
Rickettsioses are arthropod-borne diseases caused by Gram-negative obligately intracellular bacteria that primarily reside in the cytoplasm of an infected host cell [1, 2]. These diseases are prevalent in many parts of the world and include Rocky Mountain spotted fever (RMSF), bouton-neuse fever, epidemic and endemic typhus caused by Rickettsia rickettsii, R. conorii, R. prowazekii, and R. typhi, respectively. RMSF usually results in an untreated case fatality rate of ~25%, making it the most lethal tick-transmitted bacterial infection in the USA [3].
The main target is the microvascular endothelium, leading to a disseminated infection and severe complications including vasogenic cerebral edema and non-cardiogenic pulmonary edema [4-6]. These two complications are responsible for most of the morbidity and mortality of RMSF. Immunohistochemical studies performed on human tissues and animal models of rickettsioses reveal the presence of rickettsiae in the microvascular endothelium of all major organs [7-9]. The lungs show interstitial pneumonitis and presence of proteinaceous fluid in the alveolar spaces. In severe cases, diffuse alveolar damage occurs [10]. Studies from brain tissues show rickettsial organisms in the microvascular endothelium, perivascular inflammation composed of mononuclear inflammatory cells and parenchymal edema. Presently, the mechanisms responsible for increased microvascular permeability in rickettsioses are completely unknown.
The pro-inflammatory cytokines, TNF-α, IL-1β, and IFN-γ, are essential mediators of anti-rickettsial immunity partly through the induction of nitric oxide (NO) by rickettsiae-infected endothelial cells [11-14]. In mice, depletion of TNF-α and IFN-γ results in a fatal, overwhelming infection because of decreased NO production and uncontrolled rickettsial proliferation [15]. The importance of CD8+ T-lymphocytes in vivo in clearing rickettsial infections has also been well documented [16]. C3H/HeN mice depleted of CD8 +T-lymphocytes experience a fatal, overwhelming infection when infected with normally sublethal doses of R. conorii. Taken together, these results demonstrate the importance of a host inflammatory response in clearing infection. However, the relative influence of the host immune response on the endothelial dysfunction experienced during acute rickettsioses has not been studied.
Endothelial barrier function is regulated by a series of protein complexes that exist at the intercellular borders between cells. In the brain, tight and adherens junctions form the molecular barrier that regulates the flow of solutes from the blood into the parenchyma. Two proteins of particular importance are p120-catenin (p120) and β-catenin. These two proteins serve to regulate the functional interaction of VE–cadherin with the actin cytoskeleton, thereby, establishing a molecular fence at cell junctions [17, 18]. A previous study of R. conorii infection of human umbilical vein endothelial cells (HUVEC) demonstrated disassembly of adherens junction after a prolonged period of infection [19]. To our knowledge, no one has ever described the effect of R. rickettsii infection on adherens junction stability.
The present study was conducted with the purpose of creating an in vitro model to study mechanisms of permeability across rickettsiae-infected microvascular endothelial cells derived from the human brain. In this paper, we report the effects of R. rickettsii and three important cytokines, TNF-α, IFN-γ, and IL-1β, on the permeability of human brain microvascular endothelial cell monolayers. We were able to demonstrate that R. rickettsii directly causes an increase in microvascular permeability very early after invasion of the endothelium. We also demonstrated that the addition of TNF-α, but not IL-1β or IFN-γ, alone is sufficient to enhance rickettsiae-induced microvascular permeability in a dose-dependent manner. However, a low dose of all three cytokines in combination was also capable of producing greater permeability than rickettsiae alone. Rickettsial infection and cytokine stimulation were both capable of increasing NO production by a human endothelial cell line, and this was associated with decreased rickettsial titers, whereas NO produced by our cell cultures did not overtly contribute to increased microvascular permeability. Finally, we have demonstrated that both R. rickettsii and pro-inflammatory cytokines induce a loss of adherens junction assembly at intercellular borders, and this correlates with an increase in microvascular permeability.
Methods
Reagents
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one, ODQ, was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Recombinant human TNF-α, IL-1β, and IFN-γ were purchased from R&D Systems (Minneapolis, MN, USA). Unless otherwise indicated, all reagents were purchased from Sigma Chemical Company (St. Louis, MO, USA) and cell culture medium and supplements from Invitrogen (Carlsbad, CA, USA).
Rickettsial Stocks and Cell Lines
R. rickettsii (Sheila Smith strain) was originally obtained from the laboratory of Dr. David Walker and was passaged in our laboratory one time in embryonated chicken eggs. Ten percent yolk-sac stocks of R. rickettsii suspension were propagated through two passages in Vero cell monolayers, grown in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% bovine calf serum (Hyclone, Logan, UT, USA) at 34°C and purified by Renografin density gradient centrifugation as described previously [20]. Purified light-band rickettsiae were frozen in SPG (0.128 M sucrose, 0.0038 M KH2PO4, 0.0072 M K2HPO4, 0.0049 M monosodium l-glutamic acid) buffer at −80°C. Rickettsial titers of the frozen stock were determined by plaque assay on Vero cells (ATCC CCL-81, Manassas, VA, USA).
Immortalized human cerebral endothelial cells (SV-HCEC) were a kind gift from Dr. Som Dasgupta and Dr. Robert Yu of The Medical College of Georgia [21]. This cell line was maintained in M199 medium supplemented with 10% heat-inactivated fetal calf serum, insulin–transferrin–selenium (ITS) supplement and 100 μg/ml heparin. Endothelial cells were grown on rat-tail collagen I-coated plates (BD Biosciences, San Jose, CA, USA) and used between passages 15–25.
Measurements of Transendothelial Electrical Resistance by Electric Cell-Substrate Impedance Sensing
Electric cell-substrate impedance sensing (ECIS Model 1600R, Applied Biophysics, Troy, NY, USA) was utilized with 8W10E gold-coated electrodes to monitor (TER) in real time. A decrease in TER represents an increase in paracellular permeability of endothelial monolayers and will, therefore, be referred to as an increase in vascular permeability. Electrode arrays were coated with rat-tail collagen and seeded with approximately 100,000 cells per well. The electrode arrays were then connected to the detection system, and resistance was monitored until stabilization of the resistance values was observed for at least 24 h. Typical ohms values observed for stabilized monolayers were between 1,200 and 1,400 Ω. The endothelial cell monolayers were then used for the different experiments described below. Resistance measurements were taken every 2 min, and each measurement represents the average resistance of ten electrodes per well, each measuring 250 μM in diameter. Each experiment was performed in triplicate, and the values expressed as the average normalized resistance and standard deviations were calculated every 6 h.
