

Abstract
One goal of the CASP Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction is to identify the current state of the art in protein structure prediction and modeling. A fundamental principle of CASP is blind prediction on a set of relevant protein targets, i.e. the participating computational methods are tested on a common set of experimental target proteins, for which the experimental structures are not known at the time of modeling. Therefore, the CASP experiment would not have been possible without broad support of the experimental protein structural biology community. In this manuscript, several experimental groups discuss the structures of the proteins which they provided as prediction targets for CASP9, highlighting structural and functional peculiarities of these structures: the long tail fibre protein gp37 from bacteriophage T4, the cyclic GMP-dependent protein kinase Iβ (PKGIβ) dimerization/docking domain, the ectodomain of the JTB (Jumping Translocation Breakpoint) transmembrane receptor, Autotaxin (ATX) in complex with an inhibitor, the DNA-Binding J-Binding Protein 1 (JBP1) domain essential for biosynthesis and maintenance of DNA base-J (β-D-glucosyl-hydroxymethyluracil) in Trypanosoma and Leishmania, an so far uncharacterized 73 residue domain from Ruminococcus gnavus with a fold typical for PDZ-like domains, a domain from the Phycobilisome (PBS) core-membrane linker (LCM) phycobiliprotein ApcE from Synechocystis, the Heat shock protein 90 (Hsp90) activators PFC0360w and PFC0270w from Plasmodium falciparum, and 2-oxo-3-deoxygalactonate kinase from Klebsiella pneumoniae.
Keywords: CASP, protein structure, X-ray crystallography, NMR, structure prediction
Introduction
The CASP experiment would not have been possible without broad support of the experimental protein structural biology community. For objective testing and comparison of protein structure modeling methods it is essential to ensure that the prediction methods are evaluated on the same unbiased set of targets, and that the correct answer of the prediction exercise (i.e., experimental structure coordinates) is not known until after the end of the modeling routine. For rigorous assessment of the prediction results it is also important to have a large enough set of targets. These requirements are among the basic principles of CASP operation, and thus, for the CASP experiment to succeed, it is essential that organizers have access to a large pool of proteins, whose structures have not been seen by the prediction community but are expected to be publicly released in the nearest future. These soon-to-be-solved structures or structures that just have been solved but not yet deposited to protein structure databases were solicited from the experimental community and later used as prediction targets.
Over four months in the spring-summer of 2010, X-ray crystallographers and NMR spectroscopists provided CASP organizers with the sequence details of over 140 proteins they agreed to have made public not earlier than 8 weeks after the submission to CASP. One hundred and twenty nine of these proteins were selected as prediction targets for CASP9 experiment.1 At the end of the prediction season coordinates for all but 13 targets were available in time for the assessment (mid-September 2010). Over 80% of the targets were received from three structural genomic centers: Joint Center for Structural Genomics (http://www.jcsg.org/, 38 targets), Midwest Center for Structural Genomics (http://www.mcsg.anl.gov/, 28 targets) and Northeast Structural Genomics Consortium (http://www.nesg.org/, 39 targets). CASP also received contributions from the Structural Genomics Consortium (http://www.sgc.utoronto.ca/, 7 targets), New York Structural Genomics Research Center (http://www.nysgxrc.org/, 5 targets) and several individual experimental groups from around the world. A substantial share of the most interesting CASP9 targets were obtained from those smaller experimental groups and some of their contributions are also discussed here. The RCSB Protein Data Bank2 put in place a mechanism for keeping some deposited structures on hold, with the aim of making them available as targets for CASP. An option was provided in the ADIT structure deposition process to identify a structure as a CASP target deposited in the PDB. By choosing “hold for CASP” in ADIT deposition interfaces, the words “CASP target” were automatically added to the title for the entry. CASP structures could then be identified by searching the unreleased PDB entries. All CASP structures had 8 weeks “hold” for both coordinates and structure factors.
Once the experimental coordinates of the targets were released, the predictions of the participating groups were assessed for their correctness using mainly numerical criteria measuring structural similarity between the target structures and the predictions. Typically, global structure comparison measures are used as an objective way to quantify the overall accuracy of a prediction.3,4 However, from a functional perspective different regions of a protein might have different functional relevance. While for one protein the binding of a specific ligand might be crucial for its biological role, others might fulfill their function through their steric and mechanical properties. For this manuscript, the CASP organizers invited representatives from the experimental groups to discuss selected examples of proteins which they provided as prediction targets for CASP9, highlighting their structural and functional peculiarities and the challenges these might pose in the context of structure prediction.
Bacteriophage T4 long tail fibre protein gp37 (CASP ID - T0629, PDB ID - 2XGF)
Bacteriophages are the most abundant genetic entities on earth and second in mass only to bacteria; they are used in applications such as phage display, identification and control of bacteria and phage therapy. The great majority of bacteriophages have tails and belong the Caudovirales order, which is comprised of three families, the Siphoviridae, making up more than half of the Caudovirales, the Myoviridae, amounting about one quarter and the Podoviridae, comprising the rest.5 Siphoviridae have a long, flexible, non-contractile tail, Podoviridae have a short, non-contractile tail, while Myoviridae possess a tail of which the outer sheath can contract.6 Many of the bacteriophages belonging to the Caudovirales use fibre proteins for host recognition and adhesion.
Bacteriophage T4 is the archetypal Myovirus and has been studied extensively as a model system for morphogenesis of complex structures. The study of Escherichia coli infected by T4 also led to the discovery of fundamental principles such as the relationship between genes and their products, the existence of mRNA and virus-induced acquisition of metabolic function.7 In the case of T4, initial recognition of the bacterial cell to infect is performed by the long tail fibres. They reversibly bind to the outer glucose[α1-3]glucose region of the bacterial lipo-polysaccharide (LPS) or the Outer Membrane Porin C (OmpC).
The T4 base-plate is a sophisticated multi-protein complex.8 Upon receipt of the signal that at least three long tail fibres have encountered suitable receptors, a conformational change of the base-plate allows the short tail fibres, which are trimers of gene product (gp) 12, to extend. Once these short tail fibres have irreversibly bound the core region of the LPS, a further conformational change presumably allows the inner tail tube to pass through the base-plate, driven by contraction of the outer tail sheath. Phage proteins and DNA can then enter the bacteria and initiate infection, which, in favourable conditions, can lead to several hundred daughter phages and bacterial lysis within 30 minutes.
The long tail fibre can be divided in proximal and distal halves, each over 70 nm long and connected at an angle of around 160°.9 The half proximal to the phage (the thigh) is made up of a trimer of gp34, a 1289 amino acid protein of unknown structure. At the kink, or knee, a single copy of gp35 (372 residues) is located. The top of the shin is made up of a trimer of the 221-amino acid protein gp36, while the major part of the shin and the receptor-binding tip (or foot) is comprised of a parallel homo-trimer of gp37. Full-length gp37 contains 1026 residues.
For correct folding of the trimeric fibrous proteins gp12, gp34 and gp37, the phage-encoded chaperone gp57 is necessary. Gp37 needs a specific additional chaperone, gp38, for correct trimerisation and folding. By co-expression with gp57 and gp38, nearly full-length gp37 (amino acids 12-1026) has been successfully expressed in a correctly folded and soluble form.10 However, crystallisation trials of the entire protein were not successful. Therefore, expression vectors for several N-terminal deletion fragments were constructed and a C-terminal construct consisting of residues 651-1026 yielded crystallisable protein after treatment with trypsin.11 The asymmetric unit of the crystals contained a trimer of gp37(785-1026), of which residues 811-1026 for each of the three chains could be resolved at 2.2 Å resolution (PDB: 2XGF; Figure 1).
Figure 1.

Crystal structure of the receptor-binding tip of the long tail fibre protein gp37. The three protein chains of the homo-trimer are coloured differently. The wider region at the top is the collar domain. The N- and C-termini of the blue chain are indicated. The central needle domain contains seven iron ions, each coordinated octahedrally by six histidine residues (shown for Fe4 only). The receptor-binding head domain is indicated with “H”, an asterisk indicates where the blue chain passes through a loop formed by the purple chain.
