

Abstract
The role of B cells in autoimmune diseases involves different cellular functions, including the well-established secretion of autoantibodies, autoantigen presentation and ensuing reciprocal interactions with T cells, secretion of inflammatory cytokines, and the generation of ectopic germinal centers. Through these mechanisms B cells are involved both in autoimmune diseases that are traditionally viewed as antibody mediated and also in autoimmune diseases that are commonly classified as T cell mediated. This new understanding of the role of B cells opened up novel therapeutic options for the treatment of autoimmune diseases. This paper includes an overview of the different functions of B cells in autoimmunity; the involvement of B cells in systemic lupus erythematosus, rheumatoid arthritis, and type 1 diabetes; and current B-cell-based therapeutic treatments. We conclude with a discussion of novel therapies aimed at the selective targeting of pathogenic B cells.
1. Introduction
Traditionally, autoimmune disorders were classified as T cell mediated or autoantibody mediated. However the improved understanding of the complexity of the immune system has significantly influenced the way we view autoimmune diseases and their pathogeneses. Reciprocal roles of T-cell help for B cells during adaptive immune responses and B-cell help in CD4+ T-cell activation are being increasingly recognized. The observation that most autoantibodies in traditionally autoantibody-mediated diseases are of the IgG isotype and carry somatic mutations strongly suggests T-cell help in the autoimmune B-cell response. Likewise B cells function as crucial antigen presenting cells in autoimmune diseases that are traditionally viewed as T cell mediated. This paper will discuss the role of B cells in autoimmune diseases; however, it needs to be emphasized that most autoimmune diseases are driven by a dysfunction in the immune network consisting of B cells, T cells, and other immune cells.
2. B-Cell Functions in Autoimmunity
Different functions of B cells can contribute to autoimmune diseases (Figure 1):
secretion of autoantibodies;
presentation of autoantigen;
secretion of inflammatory cytokines;
modulation of antigen processing and presentation;
generation of ectopic GCs.
Figure 1.
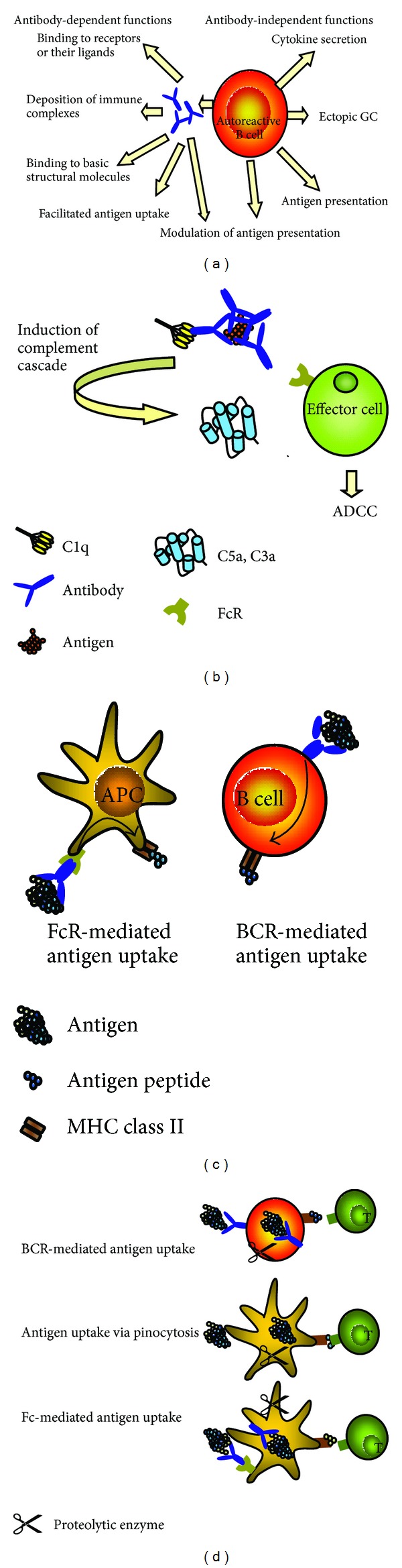
(a) B cells in autoimmune diseases. B cells have antibody-dependent and antibody-independent pathogenic functions. Secreted autoantibodies specific to receptors or receptor ligands can activate or inhibit receptor functions. Deposited immune complexes can activate complement and effector cells. Autoantibodies can bind to basic structural molecules and interfere with the synthesis of structural elements and facilitate the uptake of antigen. Independent of antibody secretion B cells secrete proinflammatory cytokines, support the formation of ectopic GCs, and serve as antigen presenting cells. Both secreted autoantibodies and BCR on B cells can modulate the processing and presentation of antigen and thereby affect the nature of presented T-cell determinants. (b) Pathogenic effects of deposited immune complexes. The Fc portion of antibodies in immune complexes can be bound by C1q of the classical complement pathway, which eventually leads to the release of C5a and C3a. These anaphylatoxins promote release of proinflammatory cytokines and serve as chemoattractants for effector cells. Moreover they induce the upregulation of activating FcR on effector cells. Binding of the Fc portion of the antibodies to FcR leads to activation of effector cells and further release of proinflammatory cytokines and proteolytic enzymes, mediators of antibody-dependent cell-mediated cytotoxicity (ADCC). (c) Effect of antibodies and antigen-specific B cells on antigen uptake. Left panel: antigen bound by antibody is taken up via FcR on APCs such as dendritic cells or macrophages. After processing, antigen is presented on MHC molecules. This FcR-mediated antigen uptake is more efficient than antigen uptake by pinocytosis. Right panel: antigen binds to the BCR of antigen-specific B cells and is internalized. B cells are highly efficient APCs in situations of low antigen concentrations. (d) Effect of antibodies and antigen-specific B cells on antigen processing and presentation. BCR-mediated antigen uptake can influence antigen processing and the nature of MHC-displayed T-cell determinants. Likewise, antigen/antibody complexes are bound by the FcR of APCs and processed in a unique fashion dependent on the epitope specificity of the bound antibody. The BCR or antibody can shield certain protein determinants from the proteolytic attack in endocytic compartments (represented as scissors in this figure). Presentation of some determinants may thereby be suppressed, while others are boosted. Thereby cryptic pathogenic peptides may be presented and stimulate autoreactive T cells.
These functions will be discussed in detail below.
2.1. Autoantibodies in Autoimmune Diseases
Autoantibodies can be detected in many autoimmune diseases. Their presence in the peripheral circulation and relative ease of detection makes them preferred markers to aid in diagnosis and prediction of autoimmune disorders. In some autoimmune diseases, the autoantibodies themselves have a pathogenic effect, as will be discussed in the following.
2.1.1. Deposition of Immune Complexes and Inflammation (Figure 1(b))
The deposition of immune complexes composed of autoantibodies and autoantigens is a prominent feature of several autoimmune diseases, including systemic lupus erythematosus, cryoglobulinemia, rheumatoid arthritis, scleroderma, and Sjögren's syndrome. The immune complexes can trigger inflammation through activation of complement and Fc-receptor-dependent effector functions [15]. In the classical complement cascade, the Fc portion of the antibody is bound by complement component C1q, which eventually triggers the activation of the anaphylatoxins C5a and C3a. C5a and to a lesser degree C3a attract effector cells such as neutrophils and NK cells and stimulate the release of proteolytic enzymes and inflammatory cytokines. Activation of complement has been consistently demonstrated in experimental models of immune-complex diseases and in the kidneys of patients with systemic lupus erythematosus and lupus nephritis [16]. The immune complexes can also directly bind to Fc-receptors on effector cells leading to antibody-dependent-cell-mediated cytotoxicity (ADCC).
2.1.2. Stimulation and Inhibition of Receptor Function
Autoantibodies can affect receptor function with different outcomes as illustrated by autoantibodies targeting the thyroid stimulating hormone (TSH) receptor. TSH receptor autoantibodies in Graves' disease stimulate receptor function, triggering the release of thyroid hormones and development of hyperthyroidism [17], while TSH receptor autoantibodies in autoimmune hypothyroidism block the binding of TSH to the receptor [18]. Inhibitory autoantibodies are also found in Myasthenia gravis, where autoantibodies bind to the nicotine ACh receptors (AChRs) and block neurotransmission at the neuromuscular junction, inducing symptoms such as muscle weakness and fatigue [19], and in multifocal motor neuropathy, where autoantibodies bind to the ganglioside GM1 and cause motor neuropathy with conduction block at multiple sites [20]. Other autoantibodies can bind receptor ligands, preventing their binding to the receptor, as seen in Graves' disease with anti-TSH autoantibodies [21]. Table 1 summarizes other examples of receptor autoantibodies, their targets, pathogenic mechanisms, and associated diseases.
Table 1.
Examples for receptor autoantibodies.
| Targeted receptor | Mechanism | Associated disease | References |
|---|---|---|---|
| Endothelial receptor type A (ETAR) | Activation | Pulmonary arterial hypertension (PAH) | [1] |
| Angiotensin II receptor (AT1R), ETAR | Activation | Systemic sclerosis | [2] |
| AT1R | Activating | Preeclampsia | [3–5] |
| α 1-adrenergic receptors (α 1-ARs) | Activating | Refractory hypertension | [3, 6, 7] |
| β 1-adrenergic receptor | Activation | Dilated cardiomyopathy (DCM), Chagas' disease | [8, 9] |
| N-methyl-D-aspartate receptor (NMDAR) | Activation | SLE | [10] |
| Glutamate receptor | Activation | SLE | [11] |
| Insulin receptor | Inhibition | Autoimmune hypoglycemia | [12] |
| Muscarinic type 3 receptor | Internalization | Sjögren's syndrome | [13] |
| NMDAR | Internalization | Anti-NMDA receptor encephalitis | [14] |
2.1.3. Facilitation of Antigen Uptake (Figure 1(c))
Autoantibodies facilitate antigen uptake by antigen presenting cells (APCs). Antigen complexed with antibodies is taken up via Fc receptors (FcRs) present on monocytes and dendritic cells [22]. This mechanism is more efficient than pinocytosis and results in 10–100-fold lower necessary antigen concentration for successful T-cell stimulation [23–26]. The importance of this mechanism has been demonstrated in a number of animal studies, where antibodies to various antigens enhanced T-cell responses to the respective antigens [27–29]. Autoantibodies can therefore break tolerance of normal T cells through their capacity to promote uptake of self-antigen by APCs via their FcRs. Indeed, autoantibodies to thyroid self-antigens dramatically enhanced uptake of thyroid peroxidase (TPO) by APCs and subsequent activation of TPO-reactive T cells [30] and blockade of FcγR markedly reduced this response [31]. Autoantibodies have also been demonstrated to facilitate the uptake of myelin by macrophages, and the removal of the Fc-portion of the antibodies prevented antigen uptake [32]. Moreover, FcγR–deficient DBA/1 mice were protected from myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (EAE), suggesting that FcR-mediated uptake of antibody-bound myelin is involved in the pathogenesis of multiple sclerosis [33]. Autoantibody-mediated antigen uptake may therefore be a critical mechanism in the pathogenesis of T-cell-mediated autoimmune diseases.