Nitrite Measurement
Nitrite was measured with the Greiss assay as described previously [14]. Briefly, 100 μl of filtered cell-culture supernatant was mixed with an equal volume of 1% sulfanilamide, 0.1% N-(1-napthyl)ethylenediamine in 2.5% phosphoric acid. A standard curve was generated using a nitrite standard from 100–0.78 μM, and absorbance was read at 540 nm.
Semi-Quantitative Polymerase Chain Reaction
DNA was isolated from infected cells using the Qiagen DNeasy tissue kit, and all samples were prepared in parallel. For all polymerase chain reactions (PCR), the following primers were used to amplify a 147-bp product of the rickettsial gltA gene: forward primer CS-5 (GAGA GAAATTATATCCAAATG-TTGAT) and reverse primer CS-6 (AGGGTCTTCGTGCATTTCTT) [22]. Rickettsial genome copies were normalized to the human β-actin gene using commercially available primers (SuperArray Biosciences). Real-time PCR was performed on an Eppendorf realplex2 instrument with SYBR Green and the following protocol: 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Relative copy numbers of rickettsial genomes were calculated between samples using the ΔΔCt method.
Fluorescent Confocal Microscopy
SV-HCEC monolayers were grown on plastic chamber slides until 100% confluent. The cells were then infected with R. rickettsii at a multiplicity of infection (MOI) of 10 and/or treated with the combination of TNF-α, IL-1β, and IFN-γ, each at a dose of 10 ng/ml. After 24 h of infection and stimulation, the cells were fixed in fresh 4% paraformaldehyde (Polysciences, Inc., Warrington, PA, USA) for 20 min at 4°C. After fixation, the cells were washed three times in phosphate-buffered saline (PBS) before being permeabilized with 0.05% Tween-20. The cells were blocked in 10% normal goat serum before overnight incubation with the following primary antibodies: mouse-anti-p120 (Invitrogen) and rabbit anti-β-catenin (Santa Cruz, Santa Cruz, CA, USA) both at a dilution of 1:100 in PBS. The cells were washed three times with PBS and incubated with goat-anti-mouse Alexa Fluor-568 and goat-anti-rabbit Alexa Fluor-488 (Invitrogen) both at a dilution of 1:1,000. The cells were mounted in VectaShield Hardset (Vector Laboratories, Burlingame, CA, USA), and images were acquired using an Olympus FV-1000 laser scanning confocal microscope.
Statistical Analysis
Sigma Stat v.2.03 was used for statistical analysis of the results. Differences were analyzed by performing t tests or Mann–Whitney rank sum test if indicated.
Results
R. rickettsii Causes Increased Microvascular Permeability in SV-HCEC
The effect of R. rickettsii infection on endothelial cell monolayer permeability has never been described. To this end, we sought to investigate the impact of R. rickettsii on human brain endothelial cells. Confluent SV-HCEC infected with R. rickettsii exhibited a dose-dependent increase in endothelial permeability reflected as a decrease in transendothelial electrical resistance. Figure 1a is a representative electric cell-substrate impedance sensing (ECIS) graph demonstrating the real-time loss of electrical resistance across an SV-HCEC monolayer. Permeability increased steadily after rickettsiae were internalized and continued to decline up to 72 h after infection. At 24 h, increases in permeability ranged from 12.3±2.7% at 1 MOI to 36.9±2.4% at 20 MOI relative to controls (Fig. 1b). At 48 h post-infection, monolayers infected with one MOI showed a 23.1±1.5% increase in permeability compared to controls. During the first 24 to 48 h, increases in permeability were more pronounced in monolayers infected with the highest doses, clearly demonstrating the dose dependence of the effect. Based on these experiments, further work described in this paper was based on an MOI of 10 plaque forming units (PFU). We found that this dose of rickettsiae consistently resulted in a 100% infection rate of endothelial cells early after infection with a true MOI of ~10 as determined by immunofluorescent staining within 6 h of infection.
Fig. 1.
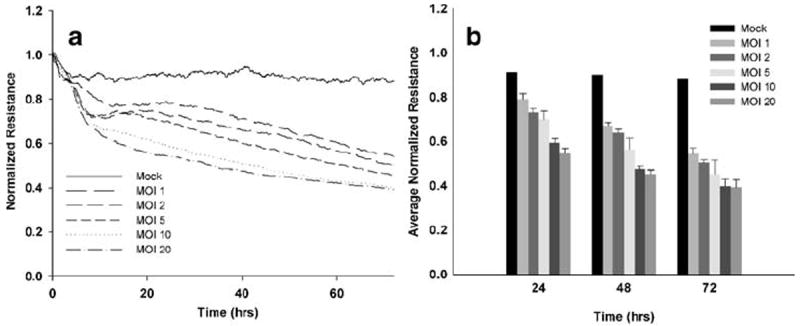
R. rickettsii caused increased microvascular permeability in human brain endothelial cells. SV-HCEC were grown to confluence on 8W10E gold-coated electrodes, and resistance was monitored by ECIS after infection with R. rickettsii at doses ranging from 1–20 MOI. A representative ECIS graph (a) and bar graphs indicating the mean ± standard deviation of three independent experiments (b).
Pro-inflammatory Cytokines Regulate Microvascular Permeability in SV-HCEC
Because TNF-α, IL-1β, and IFN-γ are well-known pro-inflammatory cytokines and have already been demonstrated to be critical to effective rickettsial immunity, we decided to determine what effect these cytokines have on uninfected endothelium. At all concentrations, IL-1β induced a rapid decline in TER by 10 h after stimulation, resulting in ~20% increase in permeability compared to untreated controls (Fig. 2a). By 24 h after stimulation, this effect ranged from a 15±5.4% increase at 0.1 ng/ml to 31.8± 13% at 1,000 ng/ml compared to controls (Fig. 2b). The effect of the lowest dose of 0.1 ng/ml was only transient, and those cells recovered to near normal levels of permeability by 72 h after stimulation with an average increase in permeability not statistically different than control cells. However, monolayers treated with higher doses showed a steady increase in permeability, which reached values between 30–45% at concentrations of 1 ng/ml (29.12±0.12%), 10 ng/ml (25.3±2.3%), 100 ng/ml (31.1±3.1%), and 1,000 ng/ml (44±7.5%), respectively, by 72 h. Overall, increasing doses of IL-1β tended to induce a progressively greater increase in permeability over control cells.
Fig. 2.

The pro-inflammatory cytokines IL-1β, TNF-α, and IFN-γ had differential effects on SV-HCEC monolayers. SV-HCEC monolayers were cultured on 8W10E ECIS arrays until confluent at which time they were stimulated with doses of cytokines ranging from 0.1–1,000 ng/ml of IL-1β (a, b), TNF-α (c, d) and IFN-γ (e). Representative ECIS graphs (a, c, e) demonstrate the time-course of responses to cytokines, whereas bar graphs (b, e) indicate the mean ± standard deviation of normalized resistance values at designated time points. *p<0.05 compared to uninfected controls.