The structure revealed a collar domain similar to that observed for gp12, which is composed of amino acids 811-861 plus a β-strand formed by the very C-terminus of the protein (residues 1016-1026). This means the N- and C-termini of the protein are close, and the intervening residues form an extensively interwoven intertwined region (residues 862-880 plus 1009-1015), a needle domain consisting of amino acids 881-933 plus 960-1008 and a head domain formed by residues 934-959. The head domain is responsible for interaction with LPS and OmpC, and thus for initial host recognition. In the needle domain, seven iron ions are coordinated in octahedral fashion by two histidine residues of each protein chain.
The extensive intertwining is probably a strategy to stabilise the thin structure, stability obviously being an important property for bacteriophage structural proteins in general and of the principal receptor-binding protein in particular. The incorporation of the iron ions likely also helps to hold the protein chains together. The collar domain may serve as the folding nucleus, as it is the only domain with extensive inter-monomer interactions. The structure also shows which residues of the receptor-binding head domain are accessible on the surface; site-directed mutagenesis, receptor-binding studies and co-crystallisation experiments should lead to a detailed view on how this protein recognises LPS and OmpC.
Logically, this structure was very hard to predict and indeed, apart from the collar domain, none of the predictions in the CASP9 experiment came close to the correct structure for the needle and head domains. However, with hindsight, perhaps some inferences could have been made from the sequence, that, combined with the knowledge of the structure of the gp10 12, gp11 13 and gp12 14 proteins, would have led to improved structure predictions.
First of all, it is known that gp37 forms needle-shaped parallel homo-trimers.9 Second, the structural homology of the collar domains in gp10, gp11 and gp12 combined with the sequence homology of the very C-terminal amino acids of gp37 with gp12 means the N- and C-termini of the structure have to be located in the same collar domain. Together, this means that the intervening residues (862-1015) must form an elongated structure in which each of the three protein chains runs from one side to the other and back. The distance the chains have to cover means that nearly all of the amino acids have to adopt an extended conformation, i.e. β-strands.
The presence of the seven metal ions could have been inferred from the seven His-X-His pair in the sequence and comparison with gp12, where a His-X-His sequence coordinates a zinc ion in the same octahedral fashion.14 Of course, how the long extended strands interact with each other in the needle domain and the interwoven fold of the head domain would have been hard to imagine, as no structural homologues exist in the PDB. Now that the structure has been determined,11 it should be possible to come up with reasonable structure predictions for the receptor-binding tips of the bacteriophage lambda fibre and other bacteriophage fibres with homologous sequences but different receptor-binding head domains.
Cyclic GMP-dependent protein kinase Iβ dimerization/docking domain reveals molecular details of isoform-specific anchoring (CASP ID - T0605, PDB ID - 3NMD)
Cyclic GMP-dependent protein kinase (PKG) is the central enzyme of nitric oxide/atrial naturetic peptide-cGMP signaling cascades, which regulate smooth muscle tone, inhibit platelet activation, and modulate neuronal function.15 PKG is the main downstream target for pharmaceutical agents that raise cellular cGMP levels to treat erectile dysfunction and cardiovascular and pulmonary disease.15 Alternative splicing produces two type I PKG isoforms (PKGIα and PKGIβ), which differ in their first ~100 amino acids, and have unique dimerization/docking (D/D) and autoinhibitory domains. The crystal structure of the coiled-coil comprising the D/D domain of PKGIβ solved by Kim and colleagues was the first for any part of PKG and provided molecular details into the mechanism of its dimerization and its interaction with binding partners.16
Coiled-coils are structural components in a diverse number of proteins including transcriptional regulators, cytoskeletal proteins, and transmembrane receptors; they often form protein docking surfaces that mediate cellular signaling and transport.17,18 They also serve as a versatile model system for studying protein-protein interaction stability and specificity, and sequence-structure relationships.19,20 Coiled-coils contain a distinct primary sequence, with a repeating pattern every seven residues. The residue positions are labeled (a-b-c-d-e-f-g), and residues in the a and d positions are typically hydrophobic and form the interaction interface between helices. The most commonly observed coiled-coils contain two parallel α-helices which oligomerize to form a left-handed supercoil, and as expected from its primary sequence, our structure of the PKGIβ D/D shows the two amphipathic helices wrapping around each other to form a parallel coiled-coil with a left-handed supercoil. A surprising feature of the PKGIβ coiled coil is that the major leucine/isoleucine repeat lies in the a position of the heptad repeat, instead of at the more common d position. The “knobs into holes” packing of residues in positions a and d within the hydrophobic dimer interface is evident, and a previous study has demonstrated the critical role that the a position leucine/isoleucine residues play in stabilizing PKGIβ dimerization.21 Another unique feature of the PKGIβ D/D domain is that basic residues in the core d positions make unique symetrical interhelical salt bridges with acidic e position residues (Figure 2). The hydrophobic tails of the d position basic residues pack into the “holes” in the adjacent helix in a dramatically bent conformation, and the d-e salt bridges appear to stabilize the bent conformation of the basic residue side chains. In typical coiled-coils, dimer formation is stabilized by interhelical salt bridges between charged residues at position g on one helix with an oppositely charged residue at position e on the neighboring helix; these interchain salt bridges are thought to mediate homo- and hetero-dimer specificity.17,22 The exact role that PKGIβ’s d-e salt bridges play in mediating dimerization has yet to be determined.
Figure 2.

A surface representation of the PKGIβ D/D domain is shown and colored according to its electrostatic potential (blue = electropositive, red = electronegative). The GKIP binding site is marked with dotted lines and residues known to mediate GKIP binding are labeled.
The isoform specific functions PKGIα and PKGIβ are, in part, mediated by interaction with cGMP-dependent protein kinase interacting proteins (GKIPs), which specifically bind each isoforms unique D/D domain.15 GKIPs for PKGIα include the myosin phosphatase targeting subunit (MYPT1) of the myosin light chain phosphatase (PP1M), GKAP-42, vimentin and the regulator of G-protein signaling-2 (RGS-2). PKGIβ specific GKIPs include the general transcriptional regulator TFII-I and inositol trisphosphate receptor-associated PKG substrate (IRAG). Detailed analysis of the interaction between PKGIβ and its GKIPs (TFII-I and IRAG) revealed a common mode of binding.23 Negatively charged residues (amino acids 26-31) in PKG Iβ interacted with basic residues on the GKIPs, and secondary structure prediction suggests that the GKIP’s basic residues lie on one face of an α-helix. The structure showed the topology of PKGIβ’s GKIP binding surface (Figure 2). Of note, alignment of the five helices within the unit cell showed that the rotomer positions of the acidic side chains in the GKIP binding surface were almost identical, suggesting that the GKIP binding surface is pre-formed.
The functional importance of the PKGI D/D domain in vivo has been explored using transgenic mice which express a dimerization deficient form of PKGIα (the first four D/D domain a position leucine/isoleucines were changed to alanines). These mice showed a number of cardiovascular abnormalities, including impaired vasodilatation, leading to systemic hypertension and cardiac hypertrophy.24 Since dimerization has been shown to be necessary for PKGIα binding to RGS-2, MYPT1, and other interacting proteins, the abnormalities are thought to be due to the loss of specific PKGIα-GKAP interactions. In the future, it would be interesting to perform a similar experiment with PKGIβ.
In CASP9, many groups have correctly identified suitable templates for target T0605 and submitted accurate models – the best ones better than 1 Å Cα RMSD. However only few predictions aimed to model the dimeric coiled-coil, while the majority of predictions for this dimerization domain were monomeric, thereby misrepresenting its biological function.
JTB: a small protein with a simple (but very difficult to predict) fold (CASP ID - T0531, PDB ID - 2KJX)
One of the most challenging prediction targets at CASP9 was T0531, a small 65 residue human protein that encompassed the ectodomain of a compact transmembrane receptor called JTB, short for Jumping Translocation Breakpoint. The name of this protein describes a rare chromosomal abnormality that afflicts the cognate human gene (on chromosome 1q21) in diverse human cancers whereby a jtb gene fragment lacking the 3’ exon (encoding the transmembrane helix and short cytoplasmic tail) ‘jumps’ onto the ends of different chromosomes where it is fused to stretches of telomeric repeats. These translocations of jtb result in multiple copies of a shortened gene that produces excess amounts of a secreted JTB ectodomain.25,26 While the link between heightened levels of circulating JTB and cancer disease processes remains under active exploration, Bazan and coworkers sought clues to the molecular function of JTB by tackling the three-dimensional structure of the free ectodomain (Rousseau, F., Pan, B., Fairbrother, W. J., Bazan, J. F. & Lingel, A. submitted to J. Mol. Biol.).