Further support for autoantibody-mediated antigen uptake as a pathogenic mechanism in autoimmunity comes from an elegant study by Harbers et al. where transgenic mice expressed ovalbumin (OVA) as “self” in both their thymus and pancreatic beta cells [34]. Presentation of OVA by dendritic cells to diabetogenic CD8+ OVA-reactive T cells was significantly stimulated by administration of antibodies specific to OVA. This response was not observed in mice lacking activating FcγR, indicating that the antibody-driven effector T-cell activation was indeed FcγR dependent.
However, autoantibodies are not always damaging to the organism, but can have protective functions [35, 36], and natural autoantibodies are commonly found in healthy individuals. Most of these antibodies are of the IgM isotype and have been speculated to have protective functions. One of these functions is the clearance of dying and aging cells and in mice natural IgM autoantibodies bind to epitopes specifically expressed on apoptotic cells [37, 38] enhancing the clearance of these cells, which may otherwise elicit a pathogenic autoimmune response [39, 40]. Lack of secreted IgM has been shown to correlate with an increase in pathogenic IgG autoantibodies and autoimmune disease possibly due to the lack of removal of apoptotic cells [41–43].
The mouse natural autoantibodies that arise without external antigen exposure are secreted from a subset of B cells, named B1 cells [44, 45], and a similar B-cell subset has been recently identified in humans [46]. In patients with SLE, higher levels of IgM associated with apoptotic cell clearance correlate with lower disease activity [47, 48], and healthy twins of SLE patients often present higher levels of these autoantibodies [49]. Another mechanism of protection by natural autoantibodies is the blockage of pathogenic autoantibodies to react with self-antigen [50], and titers of natural IgM specific to dsDNA correlated inversely with the severity of glomerulonephritis (GN) in SLE [51, 52].
Besides producing antibodies, activated B cells are also fundamental for coordinating T-cell functions as B-cell-depleted mice exhibit a dramatic decrease in numbers of CD4+ and CD8+ T cells, and a significant inhibition of memory CD8+ T cells [53, 54]. There are several antibody-independent mechanisms by which B cells can affect T cells and other immune cells as will be discussed below.
2.2. B Cells as Antigen-Presenting Cells
Especially at low antigen concentrations B cells function as superior APCs [55]. Other APCs (macrophages and dendritic cells) internalize antigen through pinocytosis, while B cells capture antigen through their antigen-specific B-cell receptors (BCRs) (Figure 1(c)). The ability of antigen-specific B cells to serve as efficient APCs has been demonstrated in several in vivo studies [56]. This mechanism is 1,000–10,000-fold more efficient than pinocytosis, and antigens can be successfully presented at very low concentrations, as those present in autoimmune diseases [57–59]. Moreover, the BCR-conferred antigen-specificity enables the B cells to focus the immune response to a specific antigen [60].
B cells serve as APCs in autoimmune diseases including rheumatoid arthritis and type 1 diabetes [61, 62]. Immunoglobulin-deficient mice in a model of autoimmune arthritis (proteoglycan-induced arthritis) did not develop arthritis. The observation that T cells isolated from proteoglycan-immunized transgenic mice that express membrane Ig (mIgM), but lack circulating antibodies, were unable to transfer disease suggested that these T cells were not adequately primed and that antigen-specific B cells may be required for this process. This was confirmed when direct targeting of proteoglycan to the BCR induced T cells competent to transfer arthritis [61].
The role of B cells as APC in type 1 diabetes is discussed in a separate chapter below.
2.3. Proinflammatory Cytokine Secretion
Activated B cells can secrete proinflammatory cytokines like interleukin-6 (IL-6), interferon-gamma (IFN-γ), IL-4, and TGF-beta [63–65]. These inflammatory mediators modulate the migration of dendritic cells, activate macrophages, exert a regulatory role on T-cell functions, and provide feedback stimulatory signals for further B-cell activation.
2.4. Modulation of Antigen Processing and Presentation
Besides facilitating antigen uptake, both membrane-bound and soluble antibodies can modulate the processing pattern of the antigen [66–69] (Figure 1(d)). Depending on the antigenic epitope recognized by the antibody or the BCR of the B cell, different T-cell determinants are presented on the MHC molecule [67, 70–73]. Indeed proteolysis of antigen-antibody complexes yielded protein fragments that were not observed in the absence of antibody [74]. This might have consequences for the ensuing T-cell response, in particular when otherwise cryptic T-cell determinants are presented. This bias in processing of antigen complexed with antibody may stem from antibody-mediated protection of distinct peptide sequences from degradation and/or sequestering of peptide sequences and interference with the loading of peptides onto MHC molecules [75].
The relevance of this mechanism in autoimmune diseases was suggested by studies showing that antibodies to thyroglobulin could augment or suppress processing and presentation of pathogenic T-cell determinants [76] and will be discussed further in the T1D chapter.
2.5. Ectopic Germinal Centers
B cells aid in the de novo generation of ectopic germinal centers (GCs) within inflamed tissues that can be observed during periods of chronic inflammation [77]. These ectopic structures are probably not a unique disease-specific occurrence, but a consequence of chronic inflammation. Activated T and B cells that infiltrate the site of chronic inflammation express membrane-bound lymphotoxin α 1 β 2 (LTα 1 β 2) [78]. High levels of LTα 1 β 2 eventually promote the differentiation of resident stromal cells into follicular dendritic cells (FDCs) and the development of ectopic GCs [79, 80]. These structures are similar to the GCs of secondary lymphoid organs and have been described in systemic lupus erythematosus, Hashimoto's thyroiditis, Graves' disease, rheumatoid arthritis, Sjögren's syndrome, multiple sclerosis, and type 1 diabetes [81–83]. The function and potential pathogenic role of ectopically formed lymphoid structures within inflamed tissues remains unclear. However, plasma cells residing within the ectopic GCs secrete autoantibodies [84], making it plausible that ectopic GCs have a role in the maintenance of immune pathology [85, 86].
Recent research has demonstrated that B cells are also involved in the inhibition of inflammatory immune responses, a function carried out by a subpopulation of B cells fittingly named regulatory B cells or Bregs.
3. IL-10 Secreting B Cells and Regulatory B Cells
A role of B cells in the inhibitory regulation of immune responses was initially suggested in autoimmune mice, where absence of B cells led to increased inflammation [87–89]. Transfer of wild-type B cells, but not IL10-negative B cells, reversed the inflammatory response [90], and IL-10 producing B cells were shown to suppress inflammation in mouse models of autoimmune diseases [91–93]. The significance of this anti-inflammatory cytokine was further supported by the finding that IL-10-deficient mice showed more severe disease accompanied with an increase in Th1 cytokine levels [88, 94, 95] and lower levels of regulatory T cells [96]. IL-10 is secreted by monocytes, Th2 T cells, regulatory T cells, and a rare subset of B cells. These IL-10 secreting B cells [97–100] can suppress CD4+ T cell responses and prevent autoimmune disease in mouse models and have been fittingly named regulatory B cells or Bregs [98–100]. The involvement of Bregs in human disease was first suggested by the observation that B-cell depletion can exacerbate Th-1-mediated autoimmune conditions such as ulcerative colitis [101] and psoriasis [102], and IL-10 producing B cells have been identified in humans [65]. For detailed discussions of Bregs please refer to other excellent reviews [99, 103].
4. B-Cell Tolerance
B-cell tolerance is established at multiple checkpoints throughout B-cell development, both in the bone marrow and the periphery. It has been estimated that 50% to 75% of newly produced human B cells are autoreactive and must be eliminated by tolerance mechanisms [104]. Induction of B-cell tolerance starts in the bone marrow. The major elimination mechanisms are receptor editing, clonal deletion, and anergy [105–107]. Defects in this early tolerance induction have been observed in subjects with rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes [53, 108–110].
Once autoreactive B cells are removed, the immature B cells leave the bone marrow and migrate to the spleen, where they may encounter autoantigen not present in the bone marrow. B cells with high avidity to autoantigen are deleted, while low-avidity or very-low avidity interactions lead to anergy or ignorance, respectively [111].
An encounter with true foreign antigen triggers the migration of the B cell to the T-cell zone of GCs, and activation by antigen-specific CD4+ T cells. During the ensuing rapid proliferation phase B cells undergo somatic hypermutation predominantly of the variable regions of their immunoglobulins. Only those B cells that express antibodies with increased affinity are selected to survive and exit the GC as antibody producing plasma cells or memory cells (for details see [112]).
4.1. Loss of Tolerance
Any of the above-discussed tolerance checkpoints can be faulted by genetic mutations allowing autoreactive B cells to survive. Some of these mutations have been identified in mouse models of autoimmune diseases with parallel findings in human disease.
Faulty negative selection at the immature B cells stage: NZM2410 mice spontaneously develop severe lupus nephritis at an early age. These mice carry the lupus susceptibility locus Sle1 containing at least three subloci, Sle1a, Sle1b, and Sle1c, involved in B-cell tolerance and activation of CD4+ T cells [113]. Using Sle1 congenic C57Bl6 mice, Kumar and colleagues [114] showed that mutations located within the Sle1 induced loss of B-cell tolerance through impaired negative selection of autoreactive B cells at the immature B-cell stage.
Increased B-cell signaling by overexpression of BCR signal-enhancing molecules or deficiency of molecules inhibiting BCR signaling: CD19 is a B-cell surface molecule that decreases the threshold for BCR stimulation. Hyperexpression of CD19 in mice led to increased levels of serum antibodies and increased B-cell activation, while the loss of CD19 reversed these phenotypes [115–119]. Deficiency of molecules that inhibit BCR-signaling, such as SHP-1 [120], Lyn [121], or FcγRIIB [122], causes increased B-cell signaling and initiates development of systemic autoimmunity in mice. The inhibitory FcγRIIB is expressed on B cells, where it regulates activating BCR signals. Lack of FcγRIIB expression leads to autoimmunity and autoimmune diseases [122–124]. The importance of FcγRIIB in human autoimmunity is exemplified by the finding that B cells from patients with lupus express lower levels of FcγRIIB on their surface due to polymorphisms in their FcγRIIB promoter [125], or the receptor itself [126, 127].
Generation of autoreactive immunoglobulins during somatic hypermutation: during affinity maturation the massive somatic hypermutations can also cause the inadvertent development of autoreactive immunoglobulins. While normally the resulting autoimmune B cells may either not receive necessary survival signals [128] or be eliminated, they accumulate in autoimmune diseases.
