

Abstract
Hepatic stellate cells (HSCs) are responsible for storing 90–95% of the retinoid present in the liver. These cells have been reported in the literature also to accumulate dietary β-carotene, but the ability of HSCs to metabolize β-carotene in situ has not been explored. To gain understanding of this, we investigated whether β-carotene-15,15′-monooxygensase (Bcmo1) and β-carotene-9′,10′-monooxygenase (Bcmo2) are expressed in HSCs. Using primary HSCs and hepatocytes purified from wild type and Bcmo1-deficient mice, we establish that Bcmo1 is highly expressed in HSCs; whereas Bcmo2 is expressed primarily in hepatocytes. We also confirmed that HSCs are an important cellular site within the liver for accumulation of dietary β-carotene. Bcmo2 expression was found to be significantly elevated for livers and hepatocytes isolated from Bcmo1-deficient compared to wild type mice. This elevation in Bcmo2 expression was accompanied by a statistically significant increase in hepatic apo-12′-carotenal levels of Bcmo1-deficient mice. Although apo-10′-carotenal, like apo-12′-carotenal, was readily detectable in livers and serum from both wild type and Bcmo1-deficient mice, we were unable to detect either apo-8′- or apo-14′-carotenals in livers or serum from the two strains. We further observed that hepatic triglyceride levels were significantly elevated in livers of Bcmo1-deficient mice fed a β-carotene-containing diet compared to mice receiving no β-carotene. Collectively, our data establish that HSCs are an important cellular site for β-carotene accumulation and metabolism within the liver.
Keywords: Vitamin A, Carotenoid, Carotene cleavage, Chylomicron, Diet
Introduction
Retinoids (vitamin A and its natural and synthetic analogs) are potent transcriptional regulators [1,2] that are proposed to modulate expression of more than 500 genes [3]. All retinoid present within mammals must be acquired from the diet either as proretinoid carotenoids that are converted within the body to retinoid or as preformed retinoid formed from carotenoids by animal species lower on the food chain [4-6]. Two structurally related proteins, β-carotene-15,15′-monooxygenase (BCMO1) and β-carotene-9′,10′-monooxygenase (BCMO2) are the sole mammalian enzymes known to cleave carotenoids [7-12]. BCMO1 catalyzes cleavage at the central double bond of proretinoid carotenoids, such as β-carotene, giving rise to retinoids; whereas, BCMO2 catalyzes eccentric cleavage of both proretinoid and non-proretinoid carotenoids giving rise to different apo-carotenal species [7-12].
It has been estimated that approximately 70% of all retinoid within the body of a healthy, well-nourished animal is stored in the liver [13,14]. Hepatic retinoid storage and metabolism are complex processes that are not well understood. However, it is well-established that both hepatocytes and the non-parenchymal hepatic stellate cells (HSCs1) (also called Ito cells, fat-storing cells and lipocytes) play an important role in these processes [13-17]. Retinoids, carotenoids, and other dietary lipids arriving in the postprandial circulation at the liver are taken up by hepatocytes [13,18,19]. Hepatocytes are the major cellular site of retinol-binding protein (RBP) synthesis within the body [20-22], but only account for about 5–10% of the total retinoid present in the liver [20,23]. The remaining 90–95% of the total retinoid present within the liver is stored in the lipid droplets of HSCs [20,23]. HSCs are also highly enriched in cellular retinol-binding protein type I (CRBPI) and lecithin:retinol acyltransferase (LRAT), proteins, respectively, involved in the intracellular transport and esterification of retinol [20,23,24].
Although it is well-established that BCMO1 and BCMO2 are both expressed in the liver [8-10,25,26], the distributions and roles of these enzymes within hepatocytes and HSCs have not been systematically explored. Lakshman and colleagues have reported, for rats given a dose of β-[14C] carotene after 2 weeks of continuous β-carotene feeding, approximately half of the β-carotene dose is recovered in isolated hepatocytes and the remainder in isolated HSCs [27]. Thus, within the intact liver, both hepatocytes and HSCs are able to acquire and accumulate carotenoid. Similarly, in vitro studies making use of GRX cells, a murine myofibroblastic cell line derived from HSCs, indicate that HSCs can take up and accumulate both β-carotene and lycopene that had been added to the culture medium [28,29]. Collectively, this published work suggests that HSCs are able to store and metabolize both proretinoid and non-proretinoid carotenoids. To better understand the roles that hepatocytes and HSCs may have in carotenoid cleavage, we undertook studies in wild type and Bcmo1-deficient (Bcmo1−/−) mice bred to be congenic in the C57Bl/6 J genetic background. Findings from these studies are presented below.
Materials and methods
Animal husbandry and dietary regimens
All mice employed in our studies were treated and maintained according to the NIH Guide for the Care and Use of Laboratory Animals, and all experimental procedures were reviewed and approved by the Columbia University Institutional Animal Care and Use Committee. A detailed description of Bcmo1-deficient mice is reported elsewhere [30]. The mice employed in our studies were derived from these original mixed genetic background mice through 10 back-crosses with C57Bl/6 J mice rendering the mutant mice congenic in the C57Bl/6 J genetic background [31]. During the breeding and lactation periods, all mice were maintained on breeder chow that contained 15 IU vitamin A/g diet (PicoLab Mouse Diet 20, PMI International). After weaning, the mice were placed on a regular chow diet which also contained 15 IU vitamin A/g diet. At 7 weeks of age, some mice (50 mice/genotype, 25 males/diet group) used in these studies were placed on one of two purified diets. One purified diet contained β-carotene beadlets (β-carotene 10% CWS, DSM Nutritional Products, Inc., Parsippany, NJ) as the only source of vitamin A (referred to in the text as “β-carotene” diet) for 30 days. This diet was formulated based on the AIN-93G formulation, but was modified to have a higher fat content (20% w/w soybean oil) and to contain 0.1 mg β-carotene/g diet as a sole source of retinoid (containing no preformed retinoid). The second diet was formulated identically to the first except it contained no β-carotene or any other source of retinoid (referred to in the text as “high fat” diet). After 30 days of maintenance on one of the purified diets, mice from each genotype were sacrificed after a 4 h fast. Serum and liver were collected, snap frozen in liquid N2, and stored in −80 °C until retinoid/β-carotene/β-apo-carotenal and triglyceride analyses were undertaken. Five mice from each group maintained on the different diets for 30 days were used for hepatocyte and HSC isolations.