JTB is a strikingly well conserved protein from nematodes to humans with a distinctive six-cysteine motif that does not match the sequence pattern of any known cysteine-rich modular fold. The predicted secondary structure of JTB (drawn from a comprehensive alignment of homolog sequences)27 confidently locates three β-strands which suggests some variant of a disulfide-bridged β-sheet fold (Figure 3 A). To resolve these structural questions, recombinant human JTB was produced in E. coli and the stable protein used for NMR analysis. The resulting solution structure of JTB (PDB file 2KJX) reveals a core, three β-strand meander fold stitched by two disulfide links (C9-C46 and C21-C57); the third conserved disulfide bridge (C24-C35) surprisingly pins together the N- and C-termini of a short helix inserted between the first and second β-strands. The βαββ JTB fold thus resembles an open hand where the β-sheet forms the palm and fingers, and the inserted helix doubles as the outstretched thumb (Figure 3 B). CONSURF analysis 28 of the JTB structure maps a hotspot-like patch of conserved residues to the concave face of the palm, and we suggest that this may form an interaction site for protein or extracellular matrix ligands (Figure 3 C); the back side of the JTB fold is notably convex and uninvitingly charged.
Figure 3.

Solution structure of the JTB ectodomain. (A) The human JTB protein chain is highlighted with conserved amino acids, and the PSIPRED secondary structure assignment (E and H, high confidence β-strand and helix, e and h, lower confidence) arrayed above the NMR-defined structure; note the unpredicted helix α1. Disulfide bridge connectivity is drawn above, with the orange circles marking the ‘helix-pinning’ disulfide link. (B) An early NMR ensemble for JTB shows the greater mobility of N- and C-termini, as well as the inserted helix α1, see PDB entry 2KJX for final ensemble. The cartoon architecture of JTB shows the antiparallel β-sheet and inserted helix marked as in (A); disulfide bridges are also pointed out. (C) Electrostatic potential surface of JTB (in the same orientation as (B)) showing the location of the putative interaction groove. (D) Superposed structure of midkine (PDB file 1MKN) onto JTB (in same pose above) with the resulting protein alignment showing concordance of core β-strands and a pair of disulfide bridges; midkine lacks the inserted helix (and orange Cys that pin the helix ends) but gains an additional (red circle) disulfide link. All molecular structures displayed with PyMOL (http://www.pymol.org).
A search for similar folds in the PDB with the SSM server 29 revealed matches to a slew of 3-β-strand meander folds (like chemokines), and a standout superposition (35 aligned Cα positions with 2.24 Å RMSD and 23% amino acid identity, suggestive of distant homology) with the N-terminal Cys-rich domain of a heparin-binding growth factor that overlays the JTB antiparallel β-sheet and its two disulfide bridges with corresponding features in midkine (PDB file IMKN);30 notably, the looped-out helix in JTB is missing from midkine (Figure 3 D).
The JTB fold architecture confounded most attempts to predict its three-dimensional structure due to a few key factors: (a) an unknown disulfide connectivity map for the conserved Cys residues; (b) a poor local alignment of the JTB species variants in the looped-out helix α1, which resulted in an unpredicted helix by programs like PSIPRED; (c) the assumption that the ~20 amino acid linker between βA and βB (which is constricted by the short helix α1 in the structure) could be used as a long overhand loop, misorienting β-strands and producing an incorrect β-sheet; and (d) a critical inability (perhaps due to the long, central linker) to locate the correct, closest template in the midkine structure. As Cys-rich domains are ubiquitous and important interaction modules in proteins—whether they are part of extracellular structures and involved in disulfide bridges, or form intracellular domains where the cysteines are metal-binding residues, fold recognition and ab initio programs have to improve their ability to predict their structures. Prediction of the correct set of disulfide-linked cystines (or nest of metal-coordinating Cys residues) is critical to defining distance constraints that potentially lead to good chain topologies and three-dimensional models of small Cys-rich modules like JTB.
The structure of Autotaxin (ATX) in complex with an inhibitor (CASP ID - T0543, PDB ID - 2XRG)
Autotaxin (ATX) is a secreted ~100 kDa extracellular glycoprotein that belongs to the enzyme family of ectonucleotide pyrophosphatase/phosphodiesterases (ENPP).31 All ENPP family members have a nucleotide pyrophosphatase activity. However, ATX is the only family member that can also hydrolyze lysophosphatidylcholine (LPC) into lysophosphatidic acid (LPA),32,33 a signaling lipid that activates many cellular pathways via the binding to specific G-protein coupled receptors.34 The LPC substrates of ATX vary in length, and in saturated and unsaturated alkyl-groups. Additionally, the lipid product LPA is a potent inhibitor of ATX.35 ATX is involved in many physiological and pathophysiological processes like inflammation, neuropathic pain, cell migration and cancer.
Since the molecular basis for the diverse substrate preferences of ATX were not well understood, and ATX is heavily investigated in industry and academia as a drug target, Perrakis and colleagues set to determine the crystal structure of ATX alone and in complex with the potent inhibitor HA155.36 Contemporary, Nishimasu and colleagues have determined the murine ATX structure in complex with LPAs of different chain lengths and alkyl chain saturations.37 All crystal structures show a compact and robust architecture for this multi-domain protein. The two N-terminal cysteine-rich somatomedin-B-like (SMB) domains and the C-terminal nuclease-like (NUC) domain flank opposing sites of the catalytic phosphodiesterase (PDE) domain. Moreover, an extended ‘lasso’ loop of about 50 residues, which starts at the end of the PDE domain, wraps tightly around the entire NUC domain, to finally enter the PDE fold from the opposite site. The crystal structures of the catalytic PDE domain revealed a hydrophobic pocket and a tunnel, both adjacent to the ATX catalytic site. The pocket is responsible for substrate and inhibitor binding, while the tunnel, which is partially formed by the SMB1 domain, is likely involved in LPA presentation to its receptors.37 The SMB domains also mediate binding to cell-surface integrins,38 giving rise to a concept where SMB-mediate binding to the cell surface and product release from the tunnel are likely important for localized LPA release in cellular micro-environments.
Both SMB1 (56-95) and SMB2 (100-140) resemble the fold of the SMB domain of vitronectin with a sequence identity of 38%; they also have 56% sequence identity to the SMB domain of ENPP1 and the RMSD between all these domains is about 1.3 Å over 40 superposed Cα atoms. The PDE domain (160-539) is related to the nucleotide pyrophosphatase of Xanthomonas axonopodis (XaNPP),39 with 32% identity and an RMSD of 1.5 Å over 335 superposed Cα atoms. The closest known structural homologue of the NUC domain (590-859) is the cyanobacterial nuclease NucA with only 19% sequence identity and an RMSD of 2.1 Å over 210 superposed Cα atoms. However, even though all individual domains of ATX resemble known folds, the relative orientations of the domains were unknown. ATX was therefore submitted as target for the CASP9, both for modeling the individual domains and the entire structure.
All full-length structure predictions have an RMSD of more than 10 Å for more than 500 superposed Cα atoms, and failed to reproduce the overall shape of Autotaxin. The individual domains are predicted well, with RMSD in the 1.5-2.5 Å region, as expected given the rather low similarity scores. The most interesting feature of ATX, is the hydrophobic pocket which accommodates the alkyl-groups of LPA (Figure 4 A), and the HA155 inhibitor with its bulky aromatic ring system (Figure 4 B). This pocket results from an 18 amino acid deletion compared to the structural homologue XaNpp and to other ENPPs. Predicting the shape of this pocket accurately would be the most important practical contribution of homology modeling, since it could have enabled structure-based inhibitor design before the structure of ATX was available. The best-ranked individual PDE prediction model, has an RMSD of 1.9 Å over 360 superposed Cαs, but predicts only a shallow pocket, unable to accommodate either the LPA substrates or HA155 (Figure 4 C-D). The second-best ranked prediction model, has an RMSD of 2.0 Å for 359 aligned Cα atoms, and would also fail to predict the substrate binding site (Figure 4 E-F). A few other models from well-established servers, all exhibited the same problems. A possible explanation of that is that the hydrophobic pocket in the PDE domain alone is a thermodynamically unstable structure, which in the PDE domain of ATX is at least partially stabilized by the adjacent NUC and SMB domain interactions: modeling the PDE domain alone would not be enough to predict the shape of the pocket following the 18-residue deletion in the template structure. Overall the modeling results on the catalytic domain of PDE emphasizes the need of further research to be able to predict ligand binding sites useful for docking studies when using structure templates of limited similarity.