Increased survival of autoreactive B cells: B-cell activation factor (BAFF) is a B-cell survival factor and overexpression of BAFF in transgenic mice led to an expansion of peripheral B cells with higher autoantibody levels and the development of a lupus-like disease in the animals [28]. Elevated serum levels of BAFF have been found in patients with rheumatoid arthritis, systemic lupus erythematosus, and primary Sjörgren's syndrome [129–131]. These observations make BAFF a potential target for therapy [132, 133]. Indeed neutralization of BAFF was shown to be associated with loss of mature B cells [134] and reduced symptoms of autoimmune diseases in animal models [135, 136].
In the following the role of B cells in autoimmune diseases will be discussed in the context of systemic lupus erythematosus, rheumatoid arthritis, and type 1 diabetes. Systemic lupus erythematosus is a classic B-cell-mediated autoimmune disease, while rheumatoid arthritis and type 1 diabetes were initially considered to be predominantly T cell mediated. However recent studies suggest a role of B cells in the pathogenesis of these autoimmune diseases, as will be discussed in detail below.
Systemic Lupus Erythematosus (SLE) is a complex autoimmune disease, characterized by hyperglobulinemia, immune complex deposition, and end organ damage. B cells have been identified as major contributors to SLE, and B-cell depletion in SLE animal models abrogated the development of disease [54, 137]. Indeed, generalized B-cell hyperactivity has been documented in several murine models of lupus [138] and is also evident in patients with lupus [139, 140], where the number of B cells at all stages of activation is increased during active disease [141]. Both the decrease in proapoptotic genes and the increase in prosurvival gene expression have been suggested to cause this prolonged half-life of B cells in SLE (see also above).
A pathogenic role of autoantibodies in SLE is supported by the observation that the passive transfer of anti-DNA antibodies induces distinct features of lupus nephritis in healthy animals [142, 143]. Autoantibodies in SLE contribute to end organ damage in glomerulonephritis (glomerular antibodies and anti-DNA antibodies) [144–146], congenital heart block (anti-Ro antibodies) [147], and thrombosis (anticardiolipin antibodies) [148]. Other autoantibodies are directed to diverse self-molecules, most notably antinuclear antibodies directed to double stranded DNA (dsDNA) [149], and small nuclear ribonucleoprotein (snRNP). However, B cells also have antibody-independent effects on the SLE pathogenesis. These functions include antigen presentation, costimulation of T cells, and secretion of proinflammatory cytokines. This role was evaluated in a set of experiments conducted by Chan and colleagues, where B cells in a SLE mouse model carried a mutation that prevented the secretion of antibodies [54]. Thus these animals had B cells but were devoid of circulating antibodies. Despite the absence of autoantibodies, the mice developed nephritis, indicating an antibody-independent effect of B cells. B-cell-deficient MRL/lpr mice remain disease-free and fail to develop activated CD8+ and CD4+ T cells found in B-cell-sufficient mice, a finding attributed to loss of B cell-CD4 T cell interactions [150].
The dual effect of IL-10 as a B-cell stimulator and inhibitor of T-cell activation is exemplified in SLE [151]. In mice models for SLE, IL-10 appears to exert mainly its above-discussed anti-inflammatory effect and IL-10-deficient mice develop a more severe disease with increased proinflammatory cytokine levels [152], while transfer of IL-10 producing B cells induced the expansion of regulatory T cells [96]. However, in human SLE IL-10 promotes disease, IL-10 serum levels are significantly elevated and correlate with disease activity [153] and IL-10 induced a significant increase of anti-DNA antibody secretion in cultured PBMCs from SLE patients [154]. This antibody secretion was significantly reduced in the presence of neutralizing IL-10-specific antibodies [155] and treatment with IL-10-specific monoclonal antibodies led to marked improvement in participants of a small clinical trial [156]. The protective effect of IL-10 in mice appears to be mediated through T-cell regulation, as IL-10 overexpression in a mouse model for lupus resulted in reduced T-cell activation, while B-cell phenotypes remained unaffected [151]. In SLE patients immune cells that normally suppress B-cell activation are defective and do not counteract the IL-10-mediated stimulation of B cells resulting in the subsequent secretion of autoantibodies [157].
Rheumatoid Arthritis (RA) is a chronic inflammation of the joint capsule (synovium) and synovial membranes, associated with proliferation of synovial fibroblasts and macrophages, leading eventually to cartilage injury and bone erosion [158]. While T cells are a major component in the pathogenesis, several observations suggest that B cells are necessary for the development of the disease, as B-cell deficiency in RA animal models abrogates disease [159, 160], and autoimmune T cells alone are not sufficient to induce disease [161]. At least two mechanisms of B-cell involvement are currently considered: the production of autoantibodies and antigen presentation. Autoantibodies in patients with RA typically target several autoantigens, including rheumatoid factor (RF), type II collagen (CII), and citrullinated proteins (ACPA). A model for the pathological role of RA-associated autoantibodies will be discussed for autoantibodies directed to CII. These autoantibodies are found in ~70% of patients with early RA [162–164] both in their serum and synovial fluids. A pathogenic role of CII-specific antibodies was indicated in an animal model termed collagen-induced arthritis (CIA), where immunization of animals with CII induced the development of CII antibodies [165] and triggered arthritic symptoms [166–168]. Moreover, arthritic symptoms were also observed after passive transfer of CII-reactive serum obtained from CIA animals [169], patients with RA [170], or monoclonal antibodies specific to CII [165, 171] to healthy recipient animals, further supporting a pathological role of CII antibodies. CII autoantibodies are thought to mediate the formation of immune complexes in the joint, followed by complement activation and inflammatory cell recruitment. After FcγR ligation, the activated cells secrete proinflammatory cytokines, further activating an immune reaction consisting of synovial macrophages and infiltrating mononuclear cells with the eventual release of tissue-degrading enzymes that cause cartilage damage [172]. CII autoantibodies may also have a direct pathogenic function, which occurs in the absence of inflammatory mediators [173]. Here the antibodies modify the synthesis of collagen fibrils effecting cartilage synthesis and stability [174–176], possibly through steric hindrance of collagen epitopes that are important for the formation of collagen fibrils [177–179].
Type 1 Diabetes (T1D) is an organ specific autoimmune disease, characterized by the destruction of the insulin-producing beta cells in the pancreas. During progression towards T1D the pancreatic islets are infiltrated by mononuclear cells consisting of CD4+ and CD8+ T cells, B cells, macrophages, and dendritic cells [180, 181]. Both CD4+ and CD8+ T cells contribute to the ultimate attack on the beta cells [182], but in recent years the pathogenic role of B cells is beginning to emerge [183, 184]. A major hallmark of the autoimmunity leading to T1D is the presence of autoantibodies to beta cell antigens. At the time of clinical diagnosis more than 90% of patients present at least one of the T1D-associated autoantibodies [185]. The four beta cell antigens most frequently targeted by autoantibodies are insulin [186], the smaller isoform of glutamate decarboxylase (GAD65) [187], protein-tyrosine-phosphatase-like protein IA-2 [188], and the zinc transporter 8 (ZnT8) [189]. These autoantigens are also targeted by autoreactive T cells, suggesting a collaborative interaction between T and B cells [190]. No direct pathogenic role has been assigned to these autoantibodies and they are generally viewed as markers only. However a potential role of GAD65Ab in enhanced antigen uptake has been suggested [191]. Stimulation of GAD65-specific T-cell clones with human recombinant GAD65 was tested in the presence of sera obtained from GAD65Ab-positive T1D patients and GAD65Ab-negative T1D patients. Only sera from GAD65Ab-positive patients significantly enhanced T-cell stimulation. Moreover, this effect was inhibited by monoclonal antibodies to the FcR, suggesting Fc-mediated uptake of GAD65 complexed with GAD65Ab as the underlying mechanism.
However, the major mechanism by which B cells contribute to T1D development is the antibody-independent presentation of beta cell antigens [190, 192, 193]. Nonobese diabetic (NOD) mice deficient of mature B cells do not develop T1D [193–199]. In the absence of B cells, NOD mice showed significantly lower numbers of CD4+ and CD8+ T cells in their insulitic lesions [62, 195, 198–200], suggesting a role of B cells in the activation of autoreactive T cells. The function of B cells as APCs was illustrated in NOD mice whose B cells were rendered MHC class II deficient [201]. Although these animals retained their ability to present antigen via dendritic cells and macrophages, they were protected from diabetes development. However, the presence of insulitis in B-cell-deficient mice [62] and the report of at least one B-cell-deficient T1D patient [202] indicate that B cells may not be absolutely essential for the development of T1D and can be substituted by other APCs. As discussed above, B cell can focus the immune response towards a specific antigen. NOD mice that expressed only B cells specific to an irrelevant antigen (Hen Egg Lysosome) did not develop an autoantigen-specific T-cell response and remained healthy, indicating that only autoantigen-specific B cells enhance the development of T1D in the NOD mouse [203]. We will discuss the role of autoantigen-specific B cells exemplified by GAD65-specific B cells. Although GAD65 levels in murine pancreatic beta cells are very low, it is a major autoantigen in the pathogenesis of T1D in the NOD mouse [204]. GAD65-specific T cells have been demonstrated in both T1D patients and the NOD mouse [205–209]. Adoptive transfer of GAD65-reactive T cells isolated from NOD mice caused recipient animals to develop T1D [207, 210], supporting the concept of diabetogenic GAD65-specific T cells in the pathogenesis of T1D. Importantly, the development of these GAD65-specific T cells depends on the presence of B cells [190, 192, 203]. The finding that reconstitution of B-cell-depleted NOD mice with B cells reinstated T1D only if the repopulating B cells were primed with GAD65 [190] suggests that B-cell-mediated presentation of GAD65 stimulates GAD65-reactive T effector cells to target pancreatic beta cells. It is however not only the antigen specificity, but also the epitope specificity of the B cells that affects the T-cell response. GAD65-specific B-cell hybridomas with different epitope specificities were tested for their capacity to stimulate GAD65-specific T-cell clones. Those T-cell clones whose epitope lays outside of the BCR epitope showed increased T-cell responses, while T-cell clones whose epitope lays inside the BCR epitope showed suppressed responses, suggesting that the BCR epitope specificity can promote the presentation of some T-cell determinants, while suppressing that of others [211, 212].
Based on the promising results of B-cell depletion in the prevention of T1D in NOD mice, the effect of B-cell depletion on human T1D was tested in a phase II multicenter clinical trial on newly diagnosed human T1D patients [213]. One year after treatment a delay in the loss of beta cell function as shown by the preservation of C-peptide was demonstrated. Moreover, patients required less insulin and had better overall blood glucose control. These results confirm that B cells contribute also to human T1D.