Hepatocyte isolation
Primary mouse hepatocytes were isolated through in situ perfusion of the liver with collagenase type I (0.75 mg/ml, Worthington Biochemical Corporation, Lakewood, NJ). Mice were anesthetized with a cocktail consisting of 1 mL ketaset, 0.5 mL xylazine, and 8.5 mL of PBS given at a dose of 100 μL/10 g body weight. The abdomen was opened to allow access to the liver. With the organs brushed aside, a catheter was angled parallel to the mouse’s body and was inserted into the inferior vena cava. The liver was first perfused at a flow rate of 5 ml/min with Hank’s balanced salt solution (without Ca++) followed by Hank’s balanced salt solution (with Ca++) containing collagenase (0.75 mg/ml), for 5 min and 15 min, respectively. Immediately after the first solution began to enter the liver, the portal vein was cut and the suprahepatic inferior vena cava was clamped. After perfusion, the partially digested liver was excised, the digest passed through a 100 μm nylon mesh to remove undigested materials, and resuspended in William’s medium containing 1% penicillin/streptomycin. Isolated hepatocytes were separated from the non-parenchymal cells and debris by centrifugation at 4 °C in the following sequence: twice for 5 min at 20g, once for 10 min at 50 g, and twice again for five min at 20 g. The supernatant was aspirated and the hepatocytes present in the pellet were resuspended in William’s medium. Cell yields were determined by counting on a hemacytometer.
Hepatic stellate cell isolation
Primary mouse HSCs were isolated by in situ liver perfusion, first with a solution containing a mixture of Pronase E (0.4 mg/ml, EMD Chemicals Inc., Gibbstown, NJ), followed by collagenase type I (0.5 mg/ml, Worthington Biochemical Corporation, Lakewood, NJ) according to established protocols [20,32,33]. HSCs were purified from the remainder of non-parenchymal cells and hepatocyte-derived debris by floatation through 9% (w/v) Nycodenz (Axis-Shield PoC AS, Oslo, Norway) in Gey’s balanced salt solution. Cell yields from each isolation were determined by counting on a hemacytometer.
Retinoid, β-carotene and β-apo-carotenal extraction and LC/MS analysis
Serum and liver samples, as well as the β-carotene containing diet, were analyzed by liquid chromatography mass spectrometry (LC/MS) for retinoid, β-carotene and β-apo-carotenal levels. Liver samples were homogenized in 10 volumes of 0.25 M sucrose solution in water. Serum, liver and samples of purified diet were extracted using a modification of the method of Kane et al. [34]. However, only the first fraction containing neutral lipids was collected and analyzed. Briefly, 1 mL of 0.025 M KOH was added to the serum, liver homogenate or a ground powder made from the diet and compounds of interest were extracted 3-times into 5 mL of hexane. The hexane extracts were combined and dried under N2. The dried extracts were subsequently redissolved in a 1:1 (v/v) mixture of methyl t-butyl ether and methanol and 20 μL of this was injected onto the HPLC for separation of retinoids, β-carotene and β-apo-carotenals. Separations were accomplished by reversed phase HPLC using a 4.6 × 150 mm 5 μm YMC C30 column (Waters Corp, Milford, MA). A ternary solvent system consisting of: solvent A, 0.1% formic acid; solvent B, 100% methanol; and solvent C, 100% methyl t-butyl ether was employed. The initial HPLC solvent consisted of 20% A/80% B/0% C and from the time of sample injection through 12 min the solvent was linearly changed to 0% A/30% B/70% C. This was followed by a 3 min re-equilibration period. The flow rate of the solvent was maintained constant at 1.8 mL/min and the column temperature at 35 °C. The UV–VIS absorbance of the eluent was monitored using a Waters 996 photodiode array detector.
The HPLC eluent was interfaced with a quadrupole/time-of-flight (QTof) mass spectrometer (Q Tof Premier, Micromass, UK) via an atmospheric pressure chemical ionization (APCI) probe. β-Carotene, apo-8′-carotenal, apo-10′-carotenal, apo-12′-carotenal, apo-14′-carotenal, and retinal were ionized in the APCI negative mode as their respective radical anions, 536.438, 416.31, 376.28, 350.26, 310.23, and 284.21 m/z. The QTof system allowed for quantitative detection with the confidence of accurate mass typically being 1 ppm. Mass spectra were acquired in V-mode (~8000 resolution) from 100–1000 m/z with a scan time of 0.5 s, peak centroiding, and enhanced duty cycle enabled for the parent m/z. At intervals of 30 s, a 0.1 s lockspray scan was acquired with leucine enkephalin as the lockspray compound (554.2615 m/z) to correct for minor deviations in calibration due to temperature fluctuations. Prior to analysis, the QTof was fully calibrated from 114 to 1473 m/z using a solution of sodium formate. The resultant MS spectra were acquired and integrated with MassLynx software, V4.1 (Micromass UK, Manchester, UK). Source parameters were: 30 μA corona current; 500 °C probe; 110 °C source block; 35 V cone; 50 L/h cone gas (N2); 400 L/h desolvation gas (N2); and collision energy 8 eV (non-fragmenting) with argon as the CID gas (4.2 × 10−3 mBar). Standard curves were generated using standards of β-carotene (Sigma), apo-8′-carotenal (Sigma), apo-12′-carotenal (Carotenature), apo-14′-carotenal (synthesized for the study), and retinal (Sigma). Apo 10′-carotenal was quantified using the standard curve made from apo-12′-carotenal. Retinol and retinyl ester standard curves were acquired separately under the same HPLC conditions but quantified using the diode array detector and absorbance at 325 nm. Recoveries for all compounds were determined to be greater than 98%.
Triglyceride measurements
Approximately 100 mg of liver was placed into 50 mL tubes containing 5 mL of 1.0 M NaCl. The samples were homogenized using a Polytron Homogenizer at 19,000 rpm for 20 s. Subsequently, 10 mL of chloroform:methanol (2:1 v/v) was added to the liver homogenates. The samples were vortexed for 1 min to ensure that they were homogenous. After centrifuging the samples for 10 min at 800g, the chloroform-containing lower phase was removed and placed into a glass tube. An additional 5 mL of chloroform:methanol was added to the remainder of the upper phase, the samples vortexed and centrifuged as above to assure complete triglyceride recovery. The pooled chloroform phases were evaporated under N2. After the chloroform had completely evaporated, 1 mL of 2% Triton X-100 in chloroform was added into the samples. The samples were mixed well and the chloroform was evaporated under N2. Triglycerides were solubilized for colorimetric assay through addition of 1 mL of deionized water into the glass tubes. Using a Matrix Plus Chemistry Reference Standard, according to the manufacturer’s instructions, a colorimetric triglyceride assay using the Thermo Scientific Infinity Triglyceride Reagent was performed in a 96 well plate for each liver extract. Color development was measured on a Multiskan Plus Microtiter Plate Reader at 520 nm.