Figure 4.
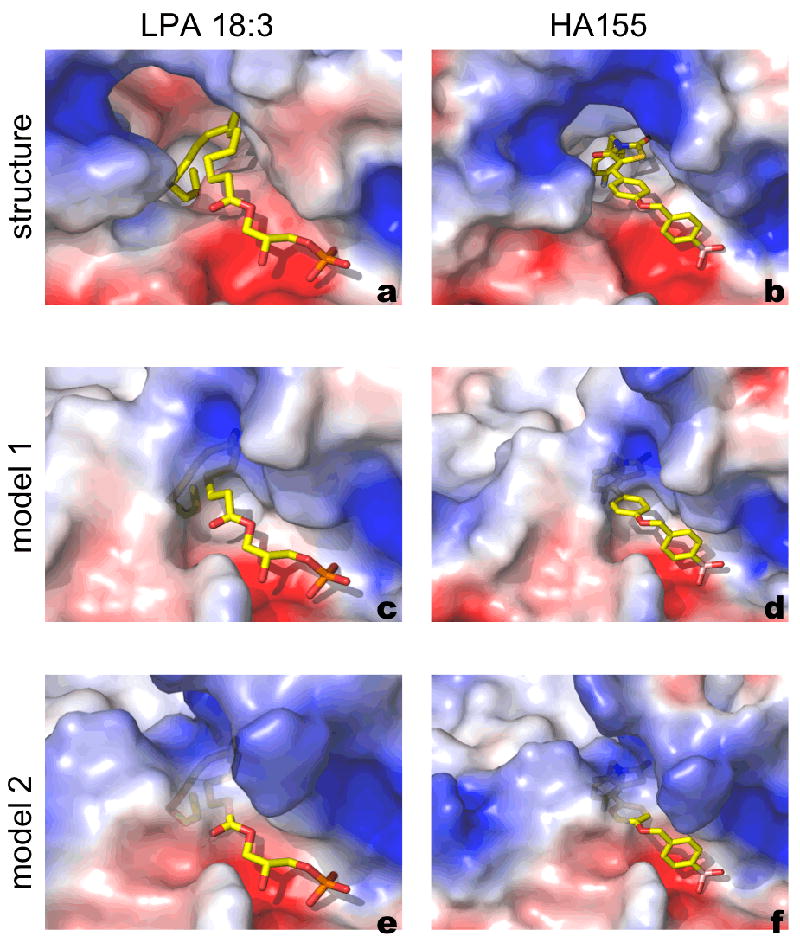
Binding poses of LPA 18:3 and HA155 to ATX structure and models. A semi-transparent surface representation focusing on the substrate binding pocket, coloured by electrostatic potential, for the crystal structure of a. murine ATX bound to LPC (PDB:3NKQ); b. the crystal structure of rat ATX bound to LPC (PDB:2XRG) to HA155 and for the top scoring models available from CASP9 (c-f). The Figure was prepared in PyMol (www.pymol.org).
Structure of the DNA-Binding JBP1 domain (CASP ID - T0561, PDB ID - 2XSE)
The J-Binding Protein 1 (JBP1) is essential for biosynthesis and maintenance of DNA base-J (β-D-glucosyl-hydroxymethyluracil), which was discovered by the Borst group in the nuclear DNA of the African trypanosome Trypanosoma brucei.40 The JBP1 protein and base J are essential for survival of pathogenic species of Leishmania, but absent from higher eukaryotes, prokaryotes and viruses, making them an attractive target for specific drug development.41
To determine exactly how JBP1 binds to J-DNA, we have performed a limited proteolysis experiment that identified a 160-residue domain that binds J-DNA, the DNA-Binding JBP1 domain (DB-JBP1). Using biophysical techniques we showed that DB-JBP1 binds to J-DNA 10,000 times better than to T-DNA, with approximately the same affinity and specificity as the full-length JBP1. In 2010 we determined the crystal structure of DB-JBP (PDB: 2XSE),42 revealing a novel “helical bouquet” fold – this 160-residue domain, which has no detectable sequence similarity with other known proteins, was submitted to the CASP-9 experiment. Based on that structure we were able to show that a single aspartate residue in the JBP1 recognition helix (Figure 5) is essential for specific J-base recognition in vitro and for the function of JBP1 in vivo.
Figure 5.

Cartoon diagram of the DB-JBP1 domain, showing the helical bouquet fold and the aspartate residue responsible for J-DNA recognition. The helices are colored in rainbow colors from the N- to the C- terminus; a few residues in the N-terminus have been omitted for clarity.
The DB-JBP1 “helical bouquet” is made by five helices, of which the four longer ones run anti-parallel in the same approximate orientation (the “flowers” of the bouquet), while one short helix is perpendicular to this arrangement, creating a “ribbon” running across the front. At the end of the ribbon helix the aspartate residue responsible for recognizing base J is located (Figure 5). Intriguingly, the loop that follows this helix and leads to the C-terminal helix of the fold, is disordered in the crystal structure and was not modeled. The helical bouquet fold is a divergent variant of the aberrant three-helical bundle helix-turn-helix (HTH) domains,43 where the ribbon helix in the DB-JBP1 helical bouquet corresponds to the recognition helix of the core three-helical bundle.
All groups in CASP9 failed to produce a model that would had been useful to derive any of the biochemical conclusions listed above. The sequence-dependent RMSD for superposed Cα atoms in the correct sequence context was more than 10 Å for all complete models. As expected, none of these models were useful for crystallographic structure solution using the molecular replacement method. Importantly, most of the top-scoring models have not built the recognition helix as a continuous helix of the right length, while they recognized most other helices of the fold but have placed them in the wrong orientation relative to each other.
Small and difficult to predict protein domains characterized by X-Ray crystallography and NMR spectroscopy at NESG (CASP ID - T0624, PDB ID - 3NRL; CASP ID - T0555, PDB ID - 2L06)
One of the more challenging prediction targets at CASP9 was T0624, an uncharacterized 73 residue domain from Ruminococcus gnavus (NESG id: UgR76, UniProt/TrEMBL: A7B1J1_RUMGN, PDB: 3NRL). This protein domain target was selected for structure determination during the second half of PSI-2 (Protein Structure Initiative – phase 2) since it possessed a predicted domain sequence that was over-represented in the genomes of human gut flora.
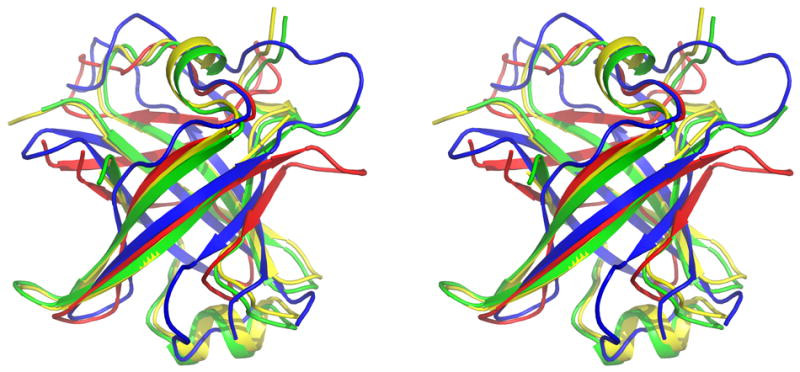
The crystal structure of UgR76 was determined at 1.9 Å by SAD methods by the Northeast Structural Genomics Consortium (NESG). The crystal structure has one globular domain of 68 residues that consists of six antiparallel β-sheets and has a fold typical for PDZ-like domains (Figure 6 A). There are two domains present in the asymmetric unit that form a tight dimer which is stabilized by the formation of number of hydrogen bonds and salt bridges as well as hydrophobic contacts. The existence of the dimer under physiological conditions has been confirmed by static light scattering experiments.
Figure 6.

(A) Crystal structure has one globular domain the of 68 residue PDZ-like domain that consists of six antiparallel β-sheets. (B) T0624’s domain fold is structurally homologous to one of the PDZ domains of the following five structures,(1) Aminopeptidase, M42 Family (3CPX, 17% identity) (2) FRV Operon protein FRVX (1XF0, 6% identity), (3) Ribosome maturation factor RIMM (3H9N, 17% identity), (4)FRV Operon protein FRVX (1Y0Y, 6% identity),(5) Elongation Factor TU (1HA3, identity 10%).
