


Abstract
The catalytic site identification web server provides the innovative capability to find structural matches to a user-specified catalytic site among all Protein Data Bank proteins rapidly (in less than a minute). The server also can examine a user-specified protein structure or model to identify structural matches to a library of catalytic sites. Finally, the server provides a database of pre-calculated matches between all Protein Data Bank proteins and the library of catalytic sites. The database has been used to derive a set of hypothesized novel enzymatic function annotations. In all cases, matches and putative binding sites (protein structure and surfaces) can be visualized interactively online. The website can be accessed at http://catsid.llnl.gov.
INTRODUCTION
Not surprisingly, in the post-genomic era, the focus on gene products and their functions has been generally determined by gene sequences and their sequence similarity to previously annotated sequences (1–10). However, many existing annotations, especially those derived solely through sequence similarity, are misleading or incorrect (11). To better understand gene products, structural genomic efforts have provided structures of thousands of proteins. Typically, for structurally similar proteins, functions are determined through template matching because proteins that adopt the same fold frequently exhibit the same function. A number of approaches use structural similarity (12–26) to determine protein function. However, sequence and structural homology are not sufficient to determine function of those proteins that have different global folds overall but similar functions. Thus, many proteins still do not have determined functions.
Several computational approaches that determine local structural similarity are able to capture functional similarity that is missed by global structural similarity algorithms (27–32). In general, these approaches emphasize the development of the local similarity-matching approach, rather than applying it to determining function. For the subset of proteins that catalyze reactions, the function of the protein can be determined by evaluating its enzymatic reaction, specifically determining the catalytic residues that perform the chemistry in the catalytic binding site. Enzymatic function is known to be shared among proteins having widely divergent sequences because key structural similarities are preserved (33). Torrance et al. (34) proposed that the spatial relationships of key ‘critical residues’ could be a method for assigning catalytic functions among disparate proteins. This hypothesis led to the development of the Catalytic Site Atlas (35), which is a compendium of catalytic sites and residues defining those sites, along with associated Enzyme Commission (EC) numbers. Many groups have consequently proposed methods that more specifically search for catalytic motifs as a way of determining function (36–40), including methods that explicitly incorporate the ligand (41).
Here, we report the catalytic site identification web server, which provides users protein annotations based on structural catalytic residues matched to known proteins with specified EC numbers. A feature of the catalytic site identification server is that it offers excellent performance in matching identified protein families through an EC number with as few as three amino acid residues.
Two main challenges need to be overcome to identify catalytic function: (i) solved structures typically are not available for the protein of interest and (ii) finding relevant structural matches with identified function may be computationally expensive. Homology modeling can address the first issue by building a 3D model of the protein from a known sequence and a homologous protein; see, e.g. (42). The catalytic site identification web server provides a way to address the second issue by scanning for structural matches in a library of catalytic sites derived from protein families whose members share catalytic function (35). The catalytic site identification web server supplements these catalytic site data with information on residue variation among catalytic site family members and also includes enzymatic site identifications from other sources [e.g. (43)].
Through a web interface, the catalytic site identification web server allows users to enter their own catalytic sites, identifies and scores potential protein matches to their catalytic sites, and allows visual inspection. Because the algorithm has been developed to generalize a catalytic site as any binding site that the user chooses to enter, the catalytic site identification web server also has the capability to rapidly scan the universe of known protein structures in Protein Data Bank (PDB) (44) for matches to any binding site. For example, after entering a specific catalytic site, the server can quickly produce a list of proteins that may have similar binding sites (anywhere on the protein) to the user-identified site, which is targeted by a specific drug candidate. The resulting list of proteins would be those proteins with a similar binding site such that the drug candidate could bind to an off-target protein, causing potential side effects (45). Additionally, allosteric binding sites on proteins, based on known binding sites, could also be identified (46). Thus, the catalytic site identification web server could be used for general binding site identification, depending on the user’s questions. These applications are the focus of further investigations.
MATERIALS AND METHODS
Search procedure
The catalytic site identification web server uses a highly efficient graph-based method to identify candidate matches to catalytic sites. The procedure treats sets of residues as nodes on a graph, with the distances between each residue its edges. To compute the distances, an atomic coordinate from the residue (usually the Cα atom of an amino acid) is chosen. A library of catalytic sites is pre-computed, and the catalytic site pattern is compared with possible patterns within the larger protein graphs. The procedure allows for residue substitutions in catalytic site identity as well. Catalytic sites are defined by the relative spatial coordinates of three or more ‘critical residues’. At least three residues are needed for the web server implementation; sites defined by fewer residues do not provide sufficient information for specificity in the search procedure. A complete description of the search algorithm is presented in a related work (47).
The web server incorporates several enhancements over the original design to search for relatively small sets of unknown targets. The present web implementation includes elements that can accommodate searches through the full PDB. Several optimizations have been developed, including pre-calculation and efficient storage of data, multithreading of the search procedure and an improved logistic regression classifier, whose descriptors are more rapidly computed such that the overall process to perform a catalytic site search against all PDB proteins can be completed in less than a minute.
The search procedure is in two stages, as shown in Figure 1. The first stage uses the rapid graph search procedure. From the graph procedure, the top 20 site-protein pairs are selected. The initial regression procedure incorporates the same distance matrix data that are used for the graph search. The output of the first stage is a refined subset of all hits that are sent to the second stage descriptor calculations, which require coordinate alignments to compute new descriptors. The output of the second stage regression is the list of candidate matches. The regression procedures are described in the next section.
Figure 1.