Glucose and insulin tolerance tests
Blood glucose levels were measured using a glycometer 0, 15, 30, 45, 60, 90, and 120 min after glucose or insulin administration. To assess glucose tolerance, a solution of glucose (0.5 g/mL in phosphate buffered saline (PBS)) at a dosage of 3 μL of glucose/g body weight was administered by intraperitoneal injection to fasted mice. To assess insulin responsiveness, insulin was given at a dose of 0.75 mU/g body weight.
RNA preparation and quantitative real time-PCR (qRT-PCR) analyses
Total RNA was isolated from liver, as well as from freshly-isolated hepatocytes and HSCs. The RNA was subjected to DNase digestion using DNase I and the RNeasy minikit (Qiagen), according to the manufacturer’s instructions. RNA was quantified at 260 nm using a Nanodrop spectrophotometer. For cDNA synthesis, 4 μg of total RNA (in a final volume of 20 μL) was denatured at 65 °C for 5 min. Subsequently, cDNA synthesis was carried out for 10 min at 25 °C and 50 min at 50 °C with reverse transcriptase (SuperScript III, Invitrogen). The reaction was stopped at 85 °C for 5 min using a thermal cycler (Perkin Elmer, Waltham, MA). Table 1 provides the sequence of primers we used for qRT-PCR analyses.
Table 1.
Primers used for qRT-PCR analyses.
| Gene | Accession number | Forward/reverse primers |
|---|---|---|
| RBP | NM_011255 | 5′-GAGTCCGTCTTCTGAGCAAC |
| 5′-CTTGAACTTGGCAGGATCT | ||
| LRAT | NM_023624 | 5′-AGTTCAAGACTAGCCTGCTCA |
| 5′-TACAAGCTGGCCTTCGAC | ||
| BCMO1 | NM_021486 | 5′-ATGAAATGTGGAGAAGACTTCTAT |
| 5′-CTTTGTCCACGACGGAT | ||
| BCMO2 | NM_133217 | 5′-AGGACCAGGGCTGTATTGTG |
| 5′-CGCTGGCTGAAGAATAGGAC | ||
| PPARα | NM_011144 | ACTGGATGACAGTGACATTTC |
| CTCCTGCAACTTCTCAATGTAG | ||
| PPARγ | NM_011146 | 5′-TCTCCAGCATTTCTGCTCCACAC |
| 5′-CAGGTTCTACTTTGATCGCACTTTGGTAT | ||
| 18S | 5′-CCATCCAATCGGTAGTAGCG | |
| 5′-GTAACCCGTTGAACCCCATT |
As a housekeeping gene to normalize expression levels, we used 18S RNA. qRT-PCR was performed in a total volume of 25 μL, including 40 ng of cDNA template, forward and reverse primers (100 nM each), and Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA) using a Stratagene Mx3005P instrument (Stratagene, La Jolla, CA). After initial enzyme activation (95 °C for 10 min), 40 cycles at 55 °C were performed for the annealing/extension steps, and fluorescence was measured. A dissociation curve program was performed after each reaction. Relative quantification of target genes was calculated based on the efficiency of each reaction and the crossing point deviation of each sample versus a control.
Western blot analysis for BCMO1
Liver, hepatocyte and hepatic stellate cell protein levels of BCMO1 were analyzed by Western blot assay, essentially as described previously [10]. For the present study, 100 μg of total protein were separated on 10% SDS–PAGE gels and transferred onto polyvinylidene fluoride membrane (Millipore Immunobilon-P Transfer Membrane). The primary antibody was described earlier [10] and consisted of a polyclonal rabbit anti-mouse BCMO1 antiserum that was diluted 1:2000. The secondary antibody was a donkey horseradish peroxidase-conjugated anti-rabbit IgG antiserum (Amersham Biosciences). Immunoblots were developed using the ECL system (Thermo Scientific).
Statistical analysis
All data are presented as means ± S.D. A Student’s t-test was used to analyze for statistically significant differences between groups. Groups were considered to be significantly different when p < 0.05.
Results
The overall goal of this investigation was to identify and characterize the cellular site(s) of carotenoid cleavage in the liver. It generally has been assumed that the hepatocyte is the predominate cell in the liver involved in carotenoid cleavage and metabolism, but there are few data confirming this possibility. It has long been established that both hepatocytes and HSCs mediate hepatic retinoid storage and metabolism [13-17]. Thus, the goal of this study was to establish whether HSCs may also play a role in carotenoid, specifically β-carotene, cleavage. As can be seen from Fig. 1, Bcmo1 mRNA and protein are expressed in both primary mouse hepatocytes and primary mouse HSCs. The data in Fig. 1 establish that Bcmo1 expression is more abundant in HSCs than in hepatocytes. Fig. 2 shows the distribution of Bcmo2 mRNA in heptocytes and HSCs. Unlike Bcmo1 expression, Bcmo2 is expressed primarily in hepatocytes with only low levels in HSCs. Interestingly, Bcmo2 mRNA levels are significantly elevated (p < 0.05) in livers, hepatocytes and HSCs from Bcmo1-deficient mice compared to wild type mice. The purities of the hepatocyte and HSC isolates used to define the cellular sites of Bcmo1 and Bcmo2 expression in the liver were assessed through measurement of markers for these cells, using retinol-binding protein (RBP), which is a hepatocyte-specific protein, and lecithin:retinol acyltransferase (LRAT), a HSC-specific protein. As can be seen in Fig. 3, the cell preparations used in these analyses were highly purified; the hepatocyte preparations were very enriched in RBP and showed low levels of LRAT expression, whereas the HSC preparations were enriched in LRAT and contained little RBP.
Fig. 1.
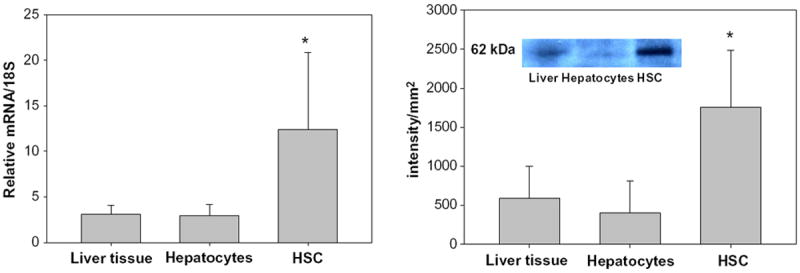
Relative levels of expression of Bcmo1 mRNA (left panel) and protein (right panel) in whole mouse liver, primary mouse hepatocytes and primary mouse HSCs. The insert in the right panel shows an immunoblot used to obtain BCMO1 protein levels for one liver, one hepatocyte and one HSC preparation. Values are given as the mean ± 1 S.D. for 5 livers or cell isolates in each group. The “*” indicates significantly different (p < 0.05) compared to the liver level.