The effects of TNF-α on uninfected SV-HCEC monolayers were more dramatic and behaved in a dose-dependent manner as demonstrated in a representative ECIS graph (Fig. 2c). Low doses of TNF-α at 0.1 ng/ml showed no significant increase in permeability compared to controls at any time point. A dose of 1 ng/ml showed a slightly higher level of permeability at 48 h (12.3±7.1%) after stimulation, but this effect was diminished by 72 h; after which, the increase in permeability was not statistically higher than control cells (Fig. 2d). Higher doses caused a steady increase in permeability over the 72-h time course ranging from 33.42±7.6% at 10 ng/ml, 49.79±7.0% at 100 ng/ml, and 64.2±3.2% at 1,000 ng/ml. The highest dose, 1,000 ng/ml, was rapidly cytotoxic and was excluded from further experiments with rickettsiae.
Stimulation of SV-HCEC with IFN-γ alone did not produce a significant increase in permeability of these cells at any time point tested (Fig. 2e).
TNF-α and IL-1β Augment Rickettsiae-Induced Microvascular Permeability in SV-HCEC
We next decided to investigate the effects of these cytokines on monolayers also infected with R. rickettsii. The activity of IL-1β on rickettsiae-infected endothelial cells produced an increase in permeability relative to cells only infected with rickettsiae (Fig. 3a). There was a dose-dependent response of rickettsiae-infected SV-HCECs to IL-1β, and by 48 h, there was a significant increase in permeability in cells treated with 10 ng/ml (13.1±1.5%) and 100 ng/ml (10.3±1.3%) of IL-1β compared to those only infected with rickettsiae. No additional effect of IL-1β was observed at any time point for doses ranging from 0.1–1 ng/ml.
Fig. 3.
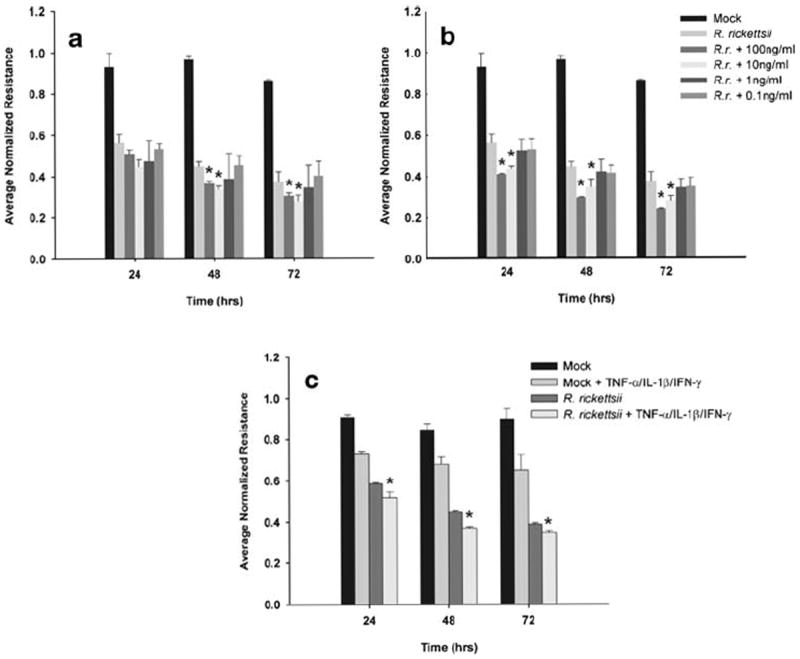
Addition of pro-inflammatory cytokines to rickettsiae-infected SV-HCEC enhanced rickettsiae-induced permeability. Cells were infected with 10 MOI of R. rickettsii and stimulated with varying concentration of IL-1β (a) or TNF-α (b) or 0.1 ng/ml of TNF-α, IL-1β, and IFN-γ in combination with and without rickettsiae (c). Data is presented as the mean ± standard deviation of three independent experiments. *p<0.05 compared to R. rickettsii-infected cells.
The addition of TNF-α to rickettsiae-infected cells elicited an additional increase in permeability at 10 ng/ml (15.9±1.1%) or 100 ng/ml (18.6±0.4%) as early as 24 h after stimulation (Fig. 3b). This trend continued for the course of the experiment with a sustained increase in permeability for those two doses above that of cells only infected with rickettsiae. Lower doses of 1 or 0.1 ng/ml had no additional impact on permeability over unstimulated infected cells.
Based on previous experiments, we decided to investigate whether low doses of cytokines acted in concert to synergistically activate endothelial cells and cause an increase in permeability. The addition of a combination of TNF-α, IL-1β, and IFN-γ to rickettsiae-infected cells at doses of 0.1 ng/ml produced a significantly greater increase in permeability compared to cells only infected with rickettsiae (Fig. 3c). This additional increase in permeability was maintained up to 72 h after the start of the experiment. Additional permutations of cytokine combinations had no additional impact on endothelial permeability (data not shown), indicating that the cumulative impact of all three cytokines is needed to produce this effect. Likewise, uninfected monolayers also demonstrated a sustained increase in permeability of approximately 20% greater than controls, although the increase in permeability was lower than either infected cells or infected cells treated with the same dose of cytokines.
Stimulation with Combinations of Pro-inflammatory Cytokines Induces an Anti-Rickettsial Effect in SV-HCEC Associated with NO Production
Because NO has been demonstrated to be an important mechanism of immunity to rickettsiae, we next sought to determine the ability of SV-HCEC to produce NO in response to cytokines and whether this has any effect on the number of viable rickettsial organisms. SV-HCEC infected with R. rickettsii and/or stimulated with 10 ng/ml of TNF-α, IL-1β, and IFN-γ produced high levels of NO as measured by nitrite in the supernatant and was first observed at 24 h after infection and stimulation (Fig. 4a). Surprisingly, R. rickettsii was alone capable of inducing NO production by SV-HCEC and was consistently higher than those cells only stimulated with cytokines. The combination of rickettsiae and cytokines produced the greatest NO production throughout the course of the experiment. We also sought to demonstrate that cytokine-stimulated endothelial cells had fewer numbers of intracellular rickettsiae compared to unstimulated controls. As expected, the addition of cytokines to rickettsiae-infected SV-HCEC resulted in a decrease in the number of viable intracellular rickettsiae (Fig. 4b). Cells treated with 0.1 ng/ml of cytokines had a decrease in the number of intracellular bacteria by approximately 23.67±1.6%. Cells treated with a higher dose of 10 ng/ml had a greater reduction in the number of intracellular rickettsiae to approximately 33.5±11.2% that of unstimulated controls at 24 h after infection. In both groups, this was a significant decrease in the number of intracellular rickettsiae compared to non-treated cells (p<0.05). However, there was not a significant difference between the two doses (p=0.11). We also tested whether inhibition of endogenous NO production influenced the number of intracellular rickettsiae. Cells treated with the nitric oxide synthase inhibitor L-NG-nitroarginine methyl ester (l-NAME) showed an overall increase in the number of rickettsiae to 22.5±10.8%, greater than untreated, infected cells and was significantly greater than either of the cytokine-treated groups (p<0.05).