Gathering the current understanding of B cells in T1D, the following mechanisms have been suggested (Figure 2). Beta cell antigen is taken up via BCR by antigen-specific B cells (1) and presented on MHC class II molecules to CD4+ T cells (2). Activated CD4+ T cells provide help to B cells (3). B cells differentiate to plasma cells and secrete autoantibodies (4). These autoantibodies form autoantigen-autoantibody complexes that bind to the FcγR on other APCs (5). This enhanced antigen presentation eventually triggers both natural killer cells and CD8+ T cells to attack the pancreatic beta cell.
Figure 2.
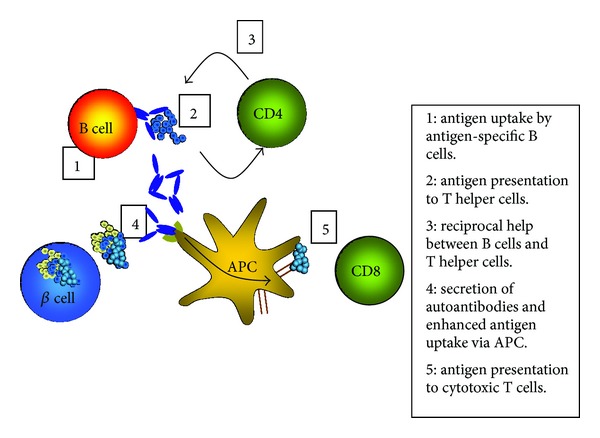
Model of pathogenic function of B cells in type 1 diabetes. Islet cell antigen released from the pancreatic beta cells is being taken up at low antigen concentrations by antigen-specific B cells, which present the antigen determinants to CD4+ T cells. T cells provide help to the B cells to eventually differentiate into antibody secreting plasma cells. Autoantibodies can now bind to the autoantigen and the resulting autoantibody/autoantigen complexes are efficiently taken up via FcR present on other APCs. This enhanced autoantigen uptake and presentation finally activates cytotoxic CD8+ T cells, which carry out the killing of the beta cells.
5. B-Cell Depletion
The growing understanding that B cells play a pathological role also in autoimmune diseases that are traditionally viewed as T cell mediated led to B-cell depletion treatment not only in diseases that are clearly B cell dominated, but also in autoimmune diseases that are traditionally viewed as T cell mediated, such as T1D.
B-cell depletion can target a number of different B-cell molecules, either with the goal of B-cell elimination, or the suppression of survival. Four major classes of B-cell targeting drugs have been evaluated for the treatment of autoimmune diseases: neutralization of survival factors BAFF and APRIL [214], killing of B cells using monoclonal antibodies directed to CD19, CD20, and CD22 [215–217], induction of apoptosis using reagents targeting the BCR itself or BCR associated transmembrane signaling proteins such as CD79 [193, 218], and ablation of the formation of ectopic GCs by antibodies against lymphotoxin-β receptor (LTβR) [219].
B-cell depletion for treatment of human autoimmune diseases is often accomplished through antibodies targeting the surface molecule CD20 (e.g., Rituximab and Ofatumumab). Treatment with these antibodies depletes B cells by a combination of antibody-mediated cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-triggered apoptosis [220] (Figure 3). The CD20 density on B cells appears to be important for CDC, since it is highly correlated with CDC [221]. CD20mAb/CD20 immune complexes aggregate in microdomains, where the antibodies' Fc regions are bound by C1q, leading to complement activation [222]. CD20 may also act as a signaling molecule to trigger apoptosis when engaged with CD20mAb [223, 224].
Figure 3.
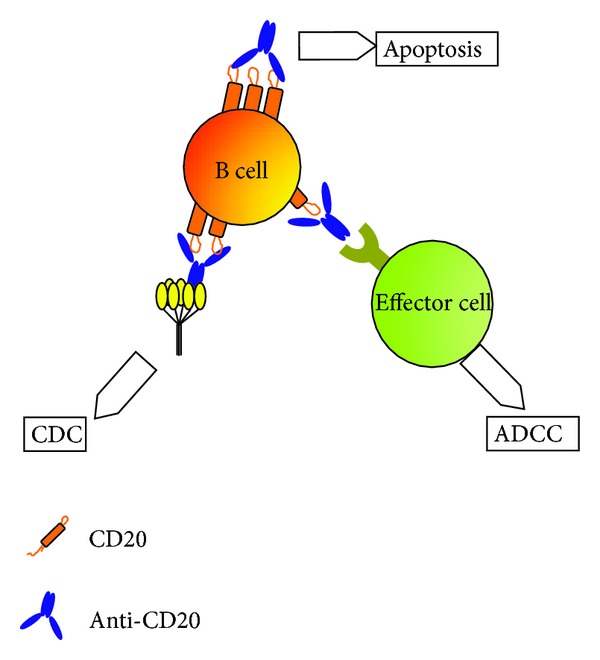
B-cell depletion with CD20 (Rituximab). Anti-CD20 mAb can direct the killing of B cells by antibody-dependent cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), or apoptosis. ADCC is triggered by the interaction between the Fc region of the antibody and the FcR on effector cells of the immune system. In CDC the Fc region is bound by the complement component C1q, which triggers a proteolytic cascade. Apoptosis occurs when CD20 molecules are cross-linked by anti-CD20 mAb in lipid rafts and activate signaling pathways leading to cell death.
B-cell depletion using Rituximab has been used for the treatment of a number of autoimmune and chronic inflammatory diseases [213, 225, 226]. Rituximab treatment results in nearly undetectable circulating B-cell levels one month after therapy and B cell counts remain low for 6–12 months [227]. Because the drug targets B cells expressing surface CD20, mature and memory CD20+CD27+ B cells in blood and primary lymphoid organs are effectively depleted, while long-lived plasma cells are not directly depleted [228], and Rituximab treatment appears not to affect circulating IgG levels [229], while reducing circulating IgM levels [230]. This effect of Rituximab is illustrated by the observation that immunization within the first 9 months after Rituximab treatment results in significantly reduced antibody responses, which develop from IgM-positive B cells [231, 232]. It is therefore of interest that for some autoimmune diseases B-cell depletion was reported to be associated with a decrease in IgG autoantibody titers [77] and specific depletion of autoreactive B cells by CD20mAb was demonstrated in mice [233]. As bone marrow stem cells and early B-cell precursors (pro-B cells) do not express CD20 [234], the new naïve B cells repopulate the B-cell compartment once the drug has cleared the system, allowing the immune response to return to normal. Disease relapses in about 50% of patients either at the time that B-cell numbers increase to pretreatment levels or within 3 months, while in other cases clinical relapse can be delayed for years [235]. Additional Rituximab courses can induce subsequent remission [236]. Multiple Rituximab courses are often associated with progressive decrease in circulating IgM [237] and IgG levels [238].
The antibody-independent effect of Rituximab treatment may be due to the elimination of B cells as APC and subsequent reduced stimulation of T cells [239, 240]. However, not all CD20+ B cells are equally affected by Rituximab treatment. B cells located in the peritoneal cavity are surprisingly resistant to depletion [241]. While these B cells express normal CD20 densities and are bound by CD20mAb, only about 50% of these cells are depleted. These location-dependent sensitivities to CD20mAb-mediated depletion could have significant consequences for therapy and may be the reason of the heterogeneity of results in human clinical trials. Other factors such as gender, age, and weight [242] and immunological profile [243] affect the outcome of Rituximab treatment. The major side effect of B-cell depletion is the risk of severe infections, which needs to be taken into consideration when evaluating the risks and benefits of B-cell depletion [244, 245].
In summary, B-cell depletion offers a promising therapy for the treatment of a variety of autoimmune diseases. The treatment is usually well tolerated; however, adverse events include infusion reactions, infections, and hypogammaglobulinemia.
6. Conclusions and Future Directions
The traditional concept of T-cell-mediated and autoantibody-mediated autoimmune diseases needs to be adjusted to reflect the interaction of different immune cells in autoimmune pathogenesis. The recognition of the contribution of B cells in the pathogenesis of autoimmune diseases, which are traditionally viewed as T cell mediated, led to promising immune-modulating therapies.
Global B-cell depletion eliminates both protective and pathogenic B cells. The success of B-cell depletion is therefore dictated by the extent of depletion of protective versus pathogenic B cells. The hopes that B-cell depletion would allow the restoration of immunological tolerance with long-term remission were not fulfilled, as is evident from the recurrence of autoimmune disease after the B-cell compartment is replenished. Selective depletion of antigen-specific B cells may provide an alternative to global B-cell depletion. This approach has the additional advantage that unlike Rituximab treatment it may also eliminate CD20-long-lived autoreactive plasma cells.
Several mechanisms are currently investigated in different in vitro and in vivo models of autoimmune diseases, a few of which will be discussed here.
Autoantigens can be fused to the IgG1 Fc domain to activate complement and FcR-dependent effector cell responses. This approach has been successfully evaluated in vitro and in vivo for the treatment of multiple sclerosis by autoantigen fused to Fc, which induced the effective and specific effector lysis of autoantigen-specific B cells [246]. An inhibitory B-cell signal can be induced by cross-linking of the autoantigen-specific BCR with the inhibitory FcγRIIb. Autoantigen fused to an FcγRIIb-binding mAb successfully reduced autoantibody levels and disease symptoms in lupus-prone MRL/lpr mice [247–249]. Autoantigen can also be coupled to an antibody specific to complement receptor 1 (CR1). CR1 negatively regulates the proliferation and differentiation of activated B cells after binding C3b [250]. In a small clinical trial SLE patients treated with dsDNA coupled to a CR1-specific monoclonal antibody showed a significant reduction of dsDNA autoantibody titers [251]. In an early study, Blank et al. employed anti-idiotypic antibodies directed to a pathogenic anti-DNA idiotype. Administration of this anti-idiotypic antibody alone or coupled to the cytotoxin saporin induced a significant reduction in anti-DNA antibody titer and diminished clinical manifestation in lupus-prone mice [252]. In a similar approach we demonstrated that GAD65Ab-specific anti-idiotypic antibodies protected NOD mice from development of T1D [253]. In addition to the direct elimination of antigen-specific B cells, autoantigen-fusion proteins can also bind pathogenic autoantibodies and route them to clearance.
Recently Bollmann proposed the targeted elimination of autoantigen-specific B cells using artificial antigens linked to magnetic nanoparticles. Here the autoantigen-specific B cells would be removed in an extracorporeal filtration method in an attempt to suppress or cure the autoimmune response [254].
The feasibility of these specific B-cell depletion approaches needs to be further evaluated; however, they offer new therapeutic options for the treatment of autoimmune diseases.