Fig. 2.
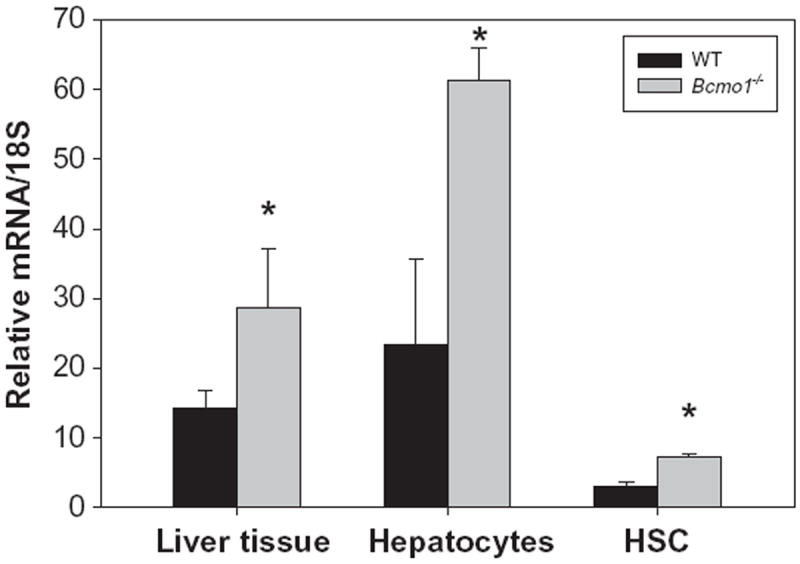
Relative expression levels of Bcmo2 mRNA in whole mouse liver, primary hepatocytes and primary HSCs for wild type (black bars) and Bcmo1-deficient (gray bars) mice. Values are given as the mean ± 1 S.D. for 5 livers, 4 hepatocyte isolates, and 4 HSC isolates. The “*” indicates significantly different (p < 0.05) compared to the liver or cells obtained from wild type mice.
Fig. 3.
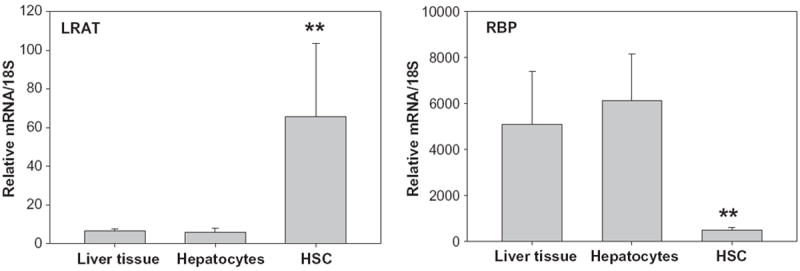
Levels of mRNA for LRAT (a marker for HSCs) and RBP (a marker for hepatocytes) in whole liver, primary hepatocytes and primary HSCs. Values are given as mean ± 1 S.D. for 5 livers or cell isolates per group. The “**” indicates significantly different (p < 0.01) compared to liver or cells from wild type mice.
To confirm the published literature that β-carotene is present in both hepatocytes and HSCs within the liver [27], wild type and Bcmo1-deficient mice were fed a purified diet rich in β-carotene (100 μg/g diet) (β-carotene diet) or the same diet lacking β-carotene for 30 days. This purified diet was formulated to be rich in fat (20% fat versus 5% fat in the standard rodent chow diet) in order to facilitate optimal β-carotene absorption, since it is well-established that dietary fat content enhances the intestinal absorption of β-carotene [35]. In order to understand how Bcmo1 expression and activity influence β-carotene distribution and levels within the liver, we employed both wild type and Bcmo1-deficient mice. We hypothesized that expression of Bcmo1 in HSCs would lower levels of intact β-carotene present in these cells by catalyzing β-carotene conversion to retinoid. After 30 days on the diets, hepatocytes and HSCs were isolated from the mice and cellular levels of retinoid and β-carotene were assessed by HPLC. An HPLC profile for an extract prepared from a hepatocyte preparation and a HSC preparation from Bcmo1-deficient mice receiving the β-carotene diet is shown in Fig. 4. This profile confirms literature reports that β-carotene can accumulate in HSCs as well as in hepatocytes.
Fig. 4.
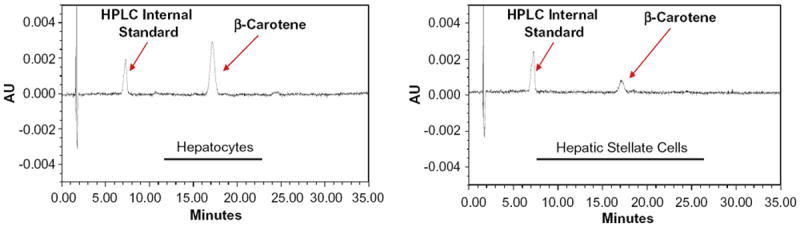
HPLC profiles employing UV–Vis detection at 450 nm showing the presence of β-carotene in extracts made using 3 × 106 isolated hepatocytes (left) and 1 × 106 isolated HSCs (right) from Bcmo1-deficient mice fed the β-carotene containing diet for 30 days. The peaks labeled β-carotene elute with a retention time (approximately 18 min) and UV–Vis spectrum identical to that of pure β-carotene. The peak labeled “HPLC Internal Standard” is a carotenoid (echinenone) that is not present in the diet.
We measured by LC/MS β-carotene, retinal, apo-8′-, apo-10′-, apo-12′ and apo-14′-carotenal concentrations in serum and liver of wild type and Bcmo1-deficient mice maintained on the β-carotene containing diet. Table 2 provides a summary of these measurements. As can be seen from Table 2, apo-10′- and apo-12′-carotenals were present in both tissues, but we were unable to detect either apo-8′- or apo-14′-carotenal in serum or liver obtained from either Bcmo1-deficient or wild type mice. Although there is a consistent trend for apo-12′- and apo-10′-carotenal levels to be higher in serum and livers of Bcmo1-deficient compared to wild type mice, this was only statistically significant (p < 0.05) for hepatic apo-12′-carotenal levels. We also checked the β-carotene-containing diet for the presence of these apo-carotenals. As seen in Table 2, the β-carotene diet contained low, but quantifiable, levels of apo-8′-, apo-10′-, apo-12′- and apo-14′-carotenal as did the β-carotene beadlets used in formulating this diet (data not shown). Retinal was detected in livers but not serum from both Bcmo1-deficient and wild type mice.