Fig. 4.
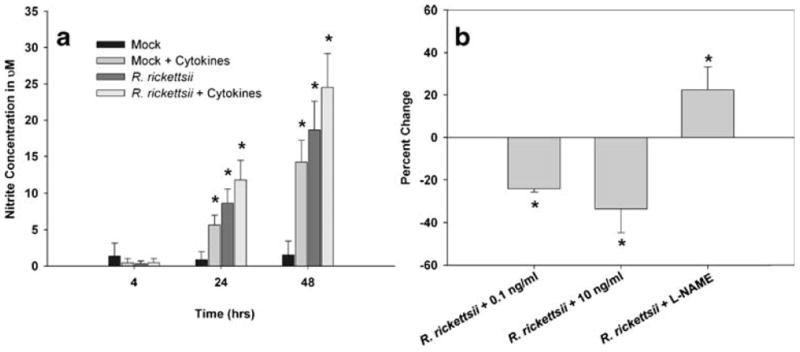
Combinations of pro-inflammatory cytokines induced an increase in microvascular permeability associated with an anti-rickettsial state in endothelial cells. SV-HCEC were infected with 10 MOI of R. rickettsii and/or stimulated with 10 ng/ml of TNF-α, IL-1β, and IFN-γ, and supernatants were collected at the given time points. The concentration of nitrite was determined using the Greiss assay (a). *p<0.05 compared to Mock-treated cells. SV-HCEC were seeded in 24-well plates and grown to confluence at which time they were infected with 10 MOI of R. rickettsii and then stimulated with 0.1 or 10 ng/ml of all three cytokines or treated with 100 uM L-NAME. The percent change in rickettsial genome copies was determined by real-time PCR using the ΔΔCt method with primers to rickettsial gltA and human β-actin (b). *p<0.05 compared to cells only infected with rickettsiae.
Rickettsiae- and Cytokine-Induced Increases in Microvascular Permeability are not Directly Attributable to NO Production
Because NO has been implicated in microvascular and endothelial barrier regulation, we sought to determine the impact of NO production by SV-HCEC, and its downstream signaling effects on microvascular permeability. The addition of the NO synthase inhibitor L-NAME resulted in decreased production of NO in response to cytokine stimulation. Likewise, the addition of L-NAME decreased the amount of NO being produced in response to rickettsial infection alone (Fig. 5a). The soluble guanalyl cyclase inhibitor ODQ had no impact of the level of NO production, as expected (Fig. 5c). Next, we determined the effect of nitric oxide on the early stages of permeability experienced during cytokine stimulation of rickettsiae-infected endothelial cells. Inhibition of NO production by rickettsiae-infected SV-HCEC had no impact on the ability of R. rickettsii to induce a loss of electrical resistance (Fig. 5b). Additionally, inhibition of NO production after cytokine stimulation had no impact on cytokine-induced microvascular permeability. Finally, inhibition of the downstream signaling activity of NO with ODQ failed to induce any abrogation of the effects of either R. rickettsii or cytokine combinations, suggesting alternative mechanisms of rickettsiae- and cytokine-induced permeability (Fig. 5d).
Fig. 5.

Rickettsiae- and cytokine-induced increases in microvascular permeability are not directly attributable to increased nitric oxide production in SV-HCEC. Nitrite concentrations were measured in cell culture supernatants of infected and uninfected cells treated with or without 10 ng/ml of cytokines or untreated plus 100 μM L-NAME (a) or 100 μM ODQ (c). Similarly treated cells were monitored by ECIS for changes in permeability in response to L-NAME (b) and ODQ (d).
Cytokines and R. rickettsii Induce the Loss of Adherens Junction Staining at Intercellular Borders
Uninfected SV-HCEC demonstrated staining of the adherens-junction proteins, β-catenin and p120, at intercellular borders consistent with a confluent monolayer (Fig. 6a,b). The staining of these two proteins overlapped, indicating their association with adherens junctions (Fig. 6c). The addition of cytokines appeared to have the greatest impact on p120 localization resulting in punctuate staining pattern not associated with cell–cell junctions (Fig. 6d–f). Additionally, β-catenin showed diminished association with intercellular junctions. Infection of SV-HCEC with R. rickettsii resulted in decreased staining of both proteins at cell–cell junctions and a clear change in cell morphology. Gaps between cells were evident, although the monolayer was still mostly intact (Fig. 6g–i). The p120 staining pattern was not nearly as punctuate as those cells stimulated with cytokines, suggesting an alternative pathway of modification. Also, the overall intensity of p120 staining decreased with very few intercellular junctions being observed. While β-catenin was still associated with intercellular junctions, there was decreased colocalization with p120, and the staining pattern was more diffuse than uninfected cells. The cells developed an elongated morphology as compared to either uninfected cells or those only treated with cytokines, which displayed a traditional cobblestone morphology characteristic of endothelium. As expected, the combination of rickettsial infection and cytokine stimulation had the greatest impact on stability of the adherens junctions, resulting in diminished staining of β-catenin and p120 at cell–cell junctions and an increased cytoplasmic localization of both proteins (Fig. 6j–l). Gaps between adjacent endothelial cells were clearly evident, and there was no clear definition of where adherens junctions were localized.
Fig. 6.