Acknowledgments
This work was supported by the National Institutes of Health (DK26190) and the Juvenile Diabetes Research Foundation.
Abbreviations
- AChR:
ACh receptor
- ACPA:
Citrullinated proteins
- ADCC:
Antibody-dependent-cell-mediated cytotoxicity
- APCs:
Antigen presenting cells
- BAFF:
B-cell activation factor
- BCR:
B-cell receptors
- Bregs:
Regulatory B cells
- CII:
Type II collagen
- CDC:
Complement-dependent cytotoxicity
- FcR:
Fc receptor
- FcγR:
Fc gamma receptor
- FDCs:
Follicular dendritic cells
- GAD65:
65 kD isoform of glutamate decarboxylase
- GC:
Germinal centers
- IA-2:
Protein-tyrosine-phosphatase-like protein
- IFN-γ:
Interferon-gamma
- LTα1β2:
Membrane-bound lymphotoxin α 1 β 2
- LTβR:
Lymphotoxin-β receptor
- MHC:
Major histocompatibility complex
- mIgM:
Membrane IgM
- NOD:
Nonobese diabetic
- OVA:
Ovalbumin
- RA:
Rheumatoid arthritis
- RF:
Rheumatoid factor
- SLE:
Systemic lupus erythematosus
- T1D:
Type 1 diabetes
- TPO:
Thyroid peroxidase
- TSH:
Thyroid stimulating hormone
- ZnT8:
Zinc transporter 8.
References
- 1.Dandel M, Wallukat G, Potapov E, Hetzer R. Role of β1-adrenoceptor autoantibodies in the pathogenesis of dilated cardiomyopathy. Immunobiology. 2011;217(5):511–520. doi: 10.1016/j.imbio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Riemekasten G, Philippe A, Näther M, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Annals of the Rheumatic Diseases. 2011;70(3):530–536. doi: 10.1136/ard.2010.135772. [DOI] [PubMed] [Google Scholar]
- 3.Xia Y, Kellems RE. Is preeclampsia an autoimmune disease? Clinical Immunology. 2009;133(1):1–12. doi: 10.1016/j.clim.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaMarca BD, Gilbert J, Granger JP. Recent progress toward the understanding of the pathophysiology of hypertension during preeclampsia. Hypertension. 2008;51(4):982–988. doi: 10.1161/HYPERTENSIONAHA.107.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenzel K, Rajakumar A, Haase H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58(1):77–84. doi: 10.1161/HYPERTENSIONAHA.111.171348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wenzel K, Wallukat G, Qadri F, et al. α1A-Adrenergic receptor-directed autoimmunity induces left ventricular damage and diastolic dysfunction in rats. PLoS One. 2010;5(2) doi: 10.1371/journal.pone.0009409.e9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luther HP, Homuth V, Wallukat G. α1-Adrenergic receptor antibodies in patients with primary hypertension. Hypertension. 1997;29(2):678–682. doi: 10.1161/01.hyp.29.2.678. [DOI] [PubMed] [Google Scholar]
- 8.Borda E, Pascual J, Cossio P. A circulating IgG in Chagas’ disease which binds to β-adrenoceptors of myocardium and modulates their activity. Clinical and Experimental Immunology. 1984;57(3):679–686. [PMC free article] [PubMed] [Google Scholar]
- 9.Jahns R, Boivin V, Schwarzbach V, Ertl G, Lohse M. Pathological autoantibodies in cardiomyopathy. Autoimmunity. 2008;41(6):454–461. doi: 10.1080/08916930802031603. [DOI] [PubMed] [Google Scholar]
- 10.Faust TW, Chang EH, Kowal C, et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(43):18569–18574. doi: 10.1073/pnas.1006980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levite M, Ganor Y. Autoantibodies to glutamate receptors can damage the brain in epilepsy, systemic lupus erythematosus and encephalitis. Expert Review of Neurotherapeutics. 2008;8(7):1141–1160. doi: 10.1586/14737175.8.7.1141. [DOI] [PubMed] [Google Scholar]
- 12.Lupsa BC, Chong AY, Cochran EK, Soos MA, Semple RK, Gorden P. Autoimmune forms of hypoglycemia. Medicine. 2009;88(3):141–153. doi: 10.1097/MD.0b013e3181a5b42e. [DOI] [PubMed] [Google Scholar]
- 13.Jin M, Hwang SM, Davies AJ, Shin Y, Bae JS. Autoantibodies in primary Sjogren’s syndrome patients induce internalization of muscarinic type 3 receptors. Biochimica et Biophysica Acta. 2012;1822:161–167. doi: 10.1016/j.bbadis.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. Journal of Neuroscience. 2010;30(17):5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin F, Chan AC. Pathogenic roles of B cells in human autoimmunity: insights from the clinic. Immunity. 2004;20(5):517–527. doi: 10.1016/s1074-7613(04)00112-8. [DOI] [PubMed] [Google Scholar]
- 16.Quigg RJ. Complement and autoimmune glomerular diseases. Current Directions in Autoimmunity. 2004;7:165–180. doi: 10.1159/000075692. [DOI] [PubMed] [Google Scholar]
- 17.Ando T, Latif R, Davies TF. Thyrotropin receptor antibodies: new insights into their actions and clinical relevance. Best Practice and Research. 2005;19(1):33–52. doi: 10.1016/j.beem.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Michalek K, Morshed SA, Latif R, Davies TF. TSH receptor autoantibodies. Autoimmunity Reviews. 2009;9(2):113–116. doi: 10.1016/j.autrev.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard FM, Jr., Lennon VA, Finley J, Matsumoto J, Elveback LR. Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Annals of the New York Academy of Sciences. 1987;505:526–538. doi: 10.1111/j.1749-6632.1987.tb51321.x. [DOI] [PubMed] [Google Scholar]
- 20.Susuki K, Yuki N, Schafer DP, et al. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Experimental Neurology. 2012;233(1):534–542. doi: 10.1016/j.expneurol.2011.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochi Y, Hamazu M, Kajita Y, Nagata A. Demonstration of anti-TSH antibody in TSH binding inhibitory immunoglobulin-positive sera of patients with Graves’ disease. Clinical Endocrinology. 2002;56(3):405–412. doi: 10.1046/j.1365-2265.2002.01489.x. [DOI] [PubMed] [Google Scholar]
- 22.Amigorena S, Bonnerot C. Fc receptor signaling and trafficking: a connection for antigen processing. Immunological Reviews. 1999;172:279–284. doi: 10.1111/j.1600-065x.1999.tb01372.x. [DOI] [PubMed] [Google Scholar]
- 23.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314(6011):537–539. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 24.Celis E, Zurawski VR, Jr., Chang TW. Regulation of T-cell function by antibodies: enhancement of the response of human T-cell clones to hepatitis B surface antigen by antigen-specific monoclonal antibodies. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(21 I):6846–6850. doi: 10.1073/pnas.81.21.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takai T. Fc receptors and their role in immune regulation and autoimmunity. Journal of Clinical Immunology. 2005;25(1):1–18. doi: 10.1007/s10875-005-0353-8. [DOI] [PubMed] [Google Scholar]
- 26.Takai T. Roles of Fc receptors in autoimmunity. Nature Reviews Immunology. 2002;2(8):580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 27.Celis E, Chang TW. Antibodies to hepatitis B surface antigen potentiate the response of human T lymphocyte clones to the same antigen. Science. 1984;224(4646):297–299. doi: 10.1126/science.6231724. [DOI] [PubMed] [Google Scholar]
- 28.Manca F, Fenoglio D, li Pira G, Kunkl A, Celada F. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. Journal of Experimental Medicine. 1991;173(1):37–48. doi: 10.1084/jem.173.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schalke BCG, Klinkert WEF, Wekerle H, Dwyer DS. Enhanced activation of a T cell line specific for acetylcholine receptor (AChR) by using anti-AChR monoclonal antibodies plus receptors. Journal of Immunology. 1985;134(6):3643–3648. [PubMed] [Google Scholar]
- 30.Nielsen CH, Leslie RGQ, Jepsen BS, Kazatchkine MD, Kaveri SV, Fischer E. Natural autoantibodies and complement promote the uptake of a self antigen, human thyroglobulin, by B cells and the proliferation of thyroglobulin-reactive CD4+ T cells in healthy individuals. European Journal of Immunology. 2001;31(9):2660–2668. doi: 10.1002/1521-4141(200109)31:9<2660::aid-immu2660>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen CH, Brix TH, Leslie RGQ, Hegedüs L. A role for autoantibodies in enhancement of pro-inflammatory cytokine responses to a self-antigen, thyroid peroxidase. Clinical Immunology. 2009;133(2):218–227. doi: 10.1016/j.clim.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 32.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992;358(6384):337–341. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 33.Abdul-Majid KB, Stefferl A, Bourquin C, et al. Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scandinavian Journal of Immunology. 2002;55(1):70–81. doi: 10.1046/j.1365-3083.2002.01024.x. [DOI] [PubMed] [Google Scholar]
- 34.Harbers SO, Crocker A, Catalano G, et al. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. Journal of Clinical Investigation. 2007;117(5):1361–1369. doi: 10.1172/JCI29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoenfeld Y, Toubi E. Protective autoantibodies: role in homeostasis, clinical importance, and therapeutic potential. Arthritis and Rheumatism. 2005;52(9):2599–2606. doi: 10.1002/art.21252. [DOI] [PubMed] [Google Scholar]
- 36.Gronwall C, Vas J. Silverman GJ protective roles of natural IgM antibodies. Frontiers in Immunology. 2012;3, article 66 doi: 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou MY, Fogelstrand L, Hartvigsen K, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. Journal of Clinical Investigation. 2009;119(5):1335–1349. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardy RR, Hayakawa K. Development of B cells producing natural autoantibodies to thymocytes and senescent erythrocytes. Springer Seminars in Immunopathology. 2005;26(4):363–375. doi: 10.1007/s00281-004-0183-1. [DOI] [PubMed] [Google Scholar]
- 39.Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38(4):259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 40.Quartier P, Potter PK, Ehrenstein MR, Walport MJ, Botto M. Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. European Journal of Immunology. 2005;35(1):252–260. doi: 10.1002/eji.200425497. [DOI] [PubMed] [Google Scholar]
- 41.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(3):1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. Journal of Experimental Medicine. 2000;191(7):1253–1258. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2009;120(5):417–426. doi: 10.1161/CIRCULATIONAHA.109.868158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Seminars in Immunopathology. 2005;26(4):347–362. doi: 10.1007/s00281-004-0182-2. [DOI] [PubMed] [Google Scholar]
- 45.Holodick NE, Tumang JR, Rothstein TL. Immunoglobulin secretion by B1 cells: differential intensity and IRF4-dependence of spontaneous IgM secretion by peritoneal and splenic B1 cells. European Journal of Immunology. 2010;40(11):3007–3016. doi: 10.1002/eji.201040545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70− . Journal of Experimental Medicine. 2011;208(1):67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grönwall C, Akhter E, Oh C, Burlingame RW, Petri M, Silverman GJ. IgM autoantibodies to distinct apoptosis-associated antigens correlate with protection from cardiovascular events and renal disease in patients with SLE. Clinical Immunology. 2012;142(3):390–398. doi: 10.1016/j.clim.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su J, Hua X, Concha H, Svenungsson E, Cederholm A, Frostegård J. Natural antibodies against phosphorylcholine as potential protective factors in SLE. Rheumatology. 2008;47(8):1144–1150. doi: 10.1093/rheumatology/ken120. [DOI] [PubMed] [Google Scholar]