Table 2.
Serum and liver levels of β-carotene, retinal, APO-8′-, APO-10′-, APO-12′- and APO-14′-carotenals for β-carotene fed wild type and Bcmo1-deficient mice.a
| β-Carotene | Retinal | Apo-8′-carotenal | Apo-10′-carotenal | Apo-12′-carotenal | Apo-14′-carotenal | |
|---|---|---|---|---|---|---|
| Serum (nM) | ||||||
| Wild type (n = 5) | 1.9 (0.9–2.6) | N.D. | N.D. | 0.2 (0.1–0.5) | 0.4 (0.1–1.1) | N.D. |
| Bcmo1−/− (n = 3) | 80.8 (73.5–89.3)b | N.D. | N.D. | 0.5 (0.4–0.7) | 0.7 (0.6–0.8) | N.D. |
| Liver (pmol/g) | ||||||
| Wild type (n = 5) | 16.8 (6.3–27.9) | 356 (198–440) | N.D. | 0.2 (0.0–0.8) | 5.9 (4.2–8.5) | N.D. |
| Bcmo1−/− (n = 4) | 381 (241–670)b | 408 (159–656) | N.D. | 0.8 (0.0–1.9) | 12.0 (7.4–16.3)c | N.D. |
| Diet (nmol/g) (n = 3) | 19.7 ± 4.2 | N.D. | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.1 |
Serum and liver concentrations are given as the mean followed in parenthesis by the range of values measured for all samples from that group. Concentrations of compounds in the diet are provided as the means ± S.D. Limits of detection for apo-carotenals range from approximately 0.1 to 0.5 nM for serum and 0.1 to 0.7 pmol/g liver. Limits of detection for retinal in serum are approximately 40–50 nM. N.D. signifies not detected.
Significantly different from corresponding wild type, p < 0.01.
Significantly different from corresponding wild type, p < 0.05.
An earlier published work demonstrated that administration of a diet containing 1 mg β-carotene/g diet to either wild type or Bcmo1-deficient mice for 7 weeks gives rise to the development of hepatic steatosis in these mice [30]. Although in the present study mice received 10-fold less β-carotene in the diet for a shorter duration, hepatic triglyceride levels were measured to assess whether excessive triglyceride accumulation had occurred. Indeed, statistically significant elevations (p < 0.01) in hepatic triglyceride levels were observed for Bcmo1-deficient mice fed the β-carotene diet compared to wild type mice. These data are provided in Fig. 5. The elevation in hepatic triglyceride levels was accompanied by an increase in hepatic expression of the gene for peroxisomal proliferators activator protein-γ (PPARγ), which has been implicated as acting critically in the development of hepatic steatosis [36], including in the Bcmo1-deficient mice [30]. This can be seen in Fig. 6. Interestingly, PPARγ expression was elevated in both the liver and isolated HSCs, but not hepatocytes obtained from Bcmo1-deficient mice. Unlike PPARγ mRNA levels, no differences in PPARα mRNA levels were observed in livers or isolated hepatocytes or HSCs obtained from wild type and Bcmo1-deficient mice fed the β-carotene diet (data not shown).
Fig. 5.
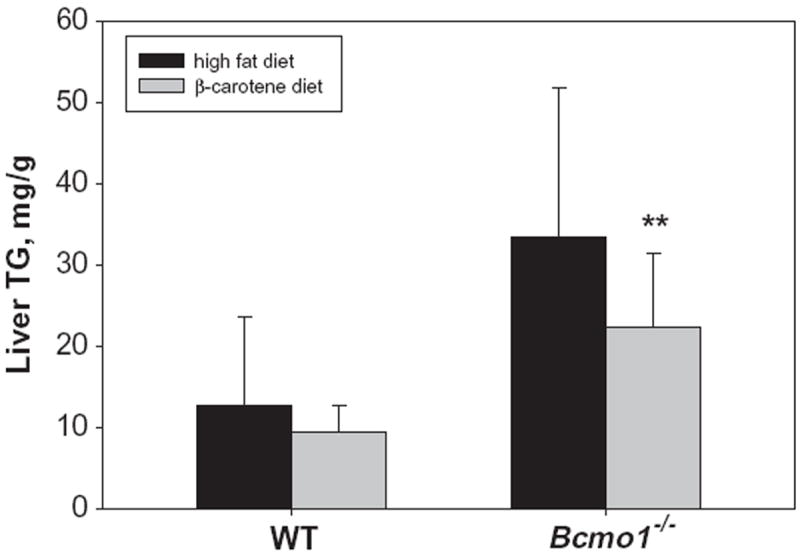
Hepatic triglyceride (TG) levels for wild type and Bcmo1-deficient (Bcmo1−/−) mice fed either a control purified diet containing 20% fat and no β-carotene (high fat diet) or the same diet containing 100 μg β-carotene/g diet (β-carotene diet). Values are given as the mean ± 1 S.D. for 5 mice in each group. The “**” indicates statistically different (p < 0.01) compared to wild type mice receiving the same diet.
Fig. 6.
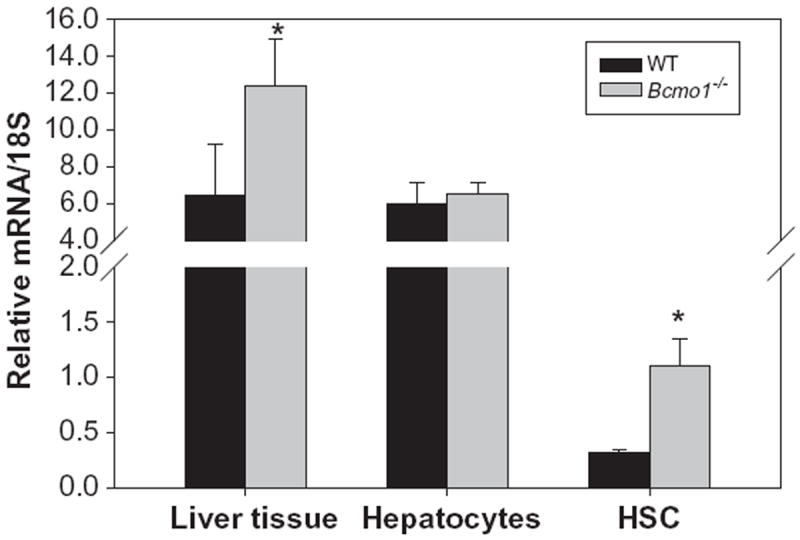
Relative levels of mRNA for PPARγ determined by qRT-PCR for whole liver (n = 6), hepatocytes (n = 4) and hepatic stellate cells (n = 4) isolated from wild type (black bars) and Bcmo1-deficient (Bcmo1−/−, gray bars) mice. The “*” indicates statistically different (p < 0.05) compared to liver or cells from wild type mice.