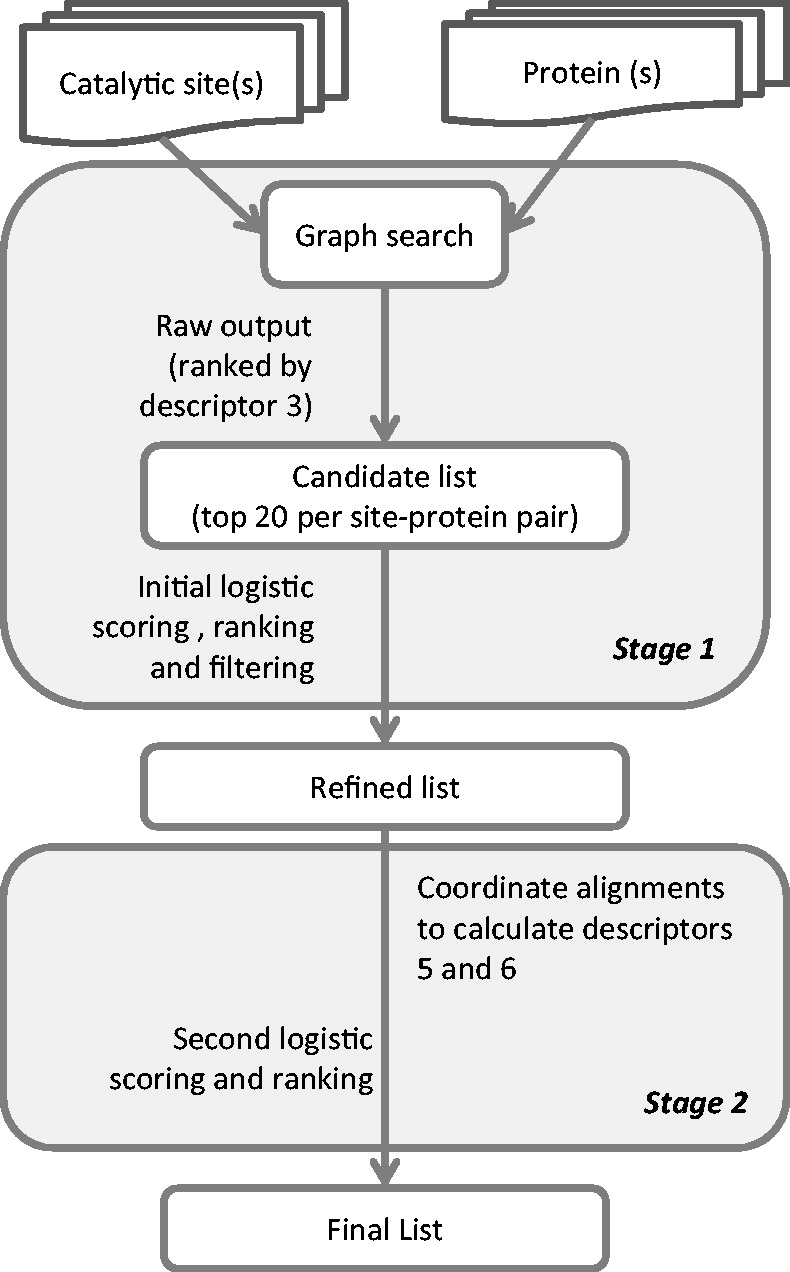
Process workflow for catalytic site identification. The logistic scoring and the descriptors are described in Table 1 and in Supplementary Methods.
Logistic regression classifiers
An important feature of the search procedure is the use of a classification procedure that allows for more systematic identification of true positives based on a set of physical descriptors. These descriptors provide information beyond that which is provided by the initial graph matching procedure and enhance the quality of the prediction considerably. We briefly discuss the specifics of our regression procedure here.
The logistic regression function, given as
| (1) |
where the variable  is a linear function of a set of descriptors
is a linear function of a set of descriptors
| (2) |
and an allowance for coefficients as a function of template size 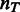 is made for the model used.
is made for the model used.
The logistic function is constructed such that a larger value indicates likelihood that the sample is a positive case (a match to the reference binding site), whereas a small number indicates a negative case. As aforementioned, the catalytic site identification web server uses two logistic classifiers. Table 1 shows the descriptors used in each classifier, along with the coefficient values that are used. The descriptors are defined in detail in the Supplementary Methods. The benchmarks and testing section describes the fitting procedure.
Table 1.
Logistic regression classifiers’ coefficient estimates and standard errors
| Descriptor | First-stage classifier | Second-stage classifier |
|
|---|---|---|---|
| Coefficient (standard error) | nT | Coefficient (standard error) | |
| Intercept | −12.19 (1.31)a | all | −5.62 (1.20)a |
| 1. Fraction residues correctly placed. (fixed distance threshold of 0.5 Å) | −6.27 (1.35)a | 3 | 1.62 (0.55)b |
| 4+ | −1.33 (1.42) | ||
| 2. Fraction residues correctly placed (relative distance threshold of 10%) | 5.39 (2.36)c | ||
| 3. Residue-pair distance difference | 0.83 (0.44)d | 3 | −0.25 (0.13)d |
| 4+ | −0.82 (0.23)a | ||
| 4. Normalized residue-pair distance difference | 1.40 (0.36)a | 3 | −0.24 (0.21) |
| 4+ | 0.20 (0.26) | ||
| 5. Position of backbone atoms | 3 | 0.96 (0.17)a | |
| 4+ | 1.14 (0.29)a | ||
| 6. Orientation of backbone atoms | 3 | 0.45 (0.12)a | |
| 4+ | 0.80 (0.16)a | ||
aSignificant at 0.1% level.
bSignificant at 1% level.
cSignficant at 5% level.
dSignificant at 10% level.
The first-stage and second-stage classifiers use different subsets of descriptors; the second-stage classifier distinguishes between coefficients applied to three-residue catalytic sites and sites having four or more residues (nT). The distance-difference descriptors enter the estimation as the transformed variable, d′ = 1/(0.1 + d), so that smaller distance differences are ‘better’ (i.e. are expected to have a positive coefficient) while avoiding singularities.
The subset of candidate matches from the first-stage ranking that proceed to the second-stage calculations is determined as follows. Catalytic site-protein pairs that score >0.06 are accepted, with the proviso that no more than 100 such pairs per protein will be accepted—unless the pair’s score is >0.35 (in which case that pair will be accepted). The proviso applies only in the case when library catalytic sites are being matched to user-specified proteins.