- 49.Silverman GJ, Srikrishnan R, Germar K, et al. Genetic imprinting of autoantibody repertoires in systemic lupus erythematosus patients. Clinical and Experimental Immunology. 2008;153(1):102–116. doi: 10.1111/j.1365-2249.2008.03680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oppezzo P, Dighiero G. Autoantibodies, tolerance and autoimmunity. Pathologie Biologie. 2003;51(5):297–304. doi: 10.1016/s0369-8114(02)00318-8. [DOI] [PubMed] [Google Scholar]
- 51.Bootsma H, Spronk PE, Hummel EJ, et al. Anti-double stranded DNA antibodies in systemic lupus erythematosus: detection and clinical relevance of IgM-class antibodies. Scandinavian Journal of Rheumatology. 1996;25(6):352–359. doi: 10.3109/03009749609065646. [DOI] [PubMed] [Google Scholar]
- 52.Witte T, Hartung K, Sachse C, et al. IgM anti-dsDNA antibodies in systemic lupus erythematosus: negative association with nephritis. Rheumatology International. 1998;18(3):85–91. doi: 10.1007/s002960050063. [DOI] [PubMed] [Google Scholar]
- 53.Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. Journal of Experimental Medicine. 2005;201(5):703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan OTM, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. Journal of Experimental Medicine. 1999;189(10):1639–1647. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chesnut RW, Grey HM. Antigen presenting cells and mechanisms of antigen presentation. Critical Reviews in Immunology. 1985;5(3):263–316. [PubMed] [Google Scholar]
- 56.Rivera A, Chen CC, Ron N, Dougherty JP, Ron Y. Role of B cells as antigen-presenting cells in vivo revisited: antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. International Immunology. 2001;13(12):1583–1593. doi: 10.1093/intimm/13.12.1583. [DOI] [PubMed] [Google Scholar]
- 57.Lanzavechia A. Antigen uptake and accumulation in antigen-specific B cells. Immunological Reviews. 1987;99(1):39–51. doi: 10.1111/j.1600-065x.1987.tb01171.x. [DOI] [PubMed] [Google Scholar]
- 58.Watts C, Davidson HW. Endocytosis and recycling of specific antigen by human B cell lines. EMBO Journal. 1988;7(7):1937–1945. doi: 10.1002/j.1460-2075.1988.tb03031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roth R, Gee RJ, Mamula MJ. B lymphocytes as autoantigen-presenting cells in the amplification of autoimmunity. Annals of the New York Academy of Sciences. 1997;815:88–104. doi: 10.1111/j.1749-6632.1997.tb52047.x. [DOI] [PubMed] [Google Scholar]
- 60.Harvey BP, Gee RJ, Haberman AM, Shlomchik MJ, Mamula MJ. Antigen presentation and transfer between B cells and macrophages. European Journal of Immunology. 2007;37(7):1739–1751. doi: 10.1002/eji.200636452. [DOI] [PubMed] [Google Scholar]
- 61.O’Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. Journal of Immunology. 2005;174(6):3781–3788. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- 62.Wong FS, Wen L, Tang M, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53(10):2581–2587. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- 63.Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nature Immunology. 2000;1(6):475–482. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 64.Lund FE, Garvy BA, Randall TD, Harris DP. Regulatory roles for cytokine-producing B cells in infection and autoimmune disease. Current Directions in Autoimmunity. 2005;8:25–54. doi: 10.1159/000082086. [DOI] [PubMed] [Google Scholar]
- 65.Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? Journal of Immunology. 2004;172(6):3422–3427. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 66.Mamula MJ. Epitope spreading: the role of self peptides and autoantigen processing by B lymphocytes. Immunological Reviews. 1998;164:231–239. doi: 10.1111/j.1600-065x.1998.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 67.Manca F, Fenoglio D, Kunkl A, Cambiaggi C, Sasso M, Celada F. Differential activation of T cell clones stimulated by macrophages exposed to antigen complexed with monoclonal antibodies. A possible influence of paratope specificity on the mode of antigen processing. Journal of Immunology. 1988;140(9):2893–2898. [PubMed] [Google Scholar]
- 68.Manca F, Kunkl A, Fenoglio D. Constraints in T-B cooperation related to epitope topology on E. coli β-galactosidase I. The fine specificity of T cells dictates the fine specificity of antibodies directed to conformation-dependent determinants. European Journal of Immunology. 1985;15(4):345–350. doi: 10.1002/eji.1830150408. [DOI] [PubMed] [Google Scholar]
- 69.Amigorena S, Bonnerot C. Role of B-cell and Fc receptors in the selection of T-cell epitopes. Current Opinion in Immunology. 1998;10(1):88–92. doi: 10.1016/s0952-7915(98)80037-x. [DOI] [PubMed] [Google Scholar]
- 70.Watts C, Lanzavecchia A. Suppressive effect of antibody on processing of T cell epitopes. Journal of Experimental Medicine. 1993;178(4):1459–1463. doi: 10.1084/jem.178.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watts C. Capture and processing of exogenous antigens for presentation on MHC molecules. Annual Review of Immunology. 1997;15:821–850. doi: 10.1146/annurev.immunol.15.1.821. [DOI] [PubMed] [Google Scholar]
- 72.Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annual Review of Immunology. 1990;8:773–793. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- 73.Berzofsky JA. T-B reciprocity. An Ia-restricted epitope-specific circuit regulating T cell-B interaction and antibody specificity. Survey of Immunologic Research. 1983;2(3):223–229. doi: 10.1007/BF02918417. [DOI] [PubMed] [Google Scholar]
- 74.Simitsek PD, Campbell DG, Lanzavecchia A, Fairweather N, Watts C. Modulation of antigen processing by bound antibodies can boost or suppress class II major histocompatibility complex presentation of different T cell determinants. Journal of Experimental Medicine. 1995;181(6):1957–1963. doi: 10.1084/jem.181.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davidson HW, Watts C. Epitope-directed processing of specific antigen by B lymphocytes. Journal of Cell Biology. 1989;109(1):85–92. doi: 10.1083/jcb.109.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dai Y, Carayanniotis KA, Eliades P, et al. Enhancing or suppressive effects of antibodies on processing of a pathogenic T cell epitope in thyroglobulin. Journal of Immunology. 1999;162(12):6987–6992. [PubMed] [Google Scholar]
- 77.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annual Review of Immunology. 2006;24:467–496. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 78.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annual Review of Immunology. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 79.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. Journal of Experimental Medicine. 1996;183(4):1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LTαβ directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. Journal of Experimental Medicine. 2003;197(9):1153–1163. doi: 10.1084/jem.20021761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hutloff A, Büchner K, Reiter K, et al. Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis and Rheumatism. 2004;50(10):3211–3220. doi: 10.1002/art.20519. [DOI] [PubMed] [Google Scholar]
- 82.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nature Immunology. 2006;7(4):344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 83.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nature Reviews Immunology. 2006;6(3):205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 84.Manzo A, Paoletti S, Carulli M, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. European Journal of Immunology. 2005;35(5):1347–1359. doi: 10.1002/eji.200425830. [DOI] [PubMed] [Google Scholar]
- 85.Astorri E, Bombardieri M, Gabba S, Peakman M, Pozzilli P, Pitzalis C. Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets. Journal of Immunology. 2010;185(6):3359–3368. doi: 10.4049/jimmunol.1001836. [DOI] [PubMed] [Google Scholar]
- 86.Nacionales DC, Weinstein JS, Yan XJ, et al. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. Journal of Immunology. 2009;182(7):4226–4236. doi: 10.4049/jimmunol.0800771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wolf SD, Dittel BN, Hardardottir F, Janeway CA., Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. Journal of Experimental Medicine. 1996;184(6):2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nature Immunology. 2002;3(10):944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 89.Gonnella PA, Waldner HP, Weiner HL. B cell-deficient (μMT) mice have alterations in the cytokine microenvironment of the gut-associated lymphoid tissue (GALT) and a defect in the low dose mechanism of oral tolerance. Journal of Immunology. 2001;166(7):4456–4464. doi: 10.4049/jimmunol.166.7.4456. [DOI] [PubMed] [Google Scholar]
- 90.Mizoguchi A, Mizoguchi E, Smith RN, Preffer FI, Bhan AK. Suppressive role of B cells in chronic colitis of T cell receptor α mutant mice. Journal of Experimental Medicine. 1997;186(10):1749–1756. doi: 10.1084/jem.186.10.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16(2):219–230. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 92.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. Journal of Experimental Medicine. 2003;197(4):489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. Journal of Clinical Immunology. 2005;25(1):29–40. doi: 10.1007/s10875-005-0355-6. [DOI] [PubMed] [Google Scholar]
- 94.Yin Z, Bahtiyar G, Zhang N, et al. IL-10 regulates murine lupus. Journal of Immunology. 2002;169(4):2148–2155. doi: 10.4049/jimmunol.169.4.2148. [DOI] [PubMed] [Google Scholar]
- 95.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. Journal of Immunology. 2001;167(2):1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 96.Watanabe R, Ishiura N, Nakashima H, et al. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. Journal of Immunology. 2010;184(9):4801–4809. doi: 10.4049/jimmunol.0902385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28(5):639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 98.Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nature Reviews Immunology. 2008;8(5):391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- 99.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunological Reviews. 2008;224(1):201–214. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 100.Mauri C, Ehrenstein MR. The “short” history of regulatory B cells. Trends in Immunology. 2008;29(1):34–40. doi: 10.1016/j.it.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 101.Goetz M, Atreya R, Ghalibafian M, Galle PR, Neurath MF. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflammatory Bowel Diseases. 2007;13(11):1365–1368. doi: 10.1002/ibd.20215. [DOI] [PubMed] [Google Scholar]