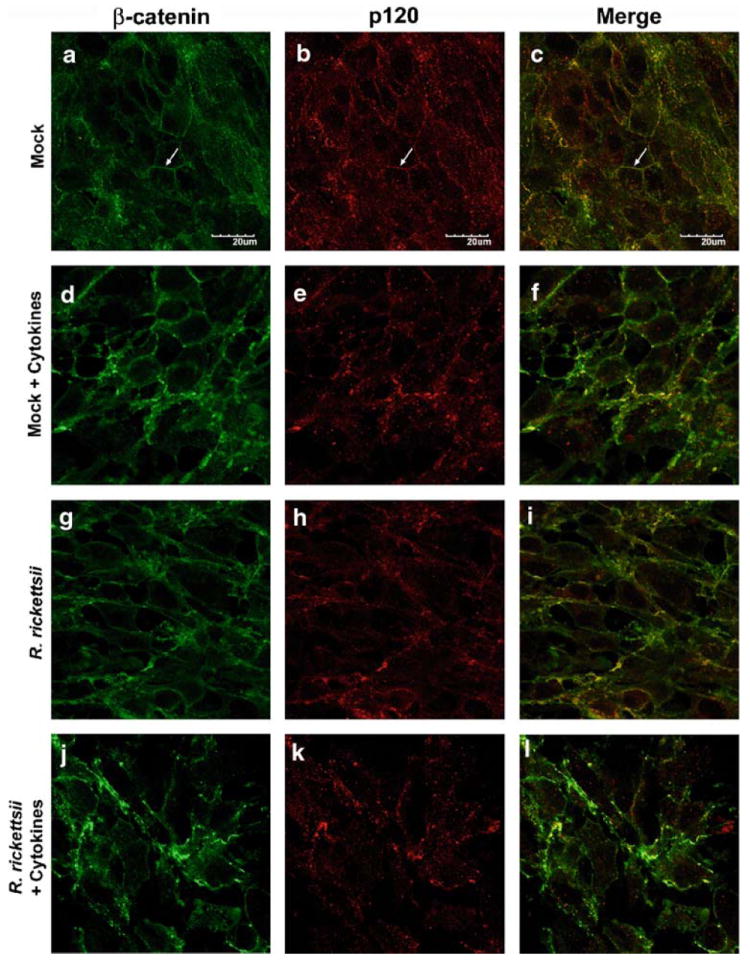
R. rickettsii and cytokines induced a loss of β-catenin (a, d, g, j) and p120 (b, e, h, k) staining at intercellular junctions. Cells were infected with 10 MOI of R. rickettsii and/or stimulated with 10 ng/ml of TNF-α, IL-1β, and IFN-γ and incubated at 37°C for 24 h. Overlaid images indicate the colocalization of β-catenin and p120 (c, f, i, l). Arrowheads indicate proper localization of adherens junctions at cell–cell contacts.
Discussion
The mechanisms of increased microvascular permeability during acute rickettsioses have not received sufficient attention to date. It is generally assumed that rickettsial pathogenesis is solely dependent on rickettsial proliferation and subsequent cell lysis. What has not been considered is the relative influence of the host’s immune response on the integrity of the microvascular barrier. We have demonstrated, in an immortalized human microvascular endothelial cell line, the ability of pro-inflammatory cytokines IL-1β, TNF-α, and IFN-γ to induce an anti-rickettsial response in the form of NO. Subsequently, we demonstrated that the cytokine stimulus but not NO is more than sufficient to induce a rapid decrease in electrical resistance across endothelial monolayers, reflecting an increase in microvascular permeability.
The role of the host immune response in the pathogenesis of acute rickettsioses is a phenomenon that has not been extensively studied. Clearly, a cellular immune response is critical to the effective clearance of rickettsial organisms from the vasculature, as has been demonstrated in the past [16]. Cytotoxic T-lymphocytes (CTL) are recruited to rickettsiae-infected endothelial cells, in part caused by the upregulation of the intercellular adhesion molecule 1 (ICAM-1) but independent of the CXCL9/10–CXCR3 chemokine system [23]. Presumably, an inflammatory state is initiated in these endothelial cells as a result of cytokine secretion by infiltrating macrophages and CD4+ and CD8+ T-lymphocytes [24, 25]. This inflammatory state is characterized by the transcriptional activation of iNOS, increased NO production, and the inhibition of rickettsial proliferation. Feng and Walker demonstrated in human endothelial cells and hepatocytes that the necessary stimuli included IFN-γ, TNF-α, IL-1β, and regulated upon activation, normal T cell expressed and secreted (RANTES), but the degree of NO production was variable between pools of HUVEC [26]. Likewise, depletion of TNF-α and IFN-γ in mice infected with R. conorii resulted in an increased proliferation of rickettsiae, and the animals are no longer able to control a sublethal infection [15].
The pleiotropic cytokine TNF-α is a vital component of a protective immune response to a number of different pathogens including rickettsiae. Its many different effects include inducing apoptotic cell death or, in other instances inducing cellular proliferation, demonstrating the wide range of often divergent effects associated with this protein. Widely produced throughout the body, TNF-α is perhaps most often associated with localized inflammatory processes, although it has been suggested that high levels of circulating cytokine are responsible for the pathological complications of Gram-negative sepsis and hantavirus pulmonary syndrome, diseases characterized by a rapid and dramatic increase in vascular permeability resulting in generalized vascular leakage and severe pulmonary edema, respectively [27, 28]. RMSF, on the other hand, to the best of out knowledge causes a much slower accumulation of interstitial fluid, suggesting that high numbers of localized inflammatory infiltrates associated with foci of rickettsial invasion might mediate the primary mechanism of fluid accumulation. TNF-α is primarily produced by activated macrophages and T-cells, which are cells known to infiltrate sites of rickettsial invasion. In this instance, the primary function of TNF-α is presumably to activate rickettsiae-infected endothelial cells to induce iNOS transcription and increase NO production to control intracellular rickettsial proliferation. However, TNF-α has also been shown to induce p42 oxidation via a NO-dependent mechanism, activate protein kinase C-α, and induce the production of prostaglandins and leukotrienes, resulting in endothelial barrier dysfunction [29-32]. Previous work in our lab demonstrated that the addition of a NO-donor to R. conorii-infected endothelial cells resulted in a stabilization of the barrier early after stimulation, suggesting that NO actually enhanced the barrier properties of endothelial cells [33]. On the other hand, work presented in this paper provides no clear indication about the role of NO in cytokine-mediated endothelial permeability, suggesting that NO-independent effects of TNF-α and/or IL-1β may be more important in regulation of endothelial barrier function. One possible explanation might be that TNF-α mediated p38 MAPK activation, resulting in redistribution of VE–cadherin as was demonstrated previously [34]. While this response is ultimately protective and survival is dependent on TNF-α activity, it is not unreasonable to conclude that the permeability-enhancing properties of TNF-α do, in fact, contribute to the pathogenesis of acute rickettsiosis, as demonstrated in this work.
IL-1β is a cytokine associated with an inflammatory state and is often regulated in conjunction with TNF-α. IL-1β demonstrates a remarkable pyrogenic action and was one of the first molecules identified to be directly responsible for causing fever, a component of the clinical triad associated with RMSF besides rash and headache. Interestingly, IL-1β generally circulates at very high levels during R. conorii infection of mice (unpublished data), suggesting that its mechanism of action in inducing vascular permeability is enhanced at sites of TNF-α production. Another intriguing response to IL-1β production is the increased transcription and secretion of IL-6 in a dose- and time-dependent manner by cultured intestinal epithelial cells [35]. IFN-γ, on the other hand, has no effect on the level of IL-6 being produced by these cells. Taken together with the observations that IL-6 is produced by rickettsiae-infected endothelial cells in the absence of additional cytokine stimulation, this may suggest a role for IL-6 in rickettsial pathogenesis [36]. In fact, lack of IL-6 was associated with decreased microvascular permeability in an experimental model of pneumococcal meningitis [37]. Likewise, IL-6 has been shown to induce a loss of TER across human umbilical vein endothelial cells in a protein kinase C (PKC)-dependent manner [38]. Therefore, it is not surprising to find that PKC is also up regulated in R. rickettsii-infected endothelial cells [39]. The exact role of IL-6 and/or PKC activation in rickettsiae-induced permeability is an area of current investigation.