The second-stage descriptors are included in the logistic regression twice: once in application to catalytic sites having three residues and once in application to catalytic sites having four or more residues. This allows training different coefficients for three-residue catalytic sites and for four-or-more-residue catalytic sites.
Web server implementation
The graph search (described earlier in the text) is coded in C++, and the auxiliary scripts for input and output processing are coded in Python and Perl. Typically, the operating system holds in cache the pre-calculated distance matrices for both the library of catalytic site templates and PDB proteins, which avoids latency caused by accessing data stored on disk without the need for any special software or hardware.
The catalytic site identification compute server (which, for institutional reasons, is a different machine than the web server host) is a small cluster of eight compute nodes, as well as a master node and a database server node. Each compute node has 24 cores. Each catalytic site identification search job (i.e. a single web-server request) currently runs on a single node, dividing the search among the 24 cores with parallel calculations dynamically scheduled with OpenMP multithreading (48). As a result, a catalytic site-protein search comparison averages ∼6 ms on the compute server. With the search comparisons divided among 24 cores, the elapsed time to search all PDB proteins with one catalytic site template, including the second-stage backbone alignments and root-mean-square distance calculations, is ∼40 s.
RESULTS
Catalytic site identification web server
The catalytic site identification web server implements three main functions: (i) search PDB for matches to a user-specified catalytic site or sites, (ii) search the web server’s library of catalytic site templates for matches to a user-specified protein structure or structures (perhaps derived by homology modeling) or (iii) browse a database of pre-calculated matches between all PDB proteins and the library of catalytic sites, including a set of hypothesized novel enzymatic function annotations. In all cases, matches and putative binding sites (protein structure and surfaces) can be visualized interactively online.
Input
Search PDB for matches to catalytic sites
Users specify a catalytic site by indicating the critical residues that comprise the site (which may include ions and cofactors). The site may be in an existing PDB protein, from which coordinate data will be extracted or in a PDB-formatted coordinate file uploaded by the user. Residue-type substitutions may be specified. For example, a site that includes a glutamic acid (Glu) may be specified so that a protein with an aspartic acid (Asp) at that location can be a match candidate. Multiple catalytic sites can be defined in a single search request. Uploaded catalytic site coordinate data files can be saved on the server for future use.
Search the server’s library of catalytic sites for matches to proteins
Users upload a PDB-formatted protein coordinate file or files that can then be used to search the library of catalytic sites for matches. Uploaded protein files can be saved on the server for future use.
Browse database of matches between PDB proteins and library catalytic sites
The user inputs a PDB code, an EC number or a partial EC number to search PDB for proteins that match the catalytic site library.
Output
In all three cases, the output is a list of matches between proteins and catalytic sites, ordered with the highest-scoring match first. The results are presented in a table format as shown in Figure 2, which is the result of browsing for catalytic sites matching PDB protein 1deo. Starting from the left, the first column, ‘View’, provides a button to open the visualizer, as described later in the text. The second column, ‘Score’, shows the resulting match score as discussed under ‘Benchmarks and testing’. Evaluation of the scoring performance indicates that matches with scores >0.02 are a good indication of a ‘positive’, i.e. a likely correct assignment of catalytic function. The third column, ‘Catalytic site’, identifies the matching structures. The final characters of the catalytic site identifier—after the last hyphen—indicate a particular binding site for multimeric proteins, which may have their catalytic function on multiple chains; inclusion of the alternate binding sites in the web server’s library allows for structural variation. The fourth and fifth columns, ‘Catalytic site EC number’ and ‘Catalytic site EC label’, indicate the catalytic function associated with the catalytic site template structure. The last two columns on the right, ‘Catalytic site UniProt EC number’ and ‘Catalytic site Uniprot EC label’, show the annotation of the catalytic site template (column 3) provided in Uniprot (49). A link allows the data to be downloaded in comma-separated-values format for import to a spreadsheet.
Figure 2.
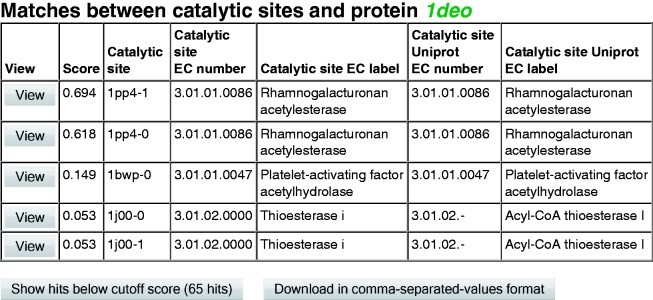
Sample output—browsing catalytic site matches to PDB 1deo. These are the top-scoring matches within a score threshold of 0.02.
Visualization
Each of the resulting matches is available for visualization on the web server with the Jmol viewer (50) with a click of the ‘View’ button. Figure 3 shows two of the catalytic site matches to protein PDB 1deo, as listed in Figure 2. These results are discussed further in the ‘Sample Uses’ section later in the text. Additional visualization options are available on the server, including whether the protein surface is shown, whether the surface is transparent or opaque and whether co-crystallized ligands are shown. Further options are available via Jmol’s menu and command line interface.
Figure 3.
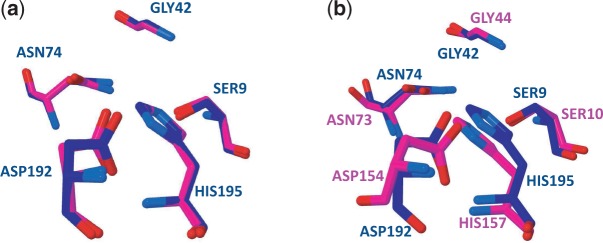
Visualization of matches between protein PDB 1deo and catalytic sites. (a) The aligned matching critical residues in protein 1deo (blue) and catalytic site 1pp4-1, 3.1.1.86, Rhamnogalacturonan acetylesterase (magenta). Residue names and numbers are identical between the protein and the catalytic site. (b) The aligned matching critical residues in protein 1deo (blue) and catalytic site 1j00-0, 3.1.2, Thioesterase I (magenta). The crystallographers’ modification of SER10 with a simulated substrate moiety is not shown here for clarity.