Since excessive hepatic triglyceride accumulation is often associated with the development of insulin resistance and the development of type II diabetes, glucose and insulin tolerance tests were also carried out in the mice. Fig. 7 shows the results of the glucose tolerance tests. The fasting serum glucose levels present in age- and gender-matched Bcmo1-deficient mice receiving the high fat diet were significantly higher (p < 0.003) (192.9 ± 43.6 mg/dl versus 112.2 ± 51.8 mg/dl) than those of the wild type group receiving the same diet. When wild type mice were fed the β-carotene diet, the fasting glucose level of these mice was found to be statistically elevated (p < 0.003) compared to wild type mice fed the β-carotene-free high fat diet. We observed no statistical significance between any of the groups with regard to the insulin response test (data not shown).
Fig. 7.
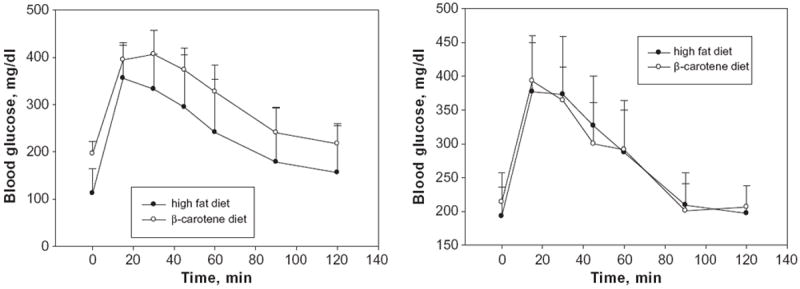
Glucose tolerance curves for wild type (left panel) and Bcmo1-deficient (right panel) mice fed either the high fat or the β-carotene diet. Values at each time are given as the mean ± 1 S.D. for 5 mice in each group. The calculated areas under the curve are not statistically different for either the wild type (right) or Bcmo1-deficient mice (left).
Discussion
The goal of these studies was to identify the cellular site(s) of β-carotene processing in the liver, after intestinal uptake and packaging of dietary β-carotene into chylomicrons. Our data (see Fig. 4) agree with earlier published reports that β-carotene can be taken up and accumulated by both hepatocytes and HSCs [27-29]. Fig. 1 establishes that Bcmo1 mRNA and protein are highly enriched in HSCs. Although hepatocytes express some Bcmo1, the HSCs express relatively high levels of Bcmo1 suggesting an important role for this enzyme in HSCs. Furthermore, these findings raise an interesting question as to how β-carotene, which must be taken up into the liver by hepatocytes (the sole hepatic cell type responsible for chylomicron clearance [19]), is then transported to HSCs. Overall, these observations were unexpected. Although the cellular distribution of β-carotene and Bcmo1 in the liver had not been extensively studied, it is generally assumed that both β-carotene and Bcmo1 are found primarily within hepatocytes, since this is the cellular site of chylomicron uptake [19]. Moreover, immunohistochemical studies of BCMO1 and BCMO2 distribution within the body reported that both of these enzymes are present within hepatocytes [25,26], although the resolution of these studies may not have been sufficient to establish HSC expression of the enzymes. Our data obtained using primary liver cell isolates, establish that HSCs are a highly enriched cellular site of Bcmo1 expression in the liver and are enriched in β-carotene, while Bcmo2 expression is localized primarily to hepatocytes.
Bcmo2 mRNA levels were found to be significantly elevated both in the intact liver, as well as in isolated hepatocytes and HSCs, in Bcmo1-deficient mice compared to wild type mice (see Fig. 2). This suggests the existence of a coordinated regulatory process that governs expression levels of Bcmo1 and Bcmo2 within liver cells. Although the regulatory processes that govern Bcmo1 gene expression are being unraveled [37,38], little information is presently available regarding the factors that may regulate Bcmo2 gene expression. Hence, it is difficult to speculate why Bcmo2 expression levels are elevated in liver cells from Bcmo1-deficient mice. It is possible that the observed elevation in expression is related either to the increased hepatic β-carotene concentration and/or to the increased levels of apo-10′- or apo-12′-carotenals themselves.
Since BCMO2 catalyzes the eccentric cleavage of β-carotene that can give rise potentially to a number of β-apo-carotenoids and mice deficient in Bcmo1 show increased levels of Bcmo2 mRNA in their livers, we wondered whether β-carotene-fed Bcmo1-deficient mice might have higher levels of β-apo-carotenal in their livers and/or serum than wild type mice. Apo-10′-carotenal and apo-12′-carotenal were the only β-apo-carotenals detected in serum and livers from both Bcmo1-deficient and wild type mice. Concentrations of apo-12′-carotenal and apo-10′-carotenal in both serum and liver tended to be higher for the Bcmo1-deficient mice but only apo-12′-carotenal levels in livers from the mutants proved to be significantly (p < 0.05) greater than those of wild type mice. This finding is consistent with the notion that elevated levels of Bcmo2 expression in livers of Bcmo1-deficient mice gives rise to increased β-apo-carotenal formation through eccentric cleavage of β-carotene. Apo-14′-carotenal and apo-8′-carotenal were not detected in any serum or liver sample for either strain but surprisingly, apo-8′-, apo-10′-, apo-12′-, and apo-14′-carotenal were all detected in the diet formulated using β-carotene beadlets and in the beadlets themselves. This suggests that apo-10′- and apo-12′-carotenals may be absorbed preferentially and that the body discriminates against accumulation of the apo-8′- and apo-14′-carotenals. Apo-14′-carotenal was the most abundant β-apo-carotenal form detected in the diet, and its absence in the circulation could suggest that biological discrimination against these two β-apo-carotenals occurs within the intestine. However, this possibility requires confirmation. As we expected based on previously published data [30,31], there was 40-times more β-carotene in serum and 20-times more β-carotene in livers of Bcmo1-deficient compared to wild type mice. Expression of Bcmo1 clearly influences the amount of β-carotene taken up from the diet and retained by the body. We did not observe a decrease in liver retinal concentrations in Bcmo1-deficient mice. We had hypothesized since retinal is the sole product of BCMO1 catalyzed cleavage of β-carotene, tissue retinal levels might be diminished when Bcmo1 is ablated. However, this was not observed. Since retinal can be formed through oxidation of retinol, as well as from β-carotene cleavage [4-6], this finding suggests that either β-carotene cleavage by BCMO1 is not required for maintaining hepatic retinal pools and/or that the liver strives to maintain retinal levels within a fairly tight concentration range, regardless of its metabolic precursor. Contrary to a recent report regarding retinal concentrations in serum [39], we were unable to detect retinal in serum from any of our mice.