These observations present a conundrum when trying to determine the mechanism of disease in acute rickettsiosis. Certainly, direct rickettsial cytotoxicity is a well-known component of rickettsial virulence as indicated by the presence of plaques in cell monolayers infected with R. rickettsii [40, 41]. Increased production of reactive oxygen species (ROS) is a recognized pathological consequence of R. rickettsii infection in human endothelial cells [42-44]. Interestingly, in one study, NO was shown to enhance hydrogen peroxide-mediated endothelial permeability associated with a significant depletion of intracellular glutathione [45]. This is similar to the effect of R. rickettsii infection on human endothelial cells characterized by decreased levels of intracellular reduced glutathione and increased levels of peroxide [46]. However, a direct connection between ROS production and increased microvascular permeability during rickettsioses has yet to be established. Our observation that R. rickettsii-infected SV-HCEC produce NO is novel and exciting. Few endothelial pathogens have been shown to induce the levels of NO seen in this study in a human-derived microvascular endothelial cell. For example, virulent Junin virus, a viral hemorrhagic fever, demonstrated the ability to induce NO production by human endothelial cells, and serum from patients acutely ill with Junin virus infection have markedly increased levels of nitrite [47]. Other factors released by endothelial cells in response to infection may also have the ability to induce NO production such as vascular endothelial growth factor, VEGF. High serum levels of VEGF has been associated with dengue virus infection, a disease characterized by general vascular dysfunction, and human endothelial cells infected with dengue virus produce VEGF [48-50]. In this work, we showed that blockade of endogenous NO allowed the rickettsiae to proliferate to higher levels. We also showed that blockade of endogenous NO had no effect on the integrity of the endothelial monolayer despite a greater number of intracellular rickettsiae. Most likely this means that the permeability-inducing effects of rickettsiae are more dependent on the initial response to invading rickettsiae.
Several important signaling pathways activated in response to R. rickettsii infection have been described, which may play an important role in microvascular barrier integrity. The p38 MAP kinase was shown to be activated via phosphorylation in response to invasion by live R. rickettsii, resulting in increased expression of IL-8 and MCP-1, two important mediators of vascular inflammation [51, 52]. p38 MAP kinase has been implicated in viral hemorrhagic fever-induced endothelial permeability and most likely contributes to rickettsiae-induced endothelial dysfunction in a similar manner [53]. Interestingly, MCP-1 is a demonstrated inducer of blood–brain barrier dysfunction both in vitro and in vivo [54, 55]. Taken together with the given observations of IL-6 and PKC activity during rickettsial infection, we can begin to see that the cellular host response to rickettsial invasion may play a much greater role in microvascular dysfunction than originally thought.
The integrity of the endothelial barrier is maintained by a complex and interwoven network of protein complexes such as the ubiquitously expressed junctions known as adherens junctions. They serve to anchor the endothelial cell to the extracellular matrix as well as neighboring cells. VE-cadherin is linked to the actin cytoskeleton by the catenin class of proteins, namely α- and β-catenin, and p120-catenin. Via confocal microscopy, we have demonstrated that β-catenin and p120 dissociate from the interendothelial cell junctions in response to inflammatory cytokine stimuli and/or infection with R. rickettsii. In contrast to the studies performed by Valbuena and Walker in which R. conorii induced adherens junction instability only after a prolonged period of infection, the effect of R. rickettsii is fast, occurring within the first 24 h of infection [19].
The data presented in this manuscript demonstrate the ability of R. rickettsii alone to cause an increase in microvascular permeability, and this response is further augmented by the presence of the pro-inflammatory cytokines TNF-α, IL-1β, and IFN-γ. These cytokines are important because of the previously demonstrated protective effect in eliminating intracellular rickettsiae. The absence of this response in vivo results in unchecked rickettsial proliferation and severe disease supporting the role for these cytokines in adaptive immunity. Taken together, it is clearly to the benefit of the host to possess this defense mechanism; however, the effects of endothelial activation more than likely contribute to the pathogenesis of RMSF. Future work may reveal novel pathways that can be modified to retain the protective effects of endothelial cell activation while reducing or ablating the potentially detrimental effects of cytokine stimulation.
Acknowledgments
This work was supported by funding from the United States Army Research and Material Command #DAMD17-02-1-0198 (J.P.O) and the NIH T32 Training Grant in Biodefense AI 060549 (M.E.W.). The authors would also like to acknowledge the W. M. Keck Center for Virus Imaging. Finally, the authors would like to thank Dr. David Walker for his critical evaluation of this work.
Contributor Information
Michael E. Woods, Department of Pathology, University of Texas Medical Branch, 301 University Blvd. Rt 0428, Galveston, TX, USA
Juan P. Olano, Email: jolano@utmb.edu, Department of Pathology, University of Texas Medical Branch, 301 University Blvd. Rt 0428, Galveston, TX, USA; Center for Biodefense and Emerging Infectious Diseases, Department of Pathology, University of Texas Medical Branch, Galveston, TX 77555-0428, USA; Department of Pathology, Center for Biodefense and Emerging Infectious Diseases, University of Texas Medical Branch, Galveston, TX 77555-0428, USA.