Benchmarks and testing
Training and test data sets
Non-overlapping samples of PDB proteins were drawn to ‘train’ the logistic regression classifier (i.e. estimate the coefficients of the logistic regression function) and to ‘test’ the classifier on data that are independent of that used in the training. The training data sample consists of approximately one-tenth of PDB proteins, filtered to include only those proteins annotated with EC numbers. Candidate matches include 53 088 catalytic site-protein pairs. Of these pairs, 52 473 matches were with catalytic site templates having three critical residues, and 720 matches were with catalytic site templates having four or more critical residues. Of the 53 088 candidate matches, 503 catalytic site-protein pairs were positives, i.e. correct matches—for all four parts of the EC number (class, subclass, sub-subclass and serial number)—between the catalytic site template EC number and the protein EC number. This definition of ‘positive’ was chosen to provide the most specific definition of ‘success’ for purposes of training the classifier. Of the positive results, 258 matches were with catalytic site templates having three critical residues; 245 matches were with catalytic site templates having four or more critical residues. The resulting trained classifier coefficients are shown in Table 1. Generally, to avoid overfitting, descriptors were retained in the regression when they contributed to a parsimonious specification according to the Akaike information criterion (51).
The test data set consists of a different sample of PDB proteins. The sample includes 27 436 catalytic site-protein pairs, 27 008 of these with three-residue catalytic site templates and 428 with four-or-more-residue catalytic site templates. There are 342 positives in the data set, 221 of these with three-residue catalytic site templates and 121 with four-or-more-residue catalytic site templates.
Receiver Operating Characteristic curves
Receiver Operating Characteristic (ROC) curves illustrate the performance of a binary classifier. The catalytic site identification classifier should distinguish matches between proteins and catalytic sites that share catalytic function (‘positives’) from matches between proteins and catalytic sites that do not share catalytic function (‘negatives’). How the classifier makes this distinction depends on the score threshold used. A high threshold may correctly identify positives (‘true positives’) and exclude negatives (‘true negatives’), but at the cost of identifying only a portion of all positives. A lower threshold will find more true positives, but at the cost of incorrectly identifying some negatives as positive (‘false positives’). An ROC curve uses proteins with known catalytic function to plot true positives as a fraction of all positives in the data set (‘true positive rate’) versus false positives as a fraction of all negatives (‘false positive rate’), both as a function of the score threshold value.
Before constructing ROC curves for the training and test data sets, ‘duplicate results’ were deleted. Occasionally, there are multiple correct matches (‘positives’), such as additional binding sites on a multimeric binding site. Similarly, there may be multiple incorrect matches (‘negatives’). To avoid overstating either the true positive rate or the false positive rate in constructing the ROC curves, only the highest-scoring of such duplicate matches is retained in the test data set. The web server also presents the results this way: only the highest scoring of such duplicate matches is presented.
The performance of the classifier on the training data set and test data set was analyzed through ROC curves and Matthews correlation coefficients (MCC). The ‘area under the curve’ (AUC) for ROC curves can serve as an indicator of the classifier’s discrimination. An ideal classifier would correctly identify all of the positives (true positive rate equals 1.0) without incorrectly identifying any negatives as positive (false positive rate equals 0). Such an ideal ROC would have AUC equal to 1.0. The MCC provides an indication of the performance of the classifier as a function of the threshold score chosen as the value to distinguish between putative positive and negative results.
The area under the ROC curve for the training data set is 0.94, as shown in Figure 4a. The AUC for the test data set is 0.89. Although the MCC for the training set shows a peak at a probability score threshold of ∼0.55, the curve is broadly flat down to a threshold value close to zero, at a true positive rate of ∼0.85 (see Figure 4b); the corresponding false positive rate at that threshold is close to zero (see Figure 4a). The data indicate that a true positive rate of 0.85 is achieved with a false positive rate of 0.004 at a threshold value of 0.02. As a practical matter, hits with a score of ∼0.02 and above appear to be of interest, as the case study below illustrates. The test set curves (Figure 5) indicate similar conclusions, though the true positive rate is somewhat lower, ∼0.79, and the false positive rate slightly higher, 0.010, at the 0.02 threshold.
Figure 4.
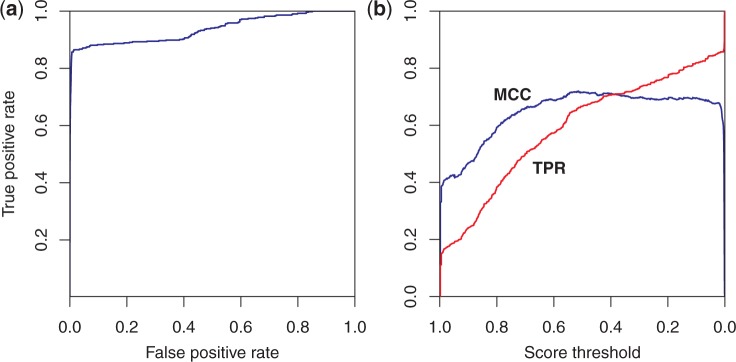
Performance of the logistic regression classifier on the training data. (a) ROC curve. AUC is 0.94. (b) MCC in blue and true positive rate (TPR) in red versus score threshold. The classifier shows good performance, as 85% of the matches are correctly identified with a false positive rate of only 0.4%.
Figure 5.