The observations that β-carotene is present in both hepatocytes and HSCs while Bcmo1 is expressed highly in HSCs raise an interesting question as to why this is the case. β-Carotene (or more generally proretinoid carotenoids) have at least two functions within the bodies of animals: to serve as precursors for retinoid synthesis and to act as antioxidants. β-Carotene present in HSCs, where Bcmo1 is highly expressed, likely serves as a ready precursor for retinoid formation. The HSCs are the major cellular site for retinoid storage in the liver and body, accounting for the majority of reti-noid present in the body of a well-nourished animal [13,14], and β-carotene cleaved by Bcmo1 likely contributes to these stores. In hepatocytes, which act critically in hepatic energy metabolism, β-carotene may act as an antioxidant, preventing the accumulation of potentially dangerous free radicals that are formed as by-products of energy metabolism. The literature indicates that much of hepatic β-carotene is associated with mitochondria, the subcellular organelle responsible for energy generation [40]. Hepatocyte β-carotene may be needed to inactivate free radicals produced through mitochondrial respiration.
The development of hepatic steatosis occurs when excessive quantities of triglyceride accumulate in liver cells, specifically hepatocytes. This condition is often associated with insulin resistance [41]. Bcmo1-deficient mice, when fed a diet containing a higher level of β-carotene (1 mg/g diet) than used in the present study, developed non-alcoholic fatty liver disease [30]. The Bcmo1-deficient mice that were employed in our studies were fed less β-carotene (100 μg/g diet) for a shorter period than in this earlier published study but still showed elevated hepatic triglyceride levels. As seen in Fig. 4, Bcmo1-deficient mice fed even this lower level of β-carotene in the diet accumulated significantly (p < 0.01) more triglyceride in their livers than wild type mice fed the same diet. This elevation in hepatic triglyceride accumulation is accompanied by an elevation of PPARγ mRNA expression in both the intact liver and isolated HSCs from Bcmo1-deficient mice (see Fig. 5). A change in hepatic PPARγ expression has been proposed to be responsible for the development of hepatic steatosis in the Bcmo1-deficient mice receiving a β-carotene diet [30]. Thus, the present data are in agreement with the earlier report that β-carotene feeding readily gives rise to excessive triglyceride accumulation in the livers of Bcmo1-deficient mice [30]. The specific molecular events underlying this observation remain to be elucidated; however, the present work and earlier published data [30] suggest that hepatic β-carotene accumulation and/or metabolism are linked to hepatic triglyceride accumulation and/or metabolism. This may have clinical relevance for human subjects consuming high amounts of β-carotene given that recently published studies have reported polymorphisms in regions of the human Bcmo1 gene that influence carotenoid metabolism [42,43]. This possibility will need further evaluation.
In summary, our data establish that Bcmo1 is highly expressed in HSCs, but Bcmo1 is also expressed in hepatocytes, albeit at relatively lower levels. Both HSCs and hepatocytes are cellular sites for dietary β-carotene accumulation in the liver. Bcmo2 is expressed primarily in hepatocytes, with much lower levels of expression detected in HSCs. Moreover, Bcmo2 expression levels are elevated in livers, hepatocytes and HSCs isolated from Bcmo1-deficient mice, raising the possibility of coordinated regulation of Bcmo1 and Bcmo2 expression within the liver. Apo-10′- and apo-12′-carotenals, but not apo-8′- or apo-14′-carotenals, could be detected in serum and livers from wild type and Bcmo1-deficient mice. Hepatic apo-12′-carotenal levels were significantly elevated in the livers of Bcmo1-deficient compared to wild type mice, possibly arising from the elevated expression of Bcmo2 in the livers of these mutant mice. These data raise the interesting possibility that the body distinguishes metabolically between different apo-carotenal species. Further investigations will be needed to establish the processes and factors responsible for this.
Acknowledgments
The authors gratefully acknowledge support from the Fulbright Faculty Development Program (IS) and the National Institutes of Health through research Grants R01 DK068347 (WSB), R01 DK079221 (WSB), R01 DK044498 (EHH), R01 HL049879 (EHH), and R01 HD042174 (LPR).
Footnotes
Abbreviations used: HSCs, Hepatic stellate cells; RBP, retinol-binding protein; CRBPI, cellular retinol-binding protein type I; LRAT, lecithin:retinol acyltransferase.