A DALI 44 search for structural homologs in PDB shows that the domain fold is structurally homologous to one of the PDZ domains of the following five structures, (1) Aminopeptidase, M42 Family (3CPX, 17% identity) (2) FRV Operon protein FRVX (1XF0, 6% identity), (3) Ribosome maturation factor RIMM (3H9N, 17% identity), (4) FRV Operon protein FRVX (1Y0Y, 6% identity), (5) Elongation Factor TU (1HA3, identity 10%). The monomeric structures superposed with a RMSD less than 3.4 Å. The overall fold of the polypeptide chain is similar though the sequence similarity between the structures are particularly low which likely contributed to the difficulty of predicting its structure. The significant difference between the structures is the in the loop region where the backbone Cα atoms differ by 5-20Å. However, pair wise structural superpositions show clear topological similarities among the core region though differences in the relative orientations of the loops are observed (Figure 6 B). PDZ domains are commonly found in a multitude of signaling pathways.45 Though this domain structure is not a novel fold, the novel loops may prove to be biomedically important as to how Ruminococcus gnavus interacts with it human host.
Another challenging prediction target from the NESG at CASP9 was T0555, a 147 residue domain from Phycobilisome (PBS) core-membrane linker (LCM) phycobiliprotein ApcE from Synechocystis sp. PCC 6803 (NESG: SgR209C, UniProt/Swiss-Prot: APCE_SYNY3, PDB: 2L06). The solution structure of the second PBS linker domain (residues 254-400) from the 896 residue ApcE was solved at the NESG by NMR. This protein target was selected for structure determination during the first half of PSI-2 since HMM analysis revealed that it possessed a BIG domain 46 (PFAM domain PF00427: PBS_linker_poly) found primarily in cyanobacteria and red algae.47
These linker domains are responsible for the structure and function of the phycobilosomes (PBS), the light-harvesting complexes that act as antennae of photosystem II and consequently allow light energy to be utilized for photosynthesis.47,48 Full-length ApcE has over 30 homologs identified in species of cyanobacteria and red algae with the same architecture, consisting of a phycopiliprotein (PBP)-like domain, which includes a phycocyanobilin chromophore binding site, followed by three PBS linker domains (Pfam 49, Cyanobase 50). ApcE, also called the anchor polypeptide, is the largest component of the PBS and the PBS linker domains are believed to stabilize the PBPs structure and determine their positions within the PBS, and additionally to modulate the spectroscopic properties of the PBS complex.47,51 All three PBS linker domains of the Synechocystis ApcE belong to the same family and share about 40% sequence identity.
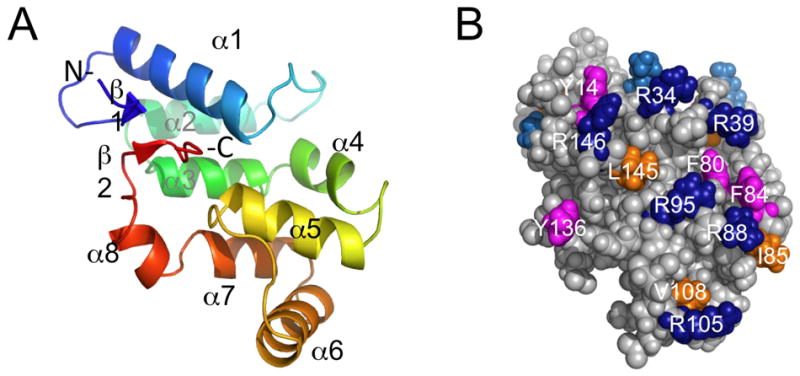
The solution NMR structure of the second PBS linker domain from ApcE is comprised of eight α-helices, and two one-residue antiparallel β-strands (β1, A15; β2, T141) that bring together the N- and C-termini (Figure 7A). The N-terminal 13 residues (P1-L13) and C-terminal 4 residues (Y144-G147 + His6 tag) are unstructured. Numbering is relative to the PBS linker polypeptide domain (P1 is P264 of full length ApcE). The overall shape of this domain is very pancake-like with a single elongated hydrophobic core. Three helices are aligned in a parallel alignment with a head-to-tail orientation on the backside of the protein (α2, α3, and α7). The front side of the protein has α 1 and α 5 in a plane with their C-termini close together. The other three helices are located around the edges of the pancake. The β-strands have contact with many of the helices (α1, α2, α3, α5, and α8) and have adjacent residues that are packed in the hydrophobic core as well (M16 and V142). The protein fold is the same as the crystal structures of the second and third PBS linker domains, subsequently solved by the NESG (PDB IDs 3OSJ and 3OHW). One difference between the NMR and crystal structures of the second PBS linker domain is a 310 helix (K43-Y45) N-terminal to α2, which is present in some of the crystal subunits and is next to a nitrate group (subunit D). An interesting difference between the second and third PBS linker domains is the absence of the antiparallel β-strands; the N-terminal residues are in an alternate conformation in the third PBS linker domain.
Figure 7. ApcE from Synechocystis.
(A) Cartoon view of the solution NMR structure of second PBS linker domain from ApcE (PDB ID, 2L06, residues 12 to 146). (B) Surface view with selected conserved surface residues colored (aromatic, magenta; ILV, orange; R, dark blue; K, blue.
The difficulty in predicting this structure may come from the large number of possible packing arrangements of the eight α-helices. Each helix has contact with 2-6 other helices (PDBsum 52). Even when the correct backbone chain meandering is predicted, the correct angle of the helices may be incorrect due to the numerous packing options. An additional complication comes from prediction of the small β-strands. In order for these strands to have the correct topology, the N- and C-termini need to end up in proximity to each other. Changes in helical tip angles, will influence the final distance between the N- and C-termini. Additionally, the favorable stabilization energy resulting from the small β-strands and their packing would not be expected to be very large. However, the placement of these β-strands may have a large effect on the 5 other helices that they contact, in particular α1, which has the most contact with these strands.
It has been suggested that PBS linker domains, which are basic, interact with acidic PBP domains via charged and hydrophobic interactions.48 The linker domain described here has many conserved solvent exposed charged and hydrophobic residues that may be important for the tight, structurally important interactions in the PBS. The second PBS linker domain is conserved among AcpE from cyanobacteria (> 70% sequence identity), and conserved surface residues are located over most of the surface as predicted by ConSurf.28 There are several conserved solvent exposed aromatic residues (Y14, Y45, F80, F84, and Y136), other hydrophobic residues (L20, I41, I85, V108, I114, L145), along with many conserved K and R residues (Figure 7 B). Prediction of the unfavorable, solvent-exposed, hydrophobic residues may be another reason that the structure of this protein was challenging to predict.
HSP90 activators PFC0360w and PFC0270w from Plasmodium falciparum (CASP ID - T0594, PDB ID - 3NI8; CASP ID - T0566, PDB ID - 3N72)
The Heat shock protein 90 (Hsp90) is a widespread molecular chaperone, assisting the maturation process of many proteins involved in cellular signaling and cell cycle regulation. Its chaperone function is dependent upon a series of conformational changes and domain rearrangements tied to an enzymatic ATPase activity. Additionally, a cohort of co-chaperones assists Hsp90 to fulfill its cellular role with several among them modulating its enzymatic activity. One of such is AHA1 (Activator of Hsp90 ATPase homolog 1), first isolated in Saccharomyces cerevisiae and the only known enhancer of the molecular chaperone’s ATPase activity. In humans and yeast, AHA1 is composed of two domains, both required for tight binding to the chaperone. The N-terminal domain is typically referred to as the AHA1-N domain and known to interact with the middle domain of Hsp90. The corresponding yeast complex has been previously captured in a crystallographic structure (PDB: 1USU 53). The C-terminal domain, known as the AHSA1 domain, also contributes by interacting with the N-terminal domain of Hsp90.54 Plasmodium parasites feature a somewhat different co-chaperone system. The gene PFC0270w encodes a protein with two AHA1-N-like domains, with the AHSA1 homologue encoded by an independent gene, namely PFC0360w.
SGC has solved the crystallographic structures of PFC0360w (PDB ID: 3NI8, Figure 8 A) and the N-terminal domain of PFC0270w (PDB ID: 3N72, Figure 8 B). They are highly similar to the AHA1-N domain of yeast and the human AHSA1 structure (PDB: 1X53), and thereby comprise relatively straight-forward template based modeling targets.