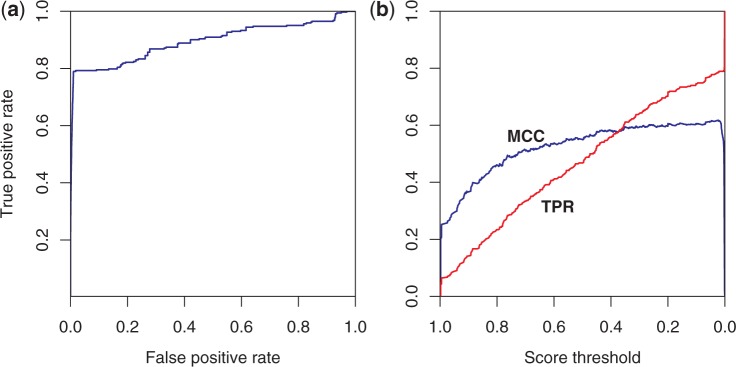
Performance of the logistic regression classifier on the test data set. (a) ROC curve. AUC is 0.89. (b) MCC in blue and true positive rate (TPR) in red versus score threshold. The classifier’s performance on the test data, which are distinct from the data used to train the classifier, is close to its performance on the training data—79% of the matches are correctly identified with a false positive rate of only 1.0%.
Sample uses
Browsing catalytic site matches to a PDB protein
The web server allows users to search its database for pre-calculated matches between library catalytic sites and PDB proteins. Figure 2 shows the results of searching for matches to PDB 1deo. The top-scoring results within a threshold score of 0.02, as suggested earlier in the text in ‘Benchmarks and testing’, are shown. Although 1deo does not have an EC annotation in its PDB file, the crystallographers have identified 1deo as a rhamnogalacturonan acetylesterase (52), which according to the IntEnz website (53) corresponds to EC 3.1.1.86. The top two matches from the catalytic site identification web server are both from PDB 1pp4, one match to each of the catalytic sites in the two chains, and correctly identify 1deo as EC 3.1.1.86. Inspection of the catalytic site in Figure 3a confirms that the residues are well aligned. The third match is to EC 3.1.1.47, which differs only in the fourth figure, indicating the substrate of the reaction is different. The 3.1.1 family of enzymes are carboxylic ester hydrolases that cleave the acetyl group from an acetylester. This chemistry is conserved in both cases, with the only difference being the particular molecule being cleaved. The final two matches are to EC 3.1.2.0000, which is a thioesterase (Thioesterase I). Thioesters are closely related to carboxylic esters. The catalytic functions are also similar with the difference being the cleavage is at a sulphur site adjacent to a carbonyl group (a thioester) rather than an oxygen site adjacent to an acetyl group. Figure 3b displays the resulting binding site match. The crystallographer’s modification of SER10 of the catalytic site, PDB 1j00, is not shown in Figure 3b to make the comparison with Figure 3a easier (54).
Browsing matches to proteins currently without annotation in PDB
The web server also allows users to search by EC number for proteins that do not have an EC annotation in their PDB file. Table 2 shows the top-scoring results of a search for matches between PDB proteins and catalytic sites in the EC 4.2.1 sub-subclass (‘Hydro-Lyases’). Although the results include only those proteins that do not have EC annotations, many proteins do have EC annotations in their UniProt record, as shown in Table 2.
Table 2.
Top 10 results of browsing protein matches to catalytic sites in EC sub-subclass 4.2.1
| Score | PDB ID | Catalytic site | Catalytic site EC number | Catalytic site EC label | Protein UniProt EC number | Protein UniProt label |
|---|---|---|---|---|---|---|
| 0.998 | 1fhu | 1fhu-MLE-0 | 4.02.01.0113 | o-Succinylbenzoate synthase (OSBS) | 4.02.01.0113 | o-Succinylbenzoate synthase |
| 0.998 | 1fhu | 1fhu-ES-0 | 4.02.01.0113 | o-Succinlybenzoate synthase (OSBS) | 4.02.01.0113 | o-Succinylbenzoate synthase |
| 0.998 | 1qrg | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.957 | 1qrf | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.948 | 1qre | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.927 | 1qrl | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.92 | 1qq0 | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.884 | 1qrm | 1qrg-0 | 4.02.01.0001 | Carbonic anhydrase | 4.02.01.0001 | Carbonate anhydrase |
| 0.548 | 1f93 | 1dco-4 | 4.02.01.0096 | Dcoh | 4.02.01.0096 | Pterin-4-alpha-carbinolamine dehydratase |
| 0.547 | 2wtb | 1dub-0 | 4.02.01.0017 | 2-enoyl-coa hydratase |
Not surprisingly, the top-scoring matches are matches between proteins and their own catalytic sites or catalytic sites from closely related proteins. In these cases, the UniProt annotations agree with the web server’s identification in all four EC figures. The UniProt confirmation of the four EC figures illustrates that the web server makes reliable functional identifications. Thus, the remaining imputed functional identifications should be worth further consideration.
The web server identifies PDB 2wtb as a ‘2-enoyl-coa hydratase’. Although 2wtb does not have an EC annotation in either PDB or UniProt, the crystallographers have identified 2wtb as having 2-trans-enoyl-CoA hydratase activity (55). Another match is with PDB 2qq3, which also does not have EC annotations and is also identified by its crystallographers as having 2-trans-enoyl-CoA hydratase activity (56). Inspection of the binding sites reveals convincing similarity between the binding sites. This example, using EC 4.2.1, reveals that the search procedure can sometimes provide an additional way of identifying similarities that can serve to complement incomplete annotations in PDB and UniProt.
Using a novel catalytic site to find matches throughout PDB
To illustrate the capability to search throughout PDB using a user-defined catalytic site, the plasminogen activator of Yersinia Pestis is used from recent studies (57,58). This protein is a member of the omptin family of proteases, and the EC number reported by the study for this site is EC 3.4.23.48, which is not currently in the server’s library of catalytic sites. The highest resolution structure for this protein is PDB 2×55 (1.85 Å), and the most recent crystal structure is PDB 4dcb. Catalytic sites were extracted from PDB 2×55 and PDB 4dcb, using residues (Asp|Asn)84, (Asp|Asn)86, (Asp|Asn)206 and His208, as identified in (58). Both sites were used to explore the effects of spatial variations.