References
- 1.Chambon P. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 2.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 3.Balmer JE, Blomhoff R. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 4.Olson JA. J Nutr. 1989;119:105–108. doi: 10.1093/jn/119.1.105. [DOI] [PubMed] [Google Scholar]
- 5.Vogel S, Gamble MV, Blaner WS. Retinoid uptake, metabolism and transport. In: Nau H, Blaner WS, editors. The Handbook of Experimental Pharmacology. The Retinoids, Springer Verlag; Heidelberg: 1999. pp. 31–96. [Google Scholar]
- 6.Ross AC. Vitamin A and Carotenoids. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, editors. Modern Nutrition in Health and Disease. tenth ed. Williams & Wilkins; Baltimore: 2006. pp. 351–375. [Google Scholar]
- 7.Wyss A, Wirtz G, Woggon W, Brugger R, Wyss M, Frielein A, Bachmann H, Hunziker W. Biochem Biophys Res Commun. 2000;271:334–336. doi: 10.1006/bbrc.2000.2619. [DOI] [PubMed] [Google Scholar]
- 8.Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, Grantt E, Cunningham FX., Jr J Biol Chem. 2000;276:6560–6565. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]
- 9.Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, von Lintig J. J Biol Chem. 2001;276:14110–14116. doi: 10.1074/jbc.M011510200. [DOI] [PubMed] [Google Scholar]
- 10.Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, Blaner WS. J Biol Chem. 2001;276:32160–32168. doi: 10.1074/jbc.M010086200. [DOI] [PubMed] [Google Scholar]
- 11.Yan W, Jang GF, Haeseleer F, Esumi N, Chang J, Kerrigan M, Campochiaro M, Campochiaro P, Palczewski K, Zack DJ. Genomics. 2001;72:193–202. doi: 10.1006/geno.2000.6476. [DOI] [PubMed] [Google Scholar]
- 12.Hu KQ, Liu C, Ernst H, Krinsky NI, Russell RM, Wang XD. J Biol Chem. 2006;282:19327–19338. doi: 10.1074/jbc.M512095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blaner WS, Olson JA. Retinol and retinoic acid metabolism. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids: Biology, Chemistry, and Medicine. second ed. Raven Press, Ltd.; New York: 1994. pp. 229–256. [Google Scholar]
- 14.Blomhoff R, Green MH, Green JB, Berg T, Norum KR. Physiol Rev. 1991;71:951–990. doi: 10.1152/physrev.1991.71.4.951. [DOI] [PubMed] [Google Scholar]
- 15.Senoo H, Kojima N, Sato NM. Vitam Horm. 2007;75:131–159. doi: 10.1016/S0083-6729(06)75006-3. [DOI] [PubMed] [Google Scholar]
- 16.Geerts A, Bleser PD, Hautekeete ML, Niki T, Wisse E. Fat-storing (Ito) cell biology. In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter D, Shafritz DA, editors. The Liver: Biology and Pathobiology. third ed. Chapter 19 Raven Press Ltd.; New York: 1994. pp. 819–837. [Google Scholar]
- 17.Friedman SL. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blomhoff R, Holte K, Naess L, Berg T. Exp Cell Res. 1994;150:186–193. doi: 10.1016/0014-4827(84)90713-4. [DOI] [PubMed] [Google Scholar]
- 19.Cooper AD. J Lipid Res. 1997;38:2173–2192. [PubMed] [Google Scholar]
- 20.Blaner WS, Hendriks HJF, Brouwer A, de Leewu AM, Knook DL, Goodman DS. J Lipid Res. 1985;26:1241–1251. [PubMed] [Google Scholar]
- 21.Weiner FR, Blaner WS, Czaja MJ, Shah A, Geerts A. Hepatology. 1992;15:336–342. doi: 10.1002/hep.1840150226. [DOI] [PubMed] [Google Scholar]
- 22.Sauvant P, Sapin V, Alexandre-Gouabau M-C, Dodeman I, Delpal S, Quadro L, Partier A, Abergel A, Colantuoni V, Rock E, Azais-Braesco V. Intern J Biochem Cell Biol. 2001;33:1000–1012. doi: 10.1016/s1357-2725(01)00066-8. [DOI] [PubMed] [Google Scholar]
- 23.Blomhoff R, Rasmussen M, Nilsson A, Norum KR, Berg T, Blaner WS, Kato M, Mertz JR, Goodman DS, Eriksson U, Peterson PA. J Biol Chem. 1985;260:13560–13565. [PubMed] [Google Scholar]
- 24.Blaner WS, Van Bennekum AM, Brouwer A, Hendriks HJF. FEBS Lett. 1990;274:89–92. doi: 10.1016/0014-5793(90)81336-m. [DOI] [PubMed] [Google Scholar]
- 25.Lindquvist A, He YG, Andersson S. J Histochem Cytochem. 2005;53:1403–1412. doi: 10.1369/jhc.5A6705.2005. [DOI] [PubMed] [Google Scholar]
- 26.Lindqvist A, Andersson S, Histochem J. Cytochem. 2004;52:491–499. doi: 10.1177/002215540405200407. [DOI] [PubMed] [Google Scholar]
- 27.Lakshman MR, Asher KA, Attlesey MG, Satchithanandam S, Mychkovsky I, Coutlakis PJ. J Lipid Res. 1989;30:1545–1550. [PubMed] [Google Scholar]
- 28.Martucci RB, Ziulkoski AL, Fortuna VA, Guaragna RM, Guma FCR, Truigo LC, Borojevic R. J Cell Biochem. 2004;92:414–423. doi: 10.1002/jcb.20073. [DOI] [PubMed] [Google Scholar]
- 29.Teodoro AJ, Perrone D, Martucci RB, Borojevic R. Eur J Nutr. 2009;48:261–268. doi: 10.1007/s00394-009-0001-6. [DOI] [PubMed] [Google Scholar]
- 30.Hessel S, Eichinger A, Isken A, Amengual J, Hunzelmann S, Hoeller U, Elste V, Hunziker W, Goralczyk R, Oberhauser V, von Lintig J, Wyss A. J Biol Chem. 2007;282:33553–33561. doi: 10.1074/jbc.M706763200. [DOI] [PubMed] [Google Scholar]
- 31.Fierce Y, De Moriais Vieira M, Piantedosi R, Wyss A, Blaner WS, Paik J. Arch Biochem Biophys. 2008;472:126–138. doi: 10.1016/j.abb.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Hepatology. 1987;7:1224–1229. doi: 10.1002/hep.1840070609. [DOI] [PubMed] [Google Scholar]
- 33.Blaner WS, Dixon JL, Moriwaki H, Martino RA, Stein O, Stein Y, Goodman DS. Eur J Biochem. 1987;164:301–307. doi: 10.1111/j.1432-1033.1987.tb11058.x. [DOI] [PubMed] [Google Scholar]
- 34.Kane MA, Chen N, Sparks S, Napoli JL. Biochem J. 2005;388:363–369. doi: 10.1042/BJ20041867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CM, Boileau AC, Boileau TW, Williams AW, Swanson KS, Heitz KA, Erdman JW., Jr J Nutr. 1999;129:2271–2277. doi: 10.1093/jn/129.12.2271. [DOI] [PubMed] [Google Scholar]
- 36.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Cell Metab. 2008;7:302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulanger A, McLemore P, Copeland NG, Gilbert DJ, Jenkins NA, Yu SS, Gentleman S, Redmond TM. FASEB J. 2003;17:1304–1306. doi: 10.1096/fj.02-0690fje. [DOI] [PubMed] [Google Scholar]
- 38.Lobo GP, Hessel S, Eichinger A, Noy N, Moise AR, Wyss A, Palczewski K, von Lintig J. FASEB J. 2010 doi: 10.1096/fj.09-150995. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, Plutzky J. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayne ST, Parker RS. Lipids. 1986;21:164–169. doi: 10.1007/BF02534440. [DOI] [PubMed] [Google Scholar]
- 41.Dixon JB, Bhathal PS, O’Brien PE. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 42.Lindqvist A, Sharvill J, Sharvill DE, Andersson S. J Nutr. 2007;137:2346–2350. doi: 10.1093/jn/137.11.2346. [DOI] [PubMed] [Google Scholar]
- 43.Leung WC, Hessel S, Meplan C, Flint J, Oberhauser V, Tourniaire F, Hesketh JE. FASEB J. 2009;23:1041–1053. doi: 10.1096/fj.08-121962. [DOI] [PubMed] [Google Scholar]