Figure 8.

(A) Structure of PFC0370w, a AHSA1 homologue and (B) Structure of N-terminal AHA1-N-like domain of PFC0270w
The 3NI8 structure features an N-terminal β-strand (β1) leading into a bent α-helix, which is followed by a convex anti-parallel β-mesh (that includes β1) and ends with a α-helix running parallel to β1. Despite relatively low sequence homology (≤ 30% identity), this structure shares the same fold with previous structures of activator chaperones (e.g. PDB ID 1X53, 1XFS, 1XUV), some of which were used as templates for the predictive model. Therefore, it is not a surprise that the latter shows excellent alignment with the crystal structure.
The 3N72 structure is a cylindrical fold with a α-helix leading into a long β-mesh and another α-helix in the C-terminus, which aligns tightly with the above-mentioned yeast homologues in the PDB, despite a relatively low sequence identity of ~ 28%. The use of 1USU and 1USV as templates resulted in predictive models deviating only slightly from the crystal structure.
2-oxo-3-deoxygalactonate kinase from Klebsiella pneumoniae (CASP ID - T0628, PDB ID - 3R1X)
The majority of organisms utilize D-galactose through the well-known Leloir pathway converting the sugar to D-glucose-1-phosphate. However, in some species galactose can also be metabolized through the so-called De Ley – Doudoroff pathway.55 This series of five reactions catalyzed by distinct enzymes converts D-galactose into pyruvate and D-glyceraldehyde 3-phosphate. In one of the steps, 2-oxo-3-deoxygalactonate (KDGal) is phosphorylated to 6-P-2-oxo-3-deoxygalactonate by its cognate kinase using ATP as a cofactor.
The MCSG selected KDGal kinase as a target for crystallographic analysis because it belongs to the medium size Pfam family (PF05035), for which no structural information was available in public databases.
Sequence analysis suggested that the protein is related to sugar kinases. Indeed, the crystal structure, determined at 2.1 Å resolution, confirmed that KDGal kinase belongs to the ASKHA (Acetate and Sugar Kinases/Hsc70/Actin) superfamily.56 The ASKHA proteins are typically composed of two closely related domains, each of which contains a common core with the topology β1β2β3α1β4α2β5α3.57 Unique features of each family representative are provided by additional elements inserted between the conserved motifs. This is also observed in the KDGal kinase structure. The C-terminal domain possesses a fully preserved ASKHA core, with an addition consisting of four extra helices (α5, 3(10)6, α7 and α8, Figure 9) inserted between the β3 - α1 elements. The N-terminal fragment of the protein contains an incomplete ASKHA core that lacks helix α2. Instead of this helix, the β4 - β5 region bears an insertion comprised of several additional secondary structure elements (3(10)2, β5, β6, α3, β7). A second insertion providing two extra β-strands (β9, β10) is present between the β5 and α3 elements.
Figure 9.

Superposition of the crystallographic model (color) of 2-oxo-3-deoxygalactonate kinase with the computed model T0628TS104_1 (grey). Green and cyan colors show the conserved βββαβαβα ASKHA core within the N- and C-terminal domains, respectively. Yellow and red colors indicate β4-β5 and β5-α3 insertions in the N-terminal domain, while the blue color shows the β3-α1 insertion in the C-terminal domain. Selected secondary structure elements form the insertions are labeled. In the right panel, the α2 helix that does not exist in the experimental structure but is present in the predicted model is marked.
Structural comparisons allowed identification of the putative active site of KDGal kinase, which is located in a deep groove between the two domains. The pocket can be divided into two sub-sites, namely the KDGal binding site and the ATP binding site. The former is localized near one of the insertions provided by the N-terminal domain. The nucleotide-binding site is formed by three ASKHA signature motifs: ADENOSINE, P1 and P2, which define fragments involved in the recognition of adenosine and phosphoryl groups. The ADENOSINE motif, which is provided by the C-terminal domain, usually forms a hydrophobic cavity suitable to accommodate the adenine ring. Interestingly, in the experimental structure of KDGal kinase, the adenine-dedicated pocket from the ADENOSINE motif is not well-defined. The P1 and P2 regions correspond to the β1 - β2 and β11 - β12 hairpins contributed by the N- and C-terminal domains, respectively. These elements represent the most conserved nucleotide-anchoring units of the ASKHA members and they are also present in the KDGal kinase structure.
In retrospect, our analysis clearly shows that the KDGal kinase contains conserved structural elements that have been observed before in many members of the ASKHA superfamily, such as the βββαβαβα core. At the same time, however, the protein presents a number of surprising features. These include the deletion of the α2 helix from the N-terminal core and several unique insertions. Clearly, these unexpected characteristics made the ab initio structure modeling more challenging. Strikingly, when the individual domains are considered, the best predictions for the C-terminal domain received higher scores than the best models for the N-terminal domain. Such results could be attributed to the fact that the C-terminal part contains a complete ASKHA core and has only one insertion. The N-terminal domain, on the other hand, is less conserved within its central portion and also contains two significant insertions. Notably, none of the four best-ranked in silico-generated models predicted secondary structure elements within these insertions, leaving them as long loops. Moreover, possibly misled by the available templates, the computed N-terminal domains contain the non-existing α2 helix. In effect, the β4 - β5 insertion could not be positioned properly. Although the localization of the β5 - α3 region was generally better predicted, failure to model the β9 - β10 hairpin has more dramatic consequences than the β4 - β5 misplacement: it means that the generated substrate-binding site is quite a distant approximation of the experimental structure. The insertion within the C-terminal domain also presented challenges. Namely, the modeled α7 helix is shifted towards the interdomain groove. Position of this helix might be crucial for ATP binding.
Overall, the CASP9 results indicate that conserved fragments of the protein can be predicted reasonably well. The top-ranked model of full-length protein is characterized by an rmsd of 2.86 Å, which could be expected taking into account that the closest homolog available at the time of competition had only 12% sequence identity (butyrate kinase58, PDB code 1X9J). This model, however, as well as other highly scored models, failed to correctly build the substrate-binding pocket. We suspect that, in this particular case, the agreement between the experimental and computed results would be better if the algorithms were able to properly assign secondary structure elements.
Conclusions
Over the last two decades, significant progress in the performance and accuracy of protein structure precision methods has been observed, as quantified by the biannual CASP experiment.59,60 While comparative methods are often able to model conserved structural features of target proteins, there is still significant need for improvement in the predictive power of modeling techniques to correctly predict the unexpected and unique features of a target protein, which define its specific molecular function and biological role. Therefore, the broad support of the CASP experiment by the experimental protein structural biology community is crucial to drive the development of improved protein structure prediction techniques. The aim of this manuscript was to illustrate structurally and functionally relevant aspects of some of the CASP9 target proteins from the experimentalist’s perspective. These insights underline on one hand the need for the CASP assessment to evaluate different features of a protein’s structure such as its oligomeric state or the accuracy of ligand binding pockets within the predicted structures.4,61 On the other side, this overview highlights that there is still ample opportunity for methods development in structure and function prediction.
Acknowledgments
M.J.v.R. thanks the Spanish Ministry of Education and Science for grant BFU2008-01588 and the European Commission for Contract NMP4-CT-2006-033256. J.M.O. thanks the Spanish Ministry of Education and Science for a José Castillejo fellowship and the Xunta de Galicia for an Angeles Alvariño fellowship. Work on PGK was supported by National Institutes of Health grants K22-CA124517 to D.E.C. and R01-GM090161 to C.K. The NESG authors thank K. Hamilton, C. Ciccosanti, D. Lee, H. Janjua, T. B. Acton and R. Xiao at the Rutgers University protein production facility for technical support, as well as J. R. Cort for NMR data collection at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, and Y. Yang for NMR data collection at at the Ohio Biomedicine Center of Excellence in Structural Biology and Metabonomics at Miami University. Work on 2-oxo-3-deoxygalactonate kinase was supported by National Institutes of Health grants GM074942, GM094585, and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357 (AJ) and by a scholarship from Foundation for Polish Science (KM). Work by the RCSB PDB was supported by NSF DBI 0829586.