The top protein matches to the input sites include the proteins from which the catalytic sites were drawn, as well as the related structure, 2×56 (see Table 3). Although the next matching protein, 1i78, is annotated with EC 3.4.21.87 in PDB, this EC number has been reassigned in UniProt as EC 3.4.23.49 (omptin) (59), a more general endopeptidase that is not specific to the plasminogen Arg560-Val561 peptide bond. This similarity is well within what would be a considered a typical correct match.
Table 3.
Top 11 matches across PDB to plasminogen activator binding sites
| Score | Catalytic site | Protein | Protein PDB EC (if known) | Protein PDB EC label |
|---|---|---|---|---|
| 0.998 | 2 × 55 | 2 × 55 | 3.4.23.48 | Plasminogen activator |
| 0.998 | 4dcb | 4dcb | 3.4.23.48 | Plasminogen activator |
| 0.962 | 2 × 55 | 2 × 56 | 3.4.23.48 | Plasminogen activator |
| 0.895 | 2 × 55 | 4dcb | 3.4.23.48 | Plasminogen activator |
| 0.893 | 4dcb | 2 × 55 | 3.4.23.48 | Plasminogen activator |
| 0.696 | 4dcb | 2 × 56 | 3.4.23.48 | Plasminogen activator |
| 0.552 | 2 × 55 | 1i78 | 3.4.21.87 | Omptin [3.4.23.49] |
| 0.368 | 4dcb | 1i78 | 3.4.21.87 | Omptin [3.4.23.49] |
| 0.063 | 2 × 55 | 3vc5 | ||
| 0.056 | 2 × 55 | 3vc6 | ||
| 0.055 | 4dcb | 1bqg | 4.2.1.40 | Glucarate dehydratase |
The next two matches, PDB 3vc5 and 3vc6, are not yet annotated in PDB. The PDB record currently reports isomerase activity. These enzymes appear to be part of the enolase (ES) superfamily (11,60), but one notable difference is the absence of a divalent metal ion, which is required for the initial proton abstraction step. The absence of this ion points to the possibility of another catalytic function for this enzyme. Viewing the aligned catalytic site (Figure 6a) shows reasonably close superposition of the catalytic residues to PDB 2×55. The amide cleavage machinery appears to be present in the 3vc5 site. However, these residues are more buried in the 3vc5 site, which suggests that endopeptidase activity (of which plasminogen activation is a specific case) is not the likely function. A different amide bond cleavage mechanism may be possible. ESs are known to host small peptide substrates, as is the case with the dipeptide isomerases (61,62). The residues appear to be reasonably placed in the known catalytic region of ESs for a mechanism of this type.
Figure 6.
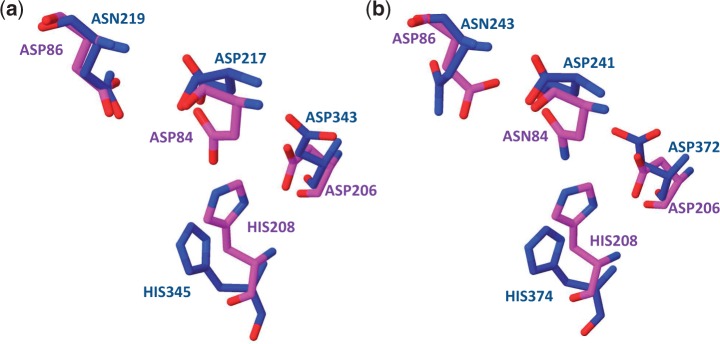
Visualization of match between plasminogen activator site and possible proteolytic sites in isomerases. (a) The aligned matching critical residues in catalytic site 2×55, 3.4.23.48, Plasminogen activator (magenta) and protein 3vc5 (blue). (b) The aligned matching critical residues in catalytic site 4dcb, 3.4.23.48, Plasminogen activator (magenta) and protein 1bqg (blue).
The next match, PDB 1bqg, has EC 4.2.1.40, ‘glucarate dehydratase’, which is also a member of the ES superfamily. For 1bqg, the residues matching those of the plasminogen activator site are in similarly good alignment (see Figure 6b) and are also in the known ES catalysis site. However, the function of 1bqg is well known to be mandelate racemase (MR) (63). The web server’s results are consistent with this: entering 1bqg as a search protein correctly returns matches to the 1ec7 ES and MR binding sites.
Since the MR functionality is well established for 1bqg, we propose that the protease site is a possible second function for 1bqg, as well as other ESs that appear in the search. Experimental verification and further study would be needed to confirm these potential function predictions for both these enzymes.
DISCUSSION
The catalytic site identification web server offers users several options for quickly exploring potential catalytic functions of novel or uncharacterized proteins, or for finding proteins that currently have not been identified as having a particular catalytic activity. In addition, because the server can generalize a catalytic site as any binding site that the user chooses to enter, the server should also have uses beyond catalytic function identification, such as in the discovery of off-target drug interactions or allosteric binding sites.
In the near term, a number of enhancements to the web server will be explored, such as additional browsing capabilities and user-customization of search parameters. In the future, we would like to increase the server’s coverage of the ‘enzymatic universe’ by adding to the server’s library of catalytic site templates. An additional goal is to extend the server’s capabilities so that it becomes an element of a protein function prediction ‘pipeline’. To allow users to start with only a protein sequence, the pipeline generates homology models of protein structure(s) based on a sequence and secondary structure template library [see (42)]. The resulting homology models are directed to the catalytic site identification server to identify potential catalytic sites and their function. These candidate catalytic sites could be verified through docking calculations, where the metabolites, suggested by the proposed EC number identifications, would each be tested for binding to the candidate sites [see (64)]. Used along with sequence-based methods, such an approach would provide independent lines of evidence that would go some way toward addressing the difficult task of protein function prediction.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Methods and Supplementary References [47,65–67].