References
- 1.Raoult D, Roux V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin Microbiol Rev. 1997;10:694–719. doi: 10.1128/cmr.10.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valbuena G, Feng HM, Walker DH. Mechanisms of immunity against rickettsiae. New perspectives and opportunities offered by unusual intracellular parasites. Microbes Infect. 2002;4:625–33. doi: 10.1016/s1286-4579(02)01581-2. [DOI] [PubMed] [Google Scholar]
- 3.Masters EJ, Olson GS, Weiner SJ, Paddock CD. Rocky Mountain spotted fever: a clinician’s dilemma. Arch Intern Med. 2003;163:769–74. doi: 10.1001/archinte.163.7.769. [DOI] [PubMed] [Google Scholar]
- 4.Walker DH. Rocky Mountain spotted fever: a seasonal alert. Clin Infect Dis. 1995;20:1111–7. doi: 10.1093/clinids/20.5.1111. [DOI] [PubMed] [Google Scholar]
- 5.Walker DH, Mattern WD. Rickettsial vasculitis. Am Heart J. 1980;100:896–906. doi: 10.1016/0002-8703(80)90072-1. [DOI] [PubMed] [Google Scholar]
- 6.Walker DH, Crawford CG, Cain BG. Rickettsial infection of the pulmonary microcirculation: the basis for interstitial pneumonitis in Rocky Mountain spotted fever. Hum Pathol. 1980;11:263–72. doi: 10.1016/s0046-8177(80)80008-6. [DOI] [PubMed] [Google Scholar]
- 7.Walker DH, Popov VL, Feng HM. Establishment of a novel endothelial target mouse model of a typhus group rickettsiosis: evidence for critical roles for gamma interferon and CD8 T lymphocytes. Lab Invest. 2000;80:1361–72. doi: 10.1038/labinvest.3780144. [DOI] [PubMed] [Google Scholar]
- 8.Feng HM, Wen J, Walker DH. Rickettsia australis infection: a murine model of a highly invasive vasculopathic rickettsiosis. Am J Pathol. 1993;142:1471–82. [PMC free article] [PubMed] [Google Scholar]
- 9.Walker DH, Popov VL, Wen J, Feng HM. Rickettsia conorii infection of C3H/HeN mice. A model of endothelial-target rickettsiosis. Lab Invest. 1994;70:358–68. [PubMed] [Google Scholar]
- 10.Roggli VL, Keener S, Bradford WD, Pratt PC, Walker DH. Pulmonary pathology of Rocky Mountain spotted fever (RMSF) in children. Pediatr Pathol. 1985;4:47–57. doi: 10.3109/15513818509025902. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Jerrells TR, Spitalny GL, Walker DH. Gamma interferon as a crucial host defense against Rickettsia conorii in vivo. Infect Immun. 1987;55:1252–5. doi: 10.1128/iai.55.5.1252-1255.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jerrells TR, Li H, Walker DH. In vivo and in vitro role of gamma interferon in immune clearance of Rickettsia species. Adv Exp Med Biol. 1988;239:193–200. doi: 10.1007/978-1-4757-5421-6_19. [DOI] [PubMed] [Google Scholar]
- 13.Feng HM, Walker DH. Interferon-gamma and tumor necrosis factor-alpha exert their antirickettsial effect via induction of synthesis of nitric oxide. Am J Pathol. 1993;143:1016–23. [PMC free article] [PubMed] [Google Scholar]
- 14.Walker DH, Popov VL, Crocquet-Valdes PA, Welsh CJ, Feng HM. Cytokine-induced, nitric oxide-dependent, intracellular anti-rickettsial activity of mouse endothelial cells. Lab Invest. 1997;76:129–138. [PubMed] [Google Scholar]
- 15.Feng HM, Popov VL, Walker DH. Depletion of gamma interferon and tumor necrosis factor alpha in mice with Rickettsia conorii-infected endothelium: impairment of rickettsicidal nitric oxide production resulting in fatal, overwhelming rickettsial disease. Infect Immun. 1994;62:1952–60. doi: 10.1128/iai.62.5.1952-1960.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker DH, Olano JP, Feng HM. Critical role of cytotoxic T lymphocytes in immune clearance of rickettsial infection. Infect Immun. 2001;69:1841–6. doi: 10.1128/IAI.69.3.1841-1846.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tinsley JH, Ustinova EE, Xu W, Yuan SY. Src-dependent, neutrophil-mediated vascular hyperpermeability and beta-catenin modification. Am J Physiol Cell Physiol. 2002;283:C1745–51. doi: 10.1152/ajpcell.00230.2002. [DOI] [PubMed] [Google Scholar]
- 18.Mehta D. p120: the guardian of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1140–2. doi: 10.1152/ajplung.00008.2004. [DOI] [PubMed] [Google Scholar]
- 19.Valbuena G, Walker DH. Changes in the adherens junctions of human endothelial cells infected with spotted fever group rickettsiae. Virchows Arch. 2005;446:379–82. doi: 10.1007/s00428-004-1165-3. [DOI] [PubMed] [Google Scholar]
- 20.Hanson BA, Wisseman CL, Jr, Waddell A, Silverman DJ. Some characteristics of heavy and light bands of Rickettsia prowazekii on Renografin gradients. Infect Immun. 1981;34:596–604. doi: 10.1128/iai.34.2.596-604.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duvar S, Suzuki M, Muruganandam A, Yu RK. Glycosphingolipid composition of a new immortalized human cerebromicrovascular endothelial cell line. J Neurochem. 2000;75:1970–6. doi: 10.1046/j.1471-4159.2000.0751970.x. [DOI] [PubMed] [Google Scholar]
- 22.Labruna MB, Whitworth T, Bouyer DH, McBride J, Camargo LM, Camargo EP, Popov V, Walker DH. Rickettsia bellii and Rickettsia amblyommii in Amblyomma ticks from the State of Rondonia, Western Amazon, Brazil. J Med Entomol. 2004;41:1073–81. doi: 10.1603/0022-2585-41.6.1073. [DOI] [PubMed] [Google Scholar]
- 23.Valbuena G, Walker DH. Effect of blocking the CXCL9/10-CXCR3 chemokine system in the outcome of endothelial-target rickettsial infections. Am J Trop Med Hyg. 2004;71:393–9. [PubMed] [Google Scholar]
- 24.Blann AD, Woywodt A, Bertolini F, Bull TM, Buyon JP, Clancy RM, Haubitz M, Hebbel RP, Lip GY, Mancuso P, Sampol J, Solovey A, Dignat-George F. Circulating endothelial cells. Biomarker of vascular disease. Thromb Haemost. 2005;93:228–35. doi: 10.1160/TH04-09-0578. [DOI] [PubMed] [Google Scholar]
- 25.Constans J, Conri C. Circulating markers of endothelial function in cardiovascular disease. Clin Chim Acta. 2006;368:33–47. doi: 10.1016/j.cca.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 26.Feng HM, Walker DH. Mechanisms of intracellular killing of Rickettsia conorii in infected human endothelial cells, hepatocytes, and macrophages. Infect Immun. 2000;68:6729–36. doi: 10.1128/iai.68.12.6729-6736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters CJ, Khan AS. Hantavirus pulmonary syndrome: the new American hemorrhagic fever. Clin Infect Dis. 2002;34:1224–31. doi: 10.1086/339864. [DOI] [PubMed] [Google Scholar]
- 28.Lorente JA, Marshall JC. Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock. 2005;24(Suppl 1):107–19. doi: 10.1097/01.shk.0000191343.21228.78. [DOI] [PubMed] [Google Scholar]