Abbreviations
- RMSD
Root mean square deviation
Footnotes
Contributions The parts of the manuscript on target T0629 were contributed by MvR, SB & JO; target T0605 by DC & CK; target T0531 by FR, AL & JB; target T0543 by JH, EC & AP; target T0561 by TH & AP; targets T0624 and T0555 by TR, SJ, JH, MK, LT, JE & GM; targets T0594 and T0566 by TH, AW, JP & RH; and target T0628 by KM & AJ. Editing, introduction, discussion and coordination by AK, JM and TS.
Contributor Information
Andriy Kryshtafovych, Genome Center, University of California, Davis, 451 Health Sciences Drive, Davis, CA 95616, USA.
John Moult, Institute for Bioscience and Biotechnology Research, Department of Cell Biology and Molecular genetics, University of Maryland, 9600 Gudelsky Drive, Rockville, MD 20850, USA.
Sergio G. Bartual, Departamento de Cristalografia y Biologia Estructural, Instituto de Quimica-Fisica Rocasolano, Consejo Superior de Investigaciones Cientificas, calle Serrano 119, E-28006 Madrid, Spain Departamento de Bioquimica y Biologia Molecular, Facultad de Farmacia, Campus Vida, Universidad de Santiago de Compostela, E-15782 Santiago de Compostela, Spain.
J. Fernando Bazan, Genentech, Departments of Protein Engineering and Structural Biology, 1 DNA Way, South San Francisco, CA 94080, USA.
Helen Berman, Rutgers, the State University of New Jersey, RCSB PDB, 610 Taylor Road Piscataway, NJ, USA 08854-8087.
Darren E. Casteel, University of California San Diego, Department of Medicine, 9500 Gilman Dr., La Jolla, CA 92093-0652, USA
Evangelos Christodoulou, Department of Biochemistry, NKI, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands.
John K. Everett, Center for Advanced Biotechnology and Medicine, Department of Molecular Biology and Biochemistry, and Northeast Structural Genomics Consortium (NESG), Rutgers, The State University of New Jersey, Piscataway, NJ, 08854, USA
Jens Hausmann, Department of Biochemistry, NKI, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands.
Tatjana Heidebrecht, Department of Biochemistry, NKI, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands.
Tanya Hills, Structural Genomics Consortium (SGC), MaRS Centre, South Tower, 101 College St., Suite 700, Toronto, ON, M5G 1L7, Canada.
Raymond Hui, Structural Genomics Consortium (SGC), MaRS Centre, South Tower, 101 College St., Suite 700, Toronto, ON, M5G 1L7, Canada.
John F. Hunt, Department of Biological Sciences, Northeast Structural Genomics Consortium (NESG), Columbia University, New York, NY 10027, USA
Seetharaman Jayaraman, Department of Biological Sciences, Northeast Structural Genomics Consortium (NESG), Columbia University, New York, NY 10027, USA.
Andrzej Joachimiak, Midwest Center for Structural Genomics (MCSG), Biosciences Division, Argonne National Laboratory, USA; Structural Biology Center, Biosciences Division, Argonne National Laboratory, USA; Department of Biochemistry and Molecular Biology, University of Chicago, USA.
Michael A. Kennedy, Department of Chemistry and Biochemistry, Northeast Structural Genomics Consortium (NESG), Miami University, Oxford, OH 45056, USA
Choel Kim, Department of Pharmacology, The Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA.
Andreas Lingel, Genentech, Departments of Protein Engineering and Structural Biology, 1 DNA Way, South San Francisco, CA 94080, USA.
Karolina Michalska, Midwest Center for Structural Genomics, Biosciences Division, Argonne National Laboratory, USA; on the leave from Adam Mickiewicz University, Poznan, Poland.
Gaetano T. Montelione, Rutgers, The State University of New Jersey, Robert Wood Johnson Medical School, and Northeast Structural Genomics Consortium (NESG), Center for Advanced Biotechnology and Medicine, and Department of Molecular Biology and Biochemistry. CABM 679 Hoes Lane, Piscataway NJ 08854, USA
José M. Otero, Departamento de Bioquimica y Biologia Molecular, Facultad de Farmacia, Campus Vida, Universidad de Santiago de Compostela, E-15782 Santiago de Compostela, Spain
Anastassis Perrakis, Department of Biochemistry, NKI, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands.
Juan C. Pizarro, Structural Genomics Consortium, MaRS Centre, South Tower, 101 College St., Suite 700, Toronto, ON, M5G 1L7, Canada
Mark J. van Raaij, Departamento de Estructura de Macromoléculas, Centro Nacional de Biotecnología - CSIC, c/Darwin 3, E-28049 Madrid, Spain Departamento de Bioquimica y Biologia Molecular, Facultad de Farmacia, Campus Vida, Universidad de Santiago de Compostela, E-15782 Santiago de Compostela, Spain.
Theresa A. Ramelot, Department of Chemistry and Biochemistry, Northeast Structural Genomics Consortium (NESG), Miami University, Oxford, Ohio 45056
Francois Rousseau, Genentech, Departments of Protein Engineering and Structural Biology, 1 DNA Way South San Francisco, CA, USA 94080.
Liang Tong, Department of Biological Sciences and Northeast Structural Genomics Consortium (NESG), Columbia University, 1212 Amsterdam Avenue, New York, NY 10027, USA.
Amy K. Wernimont, Structural Genomics Consortium (SGC), MaRS Centre, South Tower, 101 College St., Suite 700, Toronto, ON, M5G 1L7, Canada
Jasmine Young, RCSB PDB, Rutgers, the State University of New Jersey, Piscataway, NJ 08854-8087, USA.
Torsten Schwede, Biozentrum University of Basel and SIB Swiss Institute of Bioinformatics, Klingelbergstrasse 50-70, 4056 Basel, Switzerland.
References
- 1.Kinch L, Grishin N, et al. CASP9 Target Classification. Proteins. 2011 doi: 10.1002/prot.23190. THIS ISSUE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grishin N, et al. Asessment of CASP9 FM predictions. Proteins. 2011 doi: 10.1002/prot.23181. THIS ISSUE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariani V, Kiefer F, Schmidt T, Haas J, Schwede T. Assessment of template based predictions in CASP9. Proteins. 2011 doi: 10.1002/prot.23177. THIS ISSUE. [DOI] [PubMed] [Google Scholar]
- 5.Ackermann HW. Tailed bacteriophages: the order caudovirales. Adv Virus Res. 1998;51:135–201. doi: 10.1016/S0065-3527(08)60785-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossmann MG. Morphogenesis of the T4 tail and tail fibers. Virol J. 2010;7:355. doi: 10.1186/1743-422X-7-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karam G. Molecular Biology of Bacteriophage T4. Washington DC: ASM Press; 1994. [Google Scholar]
- 8.Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. The bacteriophage T4 DNA injection machine. Curr Opin Struct Biol. 2004;14(2):171–180. doi: 10.1016/j.sbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Cerritelli ME, Wall JS, Simon MN, Conway JF, Steven AC. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: a hinged viral adhesin. J Mol Biol. 1996;260(5):767–780. doi: 10.1006/jmbi.1996.0436. [DOI] [PubMed] [Google Scholar]
- 10.Bartual SG, Garcia-Doval C, Alonso J, Schoehn G, van Raaij MJ. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr Purif. 2010;70(1):116–121. doi: 10.1016/j.pep.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Bartual SG, Otero JM, Garcia-Doval C, Llamas-Saiz AL, Kahn R, Fox GC, van Raaij MJ. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc Natl Acad Sci U S A. 2010;107(47):20287–20292. doi: 10.1073/pnas.1011218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG. Evolution of bacteriophage tails: Structure of T4 gene product 10. J Mol Biol. 2006;358(3):912–921. doi: 10.1016/j.jmb.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 13.Leiman PG, Kostyuchenko VA, Shneider MM, Kurochkina LP, Mesyanzhinov VV, Rossmann MG. Structure of bacteriophage T4 gene product 11, the interface between the baseplate and short tail fibers. J Mol Biol. 2000;301(4):975–985. doi: 10.1006/jmbi.2000.3989. [DOI] [PubMed] [Google Scholar]
- 14.Thomassen E, Gielen G, Schutz M, Schoehn G, Abrahams JP, Miller S, van Raaij MJ. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J Mol Biol. 2003;331(2):361–373. doi: 10.1016/s0022-2836(03)00755-1. [DOI] [PubMed] [Google Scholar]
- 15.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casteel DE, Smith-Nguyen EV, Sankaran B, Roh SH, Pilz RB, Kim C. A crystal structure of the cyclic GMP-dependent protein kinase I{beta} dimerization/docking domain reveals molecular details of isoform-specific anchoring. J Biol Chem. 2010;285(43):32684–32688. doi: 10.1074/jbc.C110.161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. Chembiochem. 2004;5(2):170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 18.Strauss HM, Keller S. Handb Exp Pharmacol. 2008;186:461–482. doi: 10.1007/978-3-540-72843-6_19. [DOI] [PubMed] [Google Scholar]