FUNDING
Defense Threat Reduction Agency [PE0603384BP] and Laboratory Directed Research and Development [12-SI-004] at Lawrence Livermore National Laboratory. Funding for open access charge: Laboratory Directed Research and Development.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Carol Zhou and Mark Wagner for assistance in installing and testing the web server. They thank Sergio Wong and Eithon Cadag for helpful conversations and for providing case examples. Matt Jacobson provided helpful advice with the enolase example. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Release number LLNL-JRNL-618954.
REFERENCES
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 3.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology: Applications to protein modeling. J. Mol. Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 5.Sjölander K, Karplus K, Brown M, Hughey R, Krogh A, Mian IS, Haussler D. Dirichlet mixtures: a method for improved detection of weak but significant protein sequence homology. Comput. Appl. Biosci. 1996;12:327–345. doi: 10.1093/bioinformatics/12.4.327. [DOI] [PubMed] [Google Scholar]
- 6.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 7.Sankararaman S, Sjölander K. INTREPID—INformation-theoretic TREe traversal for Protein functional site Identification. Bioinformatics. 2008;24:2445–2452. doi: 10.1093/bioinformatics/btn474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glanville JG, Kirshner D, Krishnamurthy N, Sjölander K. Berkeley Phylogenomics Group web servers: resources for structural phylogenomic analysis. Nucleic Acids Res. 2007;35:W27–W32. doi: 10.1093/nar/gkm325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishnamurthy N, Brown DP, Kirshner D, Sjölander K. PhyloFacts: an online structural phylogenomic encyclopedia for protein functional and structural classification. Genome Biol. 2006;7:R83. doi: 10.1186/gb-2006-7-9-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: a Bayesian Monte Carlo approach. Mol. Biol. Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- 11.Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, Dunaway-Mariano D, Imker HJ, Jacobson MP, Minor W. The enzyme function initiative. Biochemistry. 2011;50:9950–9962. doi: 10.1021/bi201312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 14.Shindyalov IN, Bourne PE. A database and tools for 3-D protein structure comparison and alignment using the Combinatorial Extension (CE) algorithm. Nucleic Acids Res. 2001;29:228–229. doi: 10.1093/nar/29.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 16.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta. Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 19.Arakaki A, Huang Y, Skolnick J. EFICAz2: enzyme function inference by a combined approach enhanced by machine learning. BMC Bioinformatics. 2009;10:107. doi: 10.1186/1471-2105-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian W, Arakaki AK, Skolnick J. EFICAz: a comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Res. 2004;32:6226–6239. doi: 10.1093/nar/gkh956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile–profile alignments. Proc. Natl Acad. Sci. USA. 2008;105:5441. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie L, Bourne PE. A unified statistical model to support local sequence order independent similarity searching for ligand-binding sites and its application to genome-based drug discovery. Bioinformatics. 2009;25:i305–i312. doi: 10.1093/bioinformatics/btp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren J, Xie L, Li WW, Bourne PE. SMAP-WS: a parallel web service for structural proteome-wide ligand-binding site comparison. Nucleic Acids Res. 2010;38:W441–W444. doi: 10.1093/nar/gkq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankararaman S, Sha F, Kirsch JF, Jordan MI, Sjölander K. Active site prediction using evolutionary and structural information. Bioinformatics. 2010;26:617–624. doi: 10.1093/bioinformatics/btq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tseng YY, Dundas J, Liang J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J. Mol. Biol. 2009;387:451–464. doi: 10.1016/j.jmb.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marti-Renom MA, Rossi A, Al-Shahrour F, Davis FP, Pieper U, Dopazo J, Sali A. The AnnoLite and AnnoLyze programs for comparative annotation of protein structures. BMC Bioinf. 2007;8:S4. doi: 10.1186/1471-2105-8-S4-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 2002;323:387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- 28.Kleywegt GJ. Recognition of spatial motifs in protein structures. J. Mol. Biol. 1999;285:1887–1897. doi: 10.1006/jmbi.1998.2393. [DOI] [PubMed] [Google Scholar]
- 29.Spriggs RV, Artymiuk PJ, Willett P. Searching for patterns of amino acids in 3D protein structures. J. Chem. Inf. Comp. Sci. 2003;43:412–421. doi: 10.1021/ci0255984. [DOI] [PubMed] [Google Scholar]
- 30.Stark A, Russell RB. Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res. 2003;31:3341–3344. doi: 10.1093/nar/gkg506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinoshita K, Nakamura H. Identification of protein biochemical functions by similarity search using the molecular surface database eF-site. Protein Sci. 2003;12:1589–1595. doi: 10.1110/ps.0368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stivala AD, Stuckey PJ, Wirth AI. Fast and accurate protein substructure searching with simulated annealing and GPUs. BMC Bioinformatics. 2010;11:446. doi: 10.1186/1471-2105-11-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallace AC, Laskowski RA, Thornton JM. Derivation of 3D coordinate templates for searching structural databases: application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci. 1996;5:1001–1013. doi: 10.1002/pro.5560050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J. Mol. Biol. 2005;347:565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 35.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barker JA, Thornton JM. An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics. 2003;19:1644–1649. doi: 10.1093/bioinformatics/btg226. [DOI] [PubMed] [Google Scholar]
- 37.Fetrow JS, Skolnick J. Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J. Mol. Biol. 1998;281:949–968. doi: 10.1006/jmbi.1998.1993. [DOI] [PubMed] [Google Scholar]