- 102.Dass S, Vital EM, Emery P. Development of psoriasis after B cell depletion with rituximab. Arthritis and Rheumatism. 2007;56(8):2715–2718. doi: 10.1002/art.22811. [DOI] [PubMed] [Google Scholar]
- 103.Mauri C, Bosma A. Immune regulatory function of B cells. Annual Review of Immunology. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 104.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 105.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nature Immunology. 2004;5(6):645–650. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 106.Casellas R, Shih TA, Kleinewietfeld M, et al. Contribution of receptor editing to the antibody repertoire. Science. 2001;291(5508):1541–1544. doi: 10.1126/science.1056600. [DOI] [PubMed] [Google Scholar]
- 107.Hippen KL, Schram BR, Tze LE, Pape KA, Jenkins MK, Behrens TW. In vivo assessment of the relative contributions of deletion, anergy, and editing to B cell self-tolerance. Journal of Immunology. 2005;175(2):909–916. doi: 10.4049/jimmunol.175.2.909. [DOI] [PubMed] [Google Scholar]
- 108.Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. Journal of Experimental Medicine. 2005;201(10):1659–1667. doi: 10.1084/jem.20042321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Menard L, Samuels J, Ng YS, Meffre E. Inflammation-independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis and Rheumatism. 2011;63(5):1237–1245. doi: 10.1002/art.30164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Panigrahi AK, Goodman NG, Eisenberg RA, Rickels MR, Naji A, Prak ETL. RS rearrangement frequency as a marker of receptor editing in lupus and type 1 diabetes. Journal of Experimental Medicine. 2008;205(13):2985–2994. doi: 10.1084/jem.20082053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28(1):18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 112.Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27(2):190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen YF, Morel L. Genetics of T cell defects in lupus. Cellular & Molecular Immunology. 2005;2(6):403–409. [PubMed] [Google Scholar]
- 114.Kumar KR, Zhu J, Bhaskarabhatla M, Yan M, Mohan C. Enhanced expression of stem cell antigen-1 (Ly-6A/E) in lymphocytes from lupus prone mice correlates with disease severity. Journal of Autoimmunity. 2005;25(3):215–222. doi: 10.1016/j.jaut.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 115.Zhou LJ, Smith HM, Waldschmidt TJ, Schwarting R, Daley J, Tedder TF. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent B-lymphocyte development. Molecular and Cellular Biology. 1994;14(6):3884–3894. doi: 10.1128/mcb.14.6.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack of overexpress the CD19 signal transduction molecule. Immunity. 1995;3(1):39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 117.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376(6538):352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 118.Sato S, Steeber DA, Jansen PJ, Tedder TF. CD19 expression levels regulate B lymphocyte development: human CD19 restores normal function in mice lacking endogenous CD19. Journal of Immunology. 1997;158(10):4662–4669. [PubMed] [Google Scholar]
- 119.Tedder TF, Poe JC, Fujimoto M, Haas KM, Sato S. The CD19-CD21 signal transduction complex of B lymphocytes regulates the balance between health and autoimmune disease: systemic sclerosis as a model system. Current directions in autoimmunity. 2005;8:55–90. doi: 10.1159/000082087. [DOI] [PubMed] [Google Scholar]
- 120.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein- tyrosine-phosphatase deficiency. Trends in Biotechnology. 1997;15(8):302–307. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 121.Hibbs ML, Tarlinton DM, Armes J, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995;83(2):301–311. doi: 10.1016/0092-8674(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 122.Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13(2):277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 123.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nature Reviews Immunology. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 124.Boross P, Arandhara VL, Martin-Ramirez J, et al. The inhibiting Fc receptor for IgG, FcgammaRIIB, is a modifier of autoimmune susceptibility. The Journal of Immunology. 2011;187:1304–1313. doi: 10.4049/jimmunol.1101194. [DOI] [PubMed] [Google Scholar]
- 125.Blank MC, Stefanescu RN, Masuda E, et al. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Human Genetics. 2005;117(2-3):220–227. doi: 10.1007/s00439-005-1302-3. [DOI] [PubMed] [Google Scholar]
- 126.Warmerdam PAM, van de Winkel JGJ, Vlug A, Westerdaal NAC, Capel PJA. A single amino acid in the second Ig-like domain of the human Fcγ receptor II is critical for human IgG2 binding. Journal of Immunology. 1991;147(4):1338–1343. [PubMed] [Google Scholar]
- 127.Karassa FB, Trikalinos TA, Ioannidis JPA, et al. The FcγRIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney International. 2003;63(4):1475–1482. doi: 10.1046/j.1523-1755.2003.00873.x. [DOI] [PubMed] [Google Scholar]
- 128.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nature Reviews Immunology. 2005;5(11):853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 129.Rochas C, Hillion S, Saraux A, et al. Transmembrane BAFF from rheumatoid synoviocytes requires interleukin-6 to induce the expression of recombination-activating gene in B lymphocytes. Arthritis and Rheumatism. 2009;60(5):1261–1271. doi: 10.1002/art.24498. [DOI] [PubMed] [Google Scholar]
- 130.Pers JO, Daridon C, Devauchelle V, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Annals of the New York Academy of Sciences. 2005;1050:34–39. doi: 10.1196/annals.1313.004. [DOI] [PubMed] [Google Scholar]
- 131.Youinou P, Saraux A, Pers J-O. B-lymphocytes govern the pathogenesis of Sjögren's syndrome. Current Pharmaceutical Biotechnology. 2012;13(10):2071–2077. doi: 10.2174/138920112802273100. [DOI] [PubMed] [Google Scholar]
- 132.Tobón GJ, Pers JO, Youinou P, Saraux A. B cell-targeted therapies in Sjögren’s syndrome. Autoimmunity Reviews. 2010;9(4):224–228. doi: 10.1016/j.autrev.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 133.Vincent FB, Morand EF, Mackay F. BAFF and innate immunity: new therapeutic targets for systemic lupus erythematosus. Immunology and Cell Biology. 2012;90(3):293–303. doi: 10.1038/icb.2011.111. [DOI] [PubMed] [Google Scholar]
- 134.MacKay F, Schneider P. Cracking the BAFF code. Nature Reviews Immunology. 2009;9(7):491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 135.Hsu H, Khare SD, Lee F, et al. A novel modality of BAFF-specific inhibitor AMG623 peptibody reduces B-cell number and improves outcomes in murine models of autoimmune disease. Clinical and Experimental Rheumatology. 2012;30(2):197–201. [PubMed] [Google Scholar]
- 136.Jagessar SA, Heijmans N, Bauer J, et al. Antibodies against human BLyS and APRIL attenuate EAE development in marmoset monkeys. Journal of NeuroImmune Pharmacology. 2012;7(3):557–570. doi: 10.1007/s11481-012-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. Journal of Experimental Medicine. 1994;180(4):1295–1306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mohan C, Datta SK. Lupus: key pathogenic mechanisms and contributing factors. Clinical Immunology and Immunopathology. 1995;77(3):209–220. doi: 10.1006/clin.1995.1146. [DOI] [PubMed] [Google Scholar]
- 139.Fauci AS, Moutsopoulos HM. Polyclonally triggered B cells in the peripheral blood and bone marrow of normal individuals and in patients with systemic lupus erythematosus and primary Sjogren’s syndrome. Arthritis and Rheumatism. 1981;24(4):577–584. doi: 10.1002/art.1780240402. [DOI] [PubMed] [Google Scholar]
- 140.Lipsky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nature Immunology. 2001;2(9):764–766. doi: 10.1038/ni0901-764. [DOI] [PubMed] [Google Scholar]
- 141.Klinman DM, Shirai A, Ishigatsubo Y, Conover J, Steinberg AD. Quantitation of IgM- and IgG-secreting B cells in the peripheral blood of patients with systemic lupus erythematosus. Arthritis and Rheumatism. 1991;34(11):1404–1410. doi: 10.1002/art.1780341110. [DOI] [PubMed] [Google Scholar]
- 142.Tsao BP, Ohnishi K, Cheroutre H, et al. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. Journal of Immunology. 1992;149(1):350–358. [PubMed] [Google Scholar]
- 143.Vlahakos DV, Foster MH, Adams S, et al. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney International. 1992;41(6):1690–1700. doi: 10.1038/ki.1992.242. [DOI] [PubMed] [Google Scholar]
- 144.Hanrotel-Saliou C, Segalen I, le Meur Y, Youinou P, Renaudineau Y. Glomerular antibodies in lupus nephritis. Clinical Reviews in Allergy and Immunology. 2011;40(3):151–158. doi: 10.1007/s12016-010-8204-4. [DOI] [PubMed] [Google Scholar]
- 145.van Bavel CC, Fenton KA, Rekvig OP, van der Vlag J, Berden JH. Glomerular targets of nephritogenic autoantibodies in systemic lupus erythematosus. Arthritis and Rheumatism. 2008;58(7):1892–1899. doi: 10.1002/art.23626. [DOI] [PubMed] [Google Scholar]
- 146.D’Andrea DM, Coupaye-Gerard B, Kleyman TR, Foster MH, Madaio MP. Lupus autoantibodies interact directly with distinct glomerular and vascular cell surface antigens. Kidney International. 1996;49(5):1214–1221. doi: 10.1038/ki.1996.175. [DOI] [PubMed] [Google Scholar]
- 147.Tincani A, Biasini-Rebaioli C, Cattaneo R, Riboldi P. Nonorgan specific autoantibodies and heart damage. Lupus. 2005;14(9):656–659. doi: 10.1191/0961203305lu2194oa. [DOI] [PubMed] [Google Scholar]
- 148.Foster MH, Cizman B, Madaio MP. Nephritogenic autoantibodies in systemic lupus erythematosus: immunochemical properties, mechanisms of immune deposition, and genetic origins. Laboratory Investigation. 1993;69(5):494–507. [PubMed] [Google Scholar]
- 149.Hahn BH. Antibodies to DNA. New England Journal of Medicine. 1998;338(19):1359–1368. doi: 10.1056/NEJM199805073381906. [DOI] [PubMed] [Google Scholar]
- 150.Chan OTM, Shlomchik MJ. Cutting edge: B cells promote CD8+ T cell activation in MRL-Fas(lpr) mice independently of MHC class I antigen presentation. Journal of Immunology. 2000;164(4):1658–1662. doi: 10.4049/jimmunol.164.4.1658. [DOI] [PubMed] [Google Scholar]
- 151.Blenman KRM, Duan B, Xu Z, et al. IL-10 regulation of lupus in the NZM2410 murine model. Laboratory Investigation. 2006;86(11):1136–1148. doi: 10.1038/labinvest.3700468. [DOI] [PubMed] [Google Scholar]