- 19.Grigoryan G, Keating AE. Structural specificity in coiled-coil interactions. Curr Opin Struct Biol. 2008;18(4):477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu YB. Coiled-coils: stability, specificity, and drug delivery potential. Adv Drug Deliv Rev. 2002;54(8):1113–1129. doi: 10.1016/s0169-409x(02)00058-3. [DOI] [PubMed] [Google Scholar]
- 21.Richie-Jannetta R, Francis SH, Corbin JD. Dimerization of cGMP-dependent protein kinase Ibeta is mediated by an extensive amino-terminal leucine zipper motif, and dimerization modulates enzyme function. J Biol Chem. 2003;278(50):50070–50079. doi: 10.1074/jbc.M306796200. [DOI] [PubMed] [Google Scholar]
- 22.O’Shea EK, Lumb KJ, Kim PS. Peptide ‘Velcro’: design of a heterodimeric coiled coil. Curr Biol. 1993;3(10):658–667. doi: 10.1016/0960-9822(93)90063-t. [DOI] [PubMed] [Google Scholar]
- 23.Casteel DE, Boss GR, Pilz RB. Identification of the interface between cGMP-dependent protein kinase Ibeta and its interaction partners TFII-I and IRAG reveals a common interaction motif. J Biol Chem. 2005;280(46):38211–38218. doi: 10.1074/jbc.M507021200. [DOI] [PubMed] [Google Scholar]
- 24.Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, Aronovitz M, Baur W, Ohtani K, Wilkerson MK, Bonev AD, Nelson MT, Karas RH, Mendelsohn ME. High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci U S A. 2008;105(18):6702–6707. doi: 10.1073/pnas.0802128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatakeyama S, Osawa M, Omine M, Ishikawa F. JTB: a novel membrane protein gene at 1q21 rearranged in a jumping translocation. Oncogene. 1999;18(12):2085–2090. doi: 10.1038/sj.onc.1202510. [DOI] [PubMed] [Google Scholar]
- 26.Platica O, Chen S, Ivan E, Lopingco MC, Holland JF, Platica M. PAR, a novel androgen regulated gene, ubiquitously expressed in normal and malignant cells. Int J Oncol. 2000;16(5):1055–1061. doi: 10.3892/ijo.16.5.1055. [DOI] [PubMed] [Google Scholar]
- 27.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 28.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server issue):W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 30.Iwasaki W, Nagata K, Hatanaka H, Inui T, Kimura T, Muramatsu T, Yoshida K, Tasumi M, Inagaki F. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997;16(23):6936–6946. doi: 10.1093/emboj/16.23.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. 2005;30(10):542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277(42):39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 33.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158(2):227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noguchi K, Herr D, Mutoh T, Chun J. Lysophosphatidic acid (LPA) and its receptors. Curr Opin Pharmacol. 2009;9(1):15–23. doi: 10.1016/j.coph.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 35.van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem. 2005;280(22):21155–21161. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 36.Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, Schuurman K, van Tellingen O, Morris AJ, Smyth SS, Moolenaar WH, Ovaa H. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci U S A. 2010;107(16):7257–7262. doi: 10.1073/pnas.1001529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, Nureki O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat Struct Mol Biol. 2011;18(2):205–212. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- 38.Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM, van Meeteren LA, Houben AJ, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CW, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat Struct Mol Biol. 2011;18(2):198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: implications for mechanism and evolution. Biochemistry. 2006;45(32):9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 40.Gommers-Ampt JH, Van Leeuwen F, de Beer AL, Vliegenthart JF, Dizdaroglu M, Kowalak JA, Crain PF, Borst P. beta-D-glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell. 1993;75(6):1129–1136. doi: 10.1016/0092-8674(93)90322-h. [DOI] [PubMed] [Google Scholar]
- 41.Borst P, Sabatini R. Base J: discovery, biosynthesis, and possible functions. Annu Rev Microbiol. 2008;62:235–251. doi: 10.1146/annurev.micro.62.081307.162750. [DOI] [PubMed] [Google Scholar]
- 42.Heidebrecht T, Christodoulou E, Chalmers MJ, Jan S, Ter Riet B, Grover RK, Joosten RP, Littler D, van Luenen H, Griffin PR, Wentworth P, Jr, Borst P, Perrakis A. The structural basis for recognition of base J containing DNA by a novel DNA binding domain in JBP1. Nucleic Acids Res. 2011;39(13):5715–5728. doi: 10.1093/nar/gkr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29(2):231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Dessailly BH, Nair R, Jaroszewski L, Fajardo JE, Kouranov A, Lee D, Fiser A, Godzik A, Rost B, Orengo C. PSI-2: structural genomics to cover protein domain family space. Structure. 2009;17(6):869–881. doi: 10.1016/j.str.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu LN, Chen XL, Zhang YZ, Zhou BC. Characterization, structure and function of linker polypeptides in phycobilisomes of cyanobacteria and red algae: an overview. Biochim Biophys Acta. 2005;1708(2):133–142. doi: 10.1016/j.bbabio.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Sidler WA. Phycobilisome and phycobiliprotein structures. In: Bryant DA, editor. Molecular Biology of the Cyanobacteria. Dordrecht: Kluwer Academic Publishing; 1994. pp. 139–216. [Google Scholar]
- 49.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30(1):276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakao M, Okamoto S, Kohara M, Fujishiro T, Fujisawa T, Sato S, Tabata S, Kaneko T, Nakamura Y. CyanoBase: the cyanobacteria genome database update 2010. Nucleic Acids Res. 2010;38(Database issue):D379–381. doi: 10.1093/nar/gkp915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao KH, Su P, Bohm S, Song B, Zhou M, Bubenzer C, Scheer H. Reconstitution of phycobilisome core-membrane linker, LCM, by autocatalytic chromophore binding to ApcE. Biochim Biophys Acta. 2005;1706(1-2):81–87. doi: 10.1016/j.bbabio.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37(Database issue):D355–359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer P, Prodromou C, Liao C, Hu B, Roe SM, Vaughan CK, Vlasic I, Panaretou B, Piper PW, Pearl LH. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004;23(6):1402–1410. doi: 10.1038/sj.emboj.7600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, Richter K, Buchner J. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol Cell. 2010;37(3):344–354. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 55.De Ley J, Doudoroff M. The metabolism of D-galactose in Pseudomonas saccharophila. J Biol Chem. 1957;227(2):745–757. [PubMed] [Google Scholar]
- 56.Michalska K, Cuff ME, Tesar C, Feldmann B, Joachimiak A. Structure of 2-oxo-3-deoxygalactonate kinase from Klebsiella pneumoniae. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 8):678–689. doi: 10.1107/S0907444911021834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buss KA, Cooper DR, Ingram-Smith C, Ferry JG, Sanders DA, Hasson MS. Urkinase: structure of acetate kinase, a member of the ASKHA superfamily of phosphotransferases. J Bacteriol. 2001;183(2):680–686. doi: 10.1128/JB.183.2.680-686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diao J, Ma YD, Hasson MS. Open and closed conformations reveal induced fit movements in butyrate kinase 2 activation. Proteins. 2009 doi: 10.1002/prot22610. [DOI] [PubMed] [Google Scholar]
- 59.Moult J, Kryshtafovych A, Fidelis K. CASP9 results compared to those of previous CASP experiments. Proteins. 2011 doi: 10.1002/prot.23182. THIS ISSUE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kryshtafovych A, Venclovas C, Fidelis K, Moult J. Progress over the first decade of CASP experiments. Proteins. 2005;61(Suppl 7):225–236. doi: 10.1002/prot.20740. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt T, Haas J, Gallo Cassarino T, Schwede T. Assessment of ligand binding residue predictions in CASP9. Proteins. 2011 doi: 10.1002/prot.23174. THIS ISSUE. [DOI] [PMC free article] [PubMed] [Google Scholar]