- 29.Ferro TJ, Gertzberg N, Selden L, Neumann P, Johnson A. Endothelial barrier dysfunction and p42 oxidation induced by TNF-alpha are mediated by nitric oxide. Am J Physiol. 1997;272:L979–88. doi: 10.1152/ajplung.1997.272.5.L979. [DOI] [PubMed] [Google Scholar]
- 30.Ferro T, Neumann P, Gertzberg N, Clements R, Johnson A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1107–17. doi: 10.1152/ajplung.2000.278.6.L1107. [DOI] [PubMed] [Google Scholar]
- 31.Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: actions and mechanisms of action. Trends Neurosci. 1995;18:130–6. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- 32.Spellerberg B, Tuomanen EI. The pathophysiology of pneumococcal meningitis. Ann Med. 1994;26:411–8. doi: 10.3109/07853899409148362. [DOI] [PubMed] [Google Scholar]
- 33.Woods ME, Wen G, Olano JP. Nitric oxide as a mediator of increased microvascular permeability during acute rickettsioses. Ann N Y Acad Sci. 2005;1063:239–45. doi: 10.1196/annals.1355.037. [DOI] [PubMed] [Google Scholar]
- 34.Nwariaku FE, Chang J, Zhu X, Liu Z, Duffy SL, Halaihel NH, Terada L, Turnage RH. The role of p38 map kinase in tumor necrosis factor-induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock. 2002;18:82–5. doi: 10.1097/00024382-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 35.Parikh AA, Salzman AL, Fischer JE, Szabo C, Hasselgren PO. Interleukin-1 beta and interferon-gamma regulate interleukin-6 production in cultured human intestinal epithelial cells. Shock. 1997;8:249–55. doi: 10.1097/00024382-199710000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Kaplanski G, Teysseire N, Farnarier C, Kaplanski S, Lissitzky JC, Durand JM, Soubeyrand J, Dinarello CA, Bongrand P. IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway. J Clin Invest. 1995;96:2839–44. doi: 10.1172/JCI118354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paul R, Koedel U, Winkler F, Kieseier BC, Fontana A, Kopf M, Hartung HP, Pfister HW. Lack of IL-6 augments inflammatory response but decreases vascular permeability in bacterial meningitis. Brain. 2003;126:1873–82. doi: 10.1093/brain/awg171. [DOI] [PubMed] [Google Scholar]
- 38.Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002;104:118–23. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- 39.Sahni SK, Turpin LC, Brown TL, Sporn LA. Involvement of protein kinase C in Rickettsia rickettsii-induced transcriptional activation of the host endothelial cell. Infect Immun. 1999;67:6418–23. doi: 10.1128/iai.67.12.6418-6423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker DH, Cain BG. The rickettsial plaque. Evidence for direct cytopathic effect of Rickettsia rickettsii. Lab Invest. 1980;43:388–96. [PubMed] [Google Scholar]
- 41.Walker DH, Firth WT, Edgell CJ. Human endothelial cell culture plaques induced by Rickettsia rickettsii. Infect Immun. 1982;37:301–6. doi: 10.1128/iai.37.1.301-306.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silverman DJ, Santucci LA. Potential for free radical-induced lipid peroxidation as a cause of endothelial cell injury in Rocky Mountain spotted fever. Infect Immun. 1988;56:3110–5. doi: 10.1128/iai.56.12.3110-3115.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devamanoharan PS, Santucci LA, Hong JE, Tian X, Silverman DJ. Infection of human endothelial cells by Rickettsia rickettsii causes a significant reduction in the levels of key enzymes involved in protection against oxidative injury. Infect Immun. 1994;62:2619–21. doi: 10.1128/iai.62.6.2619-2621.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eremeeva ME, Silverman DJ. Effects of the antioxidant alpha-lipoic acid on human umbilical vein endothelial cells infected with Rickettsia rickettsii. Infect Immun. 1998;66:2290–9. doi: 10.1128/iai.66.5.2290-2299.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okayama N, Kevil CG, Correia L, Jourd’heuil D, Itoh M, Grisham MB, Alexander JS. Nitric oxide enhances hydrogen peroxide-mediated endothelial permeability in vitro. Am J Physiol. 1997;273:C1581–7. doi: 10.1152/ajpcell.1997.273.5.C1581. [DOI] [PubMed] [Google Scholar]
- 46.Eremeeva ME, Silverman DJ. Rickettsia rickettsii infection of the EA.hy 926 endothelial cell line: morphological response to infection and evidence for oxidative injury. Microbiology. 1998;144(Pt 8):2037–48. doi: 10.1099/00221287-144-8-2037. [DOI] [PubMed] [Google Scholar]
- 47.Gomez RM, Pozner RG, Lazzari MA, D’Atri LP, Negrotto S, Chudzinski-Tavassi AM, Berria MI, Schattner M. Endothelial cell function alteration after Junin virus infection. Thromb Haemost. 2003;90:326–33. doi: 10.1160/TH02-09-0043. [DOI] [PubMed] [Google Scholar]
- 48.Tseng CS, Lo HW, Teng HC, Lo WC, Ker CG. Elevated levels of plasma VEGF in patients with dengue hemorrhagic fever. FEMS Immunol Med Microbiol. 2005;43:99–102. doi: 10.1016/j.femsim.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Azizan A, Sweat J, Espino C, Gemmer J, Stark L, Kazanis D. Differential proinflammatory and angiogenesis-specific cytokine production in human pulmonary endothelial cells, HPMEC-ST1.6R infected with dengue-2 and dengue-3 virus. J Virol Methods. 2006;138:211–7. doi: 10.1016/j.jviromet.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Srikiatkhachorn A, Ajariyakhajorn C, Endy TP, Kalayanarooj S, Libraty DH, Green S, Ennis FA, Rothman AL. Virus-induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic fever. J Virol. 2007;81:1592–600. doi: 10.1128/JVI.01642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clifton DR, Rydkina E, Huyck H, Pryhuber G, Freeman RS, Silverman DJ, Sahni SK. Expression and secretion of chemotactic cytokines IL-8 and MCP-1 by human endothelial cells after Rickettsia rickettsii infection: regulation by nuclear transcription factor NF-kappaB. Int J Med Microbiol. 2005;295:267–78. doi: 10.1016/j.ijmm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Rydkina E, Silverman DJ, Sahni SK. Activation of p38 stress-activated protein kinase during Rickettsia rickettsii infection of human endothelial cells: role in the induction of chemokine response. Cell Microbiol. 2005;7:1519–30. doi: 10.1111/j.1462-5822.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 53.Chiang ET, Persaud-Sawin DA, Kulkarni S, Garcia JG, Imani F. Bluetongue virus and double-stranded RNA increase human vascular permeability: role of p38 MAPK. J Clin Immunol. 2006;26:406–16. doi: 10.1007/s10875-006-9024-4. [DOI] [PubMed] [Google Scholar]
- 54.Song L, Pachter JS. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res. 2004;67:78–89. doi: 10.1016/j.mvr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25:593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]