- 38.Konc J, Janezic D. ProBiS: a web server for detection of structurally similar protein binding sites. Nucleic Acids Res. 2010;38:W436–W440. doi: 10.1093/nar/gkq479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ausiello G, Via A, Helmer-Citterich M. Query3d: a new method for high-throughput analysis of functional residues in protein structures. BMC Bioinformatics. 2005;6:S5. doi: 10.1186/1471-2105-6-S4-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li GH, Huang JF. CMASA: an accurate algorithm for detecting local protein structural similarity and its application to enzyme catalytic site annotation. BMC Bioinformatics. 2010;11:439. doi: 10.1186/1471-2105-11-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anand P, Yeturu K, Chandra N. PocketAnnotate: towards site-based function annotation. Nucleic Acids Res. 2012;40:W400–W408. doi: 10.1093/nar/gks421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zemla A, Zhou CE, Slezak T, Kuczmarski T, Rama D, Torres C, Sawicka D, Barsky D. AS2TS system for protein structure modeling and analysis. Nucleic Acids Res. 2005;33:W111–W115. doi: 10.1093/nar/gki457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng EC, Polacco BJ, Babbitt PC. Superfamily active site templates. Proteins: Struct. Funct. Bioinf. 2004;55:962–976. doi: 10.1002/prot.20099. [DOI] [PubMed] [Google Scholar]
- 44.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie L, Xie L, Bourne PE. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011;21:189–199. doi: 10.1016/j.sbi.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 47.Nilmeier JP, Kirshner DA, Wong SE, Lightstone FC. Rapid catalytic template searching as an enzyme function prediction procedure. PLoS One. 2013;8:e62535. doi: 10.1371/journal.pone.0062535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. OpenMP. The OpenMP API specification for parallel programming. openmp.org (6 February 2013, date last accessed)
- 49.UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jmol. Jmol: an open-source Java viewer for chemical structures in 3D. jmol.org (23 January 2013, date last accessed)
- 51.Gagne P, Dayton CM. Best regression model using information criteria. J. Mod. Appl. Stat. Methods. 2002;1:479–488. [Google Scholar]
- 52.Mølgaard A, Kauppinen S, Larsen S. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure. 2000;8:373–383. doi: 10.1016/s0969-2126(00)00118-0. [DOI] [PubMed] [Google Scholar]
- 53.Fleischmann A, Darsow M, Degtyarenko K, Fleischmann W, Boyce S, Axelsen KB, Bairoch A, Schomburg D, Tipton KF, Apweiler R. IntEnz, the integrated relational enzyme database. Nucleic Acids Res. 2004;32:D434–D437. doi: 10.1093/nar/gkh119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lo YC, Lin SC, Shaw JF, Liaw YC. Crystal structure of Escherichia coli thioesterase Protease lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J. Mol. Biol. 2003;330:539–551. doi: 10.1016/s0022-2836(03)00637-5. [DOI] [PubMed] [Google Scholar]
- 55.Arent S, Christensen CE, Pye VE, Nørgaard A, Henriksen A. The multifunctional protein in peroxisomal β-oxidation: structure and substrate specificity of the arabidopsis thaliana protein MFP2. J. Biol. Chem. 2010;285:24066–24077. doi: 10.1074/jbc.M110.106005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeyakanthan J, Kanaujia SP, Sekar K, Ebihara A, Shinkai A, Kuramitsu S, Yokoyama S. Crystal structure of Enoyl-CoA hydrates subunit I (gk_2039) other form from. Geobacillus Kaustophilus HTA426. 2007 PDB ID 2QQ3. [Google Scholar]
- 57.Eren E, Murphy M, Goguen J, Van den Berg B. An active site water network in the plasminogen activator pla from Yersinia pestis. Structure. 2010;18:809–818. doi: 10.1016/j.str.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 58.Eren E, van den Berg B. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J. Biol. Chem. 2012;287:23971–23976. doi: 10.1074/jbc.M112.376418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schomburg I, Chang A, Placzek S, Söhngen C, Rother M, Lang M, Munaretto C, Ulas S, Stelzer M, Grote A. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 2013;41:D764–D772. doi: 10.1093/nar/gks1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerlt JA, Babbitt PC, Jacobson MP, Almo SC. Divergent evolution in enolase superfamily: strategies for assigning functions. J. Biol. Chem. 2012;287:29–34. doi: 10.1074/jbc.R111.240945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lukk T, Sakai A, Kalyanaraman C, Brown SD, Imker HJ, Song L, Fedorov AA, Fedorov EV, Toro R, Hillerich B, et al. Homology models guide discovery of diverse enzyme specificities among dipeptide epimerases in the enolase superfamily. Proc. Natl Acad. Sci. USA. 2012;109:4122–4127. doi: 10.1073/pnas.1112081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song L, Kalyanaraman C, Fedorov AA, Fedorov EV, Glasner ME, Brown S, Imker HJ, Babbitt PC, Almo SC, Jacobson MP. Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nature Chem. Biol. 2007;3:486–491. doi: 10.1038/nchembio.2007.11. [DOI] [PubMed] [Google Scholar]
- 63.Gulick AM, Palmer DR, Babbitt PC, Gerlt JA, Rayment I. Evolution of enzymatic activities in the enolase superfamily: crystal structure of (D)-glucarate dehydratase from Pseudomonas putida. Biochemistry. 1998;37:14358–14368. doi: 10.1021/bi981123n. [DOI] [PubMed] [Google Scholar]
- 64.Zhang X, Wong SE, Lightstone FC. Message passing interface and multithreading hybrid for parallel molecular docking of large databases on petascale high performance computing machines. J. Comput. Chem. 2013;34:915–927. doi: 10.1002/jcc.23214. [DOI] [PubMed] [Google Scholar]
- 65.Coutsias EA, Seok C, Dill KA. Using quaternions to calculate RMSD. J. Comput. Chem. 2004;25:1849–1857. doi: 10.1002/jcc.20110. [DOI] [PubMed] [Google Scholar]
- 66.Liu P, Agrafiotis DK, Theobald DL. Fast determination of the optimal rotational matrix for macromolecular superpositions. J. Comput. Chem. 2010;31:1561–1563. doi: 10.1002/jcc.21439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Theobald DL. Rapid calculation of RMSDs using a quaternion-based characteristic polynomial. Acta Crystallogr. A. 2005;61:478–480. doi: 10.1107/S0108767305015266. [DOI] [PubMed] [Google Scholar]