- 152.Pathak S, Mohan C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Research and Therapy. 2011;13(5) doi: 10.1186/ar3465.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Capper ER, Maskill JK, Gordon C, Blakemore AIF. Interleukin (IL)-10, IL-1ra and IL-12 profiles in active and quiescent systemic lupus erythematosus: could longitudinal studies reveal patient subgroups of differing pathology? Clinical and Experimental Immunology. 2004;138(2):348–356. doi: 10.1111/j.1365-2249.2004.02607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tyrrell-Price J, Lydyard PM, Isenberg DA. The effect of interleukin-10 and of interleukin-12 on the in vitro production of anti-double-stranded DNA antibodies from patients with systemic lupus erythematosus. Clinical and Experimental Immunology. 2001;124(1):118–125. doi: 10.1046/j.1365-2249.2001.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kanda N, Tsuchida T, Tamaki K. Estrogen enhancement of anti-double-stranded dna antibody and immunoglobulin g production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis and Rheumatism. 1999;42(2):328–337. doi: 10.1002/1529-0131(199902)42:2<328::AID-ANR16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 156.Llorente L, Richaud-Patin Y, Garcia-Padilla C, et al. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic Lupus erythematosus. Arthritis and Rheumatism. 2000;43(8):1790–1800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 157.Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. Journal of Clinical Pathology. 2003;56(7):481–490. doi: 10.1136/jcp.56.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85(3):307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 159.Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA) Clinical and Experimental Immunology. 1998;111(3):521–526. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yanaba K, Hamaguchi Y, Venturi GM, Steeber DA, St Clair EW, Tedder TF. B cell depletion delays collagen-induced arthritis in mice: arthritis induction requires synergy between humoral and cell-mediated immunity. Journal of Immunology. 2007;179(2):1369–1380. doi: 10.4049/jimmunol.179.2.1369. [DOI] [PubMed] [Google Scholar]
- 161.Luross JA, Williams NA. The genetic and immunopathological processes underlying collagen-induced arthritis. Immunology. 2001;103(4):407–416. doi: 10.1046/j.1365-2567.2001.01267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fujii K, Tsuji M, Kitamura A, Murota K. The diagnostic significance of anti-Type II collagen antibody assay in rheumatoid arthritis. International Orthopaedics. 1992;16(3):272–276. doi: 10.1007/BF00182710. [DOI] [PubMed] [Google Scholar]
- 163.Cook AD, Gray R, Ramshaw J, Mackay IR, Rowley MJ. Antibodies against the CB10 fragment of type II collagen in rheumatoid arthritis. Arthritis Research & Therapy. 2004;6(5):R477–483. doi: 10.1186/ar1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cook AD, Rowley MJ, Stockman A, Muirden KD, Mackay IR. Specificity of antibodies to type II collagen in early rheumatoid arthritis. Journal of Rheumatology. 1994;21(7):1186–1191. [PubMed] [Google Scholar]
- 165.Holmdahl R, Rubin K, Klareskog L. Characterization of the antibody response in mice with type II collagen-induced arthritis, using monoclonal anti-type II collagen antibodies. Arthritis and Rheumatism. 1986;29(3):400–410. doi: 10.1002/art.1780290314. [DOI] [PubMed] [Google Scholar]
- 166.Holmdahl R, Nordling C, Larsson P, et al. Collagen induced arthritis: an experiment model for rheumatoid arthritis with involvement of both DTH and immune complex mediated mechanisms. Clinical and Experimental Rheumatology. 1989;7(3):S51–S55. [PubMed] [Google Scholar]
- 167.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. Journal of Experimental Medicine. 1977;146(3):857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Courtenay JS, Dallman MJ, Dayan AD. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283(5748):666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 169.Stuart JM, Cremer MA, Townes AS, Kang AH. Type II collagen-induced arthritis in rats. Passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. Journal of Experimental Medicine. 1982;155(1):1–16. doi: 10.1084/jem.155.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wooley PH, Luthra HS, Singh SK. Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clinic Proceedings. 1984;59(11):737–743. doi: 10.1016/s0025-6196(12)65583-9. [DOI] [PubMed] [Google Scholar]
- 171.Nandakumar KS, Andrén M, Martinsson P, et al. Induction of arthritis by single monoclonal IgG anti-collagen type II antibodies and enhancement of arthritis in mice lacking inhibitory FcγRIIB. European Journal of Immunology. 2003;33(8):2269–2277. doi: 10.1002/eji.200323810. [DOI] [PubMed] [Google Scholar]
- 172.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 173.Nandakumar KS, Bajtner E, Hill L, et al. Arthritogenic antibodies specific for a major type II collagen triple-helical epitope bind and destabilize cartilage independent of inflammation. Arthritis and Rheumatism. 2008;58(1):184–196. doi: 10.1002/art.23049. [DOI] [PubMed] [Google Scholar]
- 174.Amirahmadi SF, Minh HP, Gray RE, et al. An arthritogenic monoclonal antibody to type II collagen, CII-C1, impairs cartilage formation by cultured chondrocytes. Immunology and Cell Biology. 2004;82(4):427–434. doi: 10.1111/j.0818-9641.2004.01267.x. [DOI] [PubMed] [Google Scholar]
- 175.Gray RE, Seng N, Mackay IR, Rowley MJ. Measurement of antibodies to collagen II by inhibition of collagen fibril formation in vitro. Journal of Immunological Methods. 2004;285(1):55–61. doi: 10.1016/j.jim.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 176.Crombie DE, Turer M, Zuasti BB, et al. Destructive effects of murine arthritogenic antibodies to type II collagen on cartilage explants in vitro. Arthritis Research & Therapy. 2005;7(5):R927–937. doi: 10.1186/ar1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Amirahmadi SF, Whittingham S, Crombie DE, et al. Arthritogenic anti-type II collagen antibodies are pathogenic for cartilage-derived chondrocytes independent of inflammatory cells. Arthritis and Rheumatism. 2005;52(6):1897–1906. doi: 10.1002/art.21097. [DOI] [PubMed] [Google Scholar]
- 178.Rowley MJ, Nandakumar KS, Holmdahl R. The role of collagen antibodies in mediating arthritis. Modern Rheumatology. 2008;18(5):429–441. doi: 10.1007/s10165-008-0080-x. [DOI] [PubMed] [Google Scholar]
- 179.Nandakumar KS. Pathogenic antibody recognition of cartilage. Cell and Tissue Research. 2010;339(1):213–220. doi: 10.1007/s00441-009-0816-8. [DOI] [PubMed] [Google Scholar]
- 180.Signore A, Pozzilli P, Gale EAM, Andreani D, Beverley PCL. The natural history of lymphocyte subsets infiltrating the pancreas of NOD mice. Diabetologia. 1989;32(5):282–289. doi: 10.1007/BF00265543. [DOI] [PubMed] [Google Scholar]
- 181.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clinical and Experimental Immunology. 2009;155(2):173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Advances in Immunology. 2005;87:123–162. doi: 10.1016/S0065-2776(05)87004-X. [DOI] [PubMed] [Google Scholar]
- 183.Wong FS, Wen L. B cells in autoimmune diabetes. The Review of Diabetic Studies. 2005;2:121–135. doi: 10.1900/RDS.2005.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.O’Neill SK, Liu E, Cambier JC. Change you can B(cell)eive in: recent progress confirms a critical role for B cells in type 1 diabetes. Current Opinion in Endocrinology, Diabetes and Obesity. 2009;16(4):293–298. doi: 10.1097/MED.0b013e32832e06a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bingley PJ. Clinical applications of diabetes antibody testing. Journal of Clinical Endocrinology and Metabolism. 2010;95(1):25–33. doi: 10.1210/jc.2009-1365. [DOI] [PubMed] [Google Scholar]
- 186.Palmer JP, Asplin CM, Clemons P. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983;222(4630):1337–1339. doi: 10.1126/science.6362005. [DOI] [PubMed] [Google Scholar]
- 187.Baekkeskov S, Aanstoot H-J, Christgau S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutatmic acid decarboxylase. Nature. 1990;347(6289):151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 188.Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-M(r) tryptic fragments of islet antigens in insulin-dependent diabetes to the protein tyrosine phosphatase- like molecule IA-2 (ICA512) Journal of Clinical Investigation. 1995;96(3):1506–1511. doi: 10.1172/JCI118188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):17040–17045. doi: 10.1073/pnas.0705894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. Journal of Immunology. 1998;161(8):3912–3918. [PubMed] [Google Scholar]
- 191.Reijonen H, Daniels TL, Lernmark A, Nepom GT. GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes. 2000;49(10):1621–1626. doi: 10.2337/diabetes.49.10.1621. [DOI] [PubMed] [Google Scholar]
- 192.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. Journal of Immunology. 1998;161(3):1163–1168. [PubMed] [Google Scholar]
- 193.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46(6):941–946. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 194.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Igμ(null) mice. Journal of Experimental Medicine. 1996;184(5):2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hu CY, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. Journal of Clinical Investigation. 2007;117(12):3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fiorina P, Vergani A, Dada S, et al. Targeting CD22 reprograms b-cells and reverses autoimmune diabetes. Diabetes. 2008;57(11):3013–3024. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zekavat G, Rostami SY, Badkerhanian A, et al. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. Journal of Immunology. 2008;181(11):8133–8144. doi: 10.4049/jimmunol.181.11.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in FcγR effector functions. Journal of Immunology. 2008;180(5):2863–2875. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 199.Mariño E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4+CD25+ T-cells control autoimmunity in the absence of B-cells. Diabetes. 2009;58(7):1568–1577. doi: 10.2337/db08-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Brodie GM, Wallberg M, Santamaria P, Wong FS, Green EA. B-cells promote intra-islet CD8+ cytotoxic T-cell survival to enhance type 1 diabetes. Diabetes. 2008;57(4):909–917. doi: 10.2337/db07-1256. [DOI] [PubMed] [Google Scholar]
- 201.Noorchashm H, Lieu YK, Noorchashm N, et al. I-A(g7)-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet β cells of nonobese diabetic mice. Journal of Immunology. 1999;163(2):743–750. [PubMed] [Google Scholar]
- 202.Martin S, Wolf-Eichbaum D, Duinkerken G, et al. Development of type 1 diabetes despite severe hereditary B-cell deficiency. New England Journal of Medicine. 2001;345(14):1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 203.Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. European Journal of Immunology. 2002;32(12):3657–3666. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 204.Kim J, Richter W, Aanstoot HJ, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42(12):1799–1808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- 205.Öling V, Reijonen H, Simell O, Knip M, Ilonen J. Autoantigen-specific memory CD4+ T cells are prevalent early in progression to Type 1 diabetes. Cellular Immunology. 2012;273(2):133–139. doi: 10.1016/j.cellimm.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 206.Monti P, Scirpoli M, Rigamonti A, et al. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. Journal of Immunology. 2007;179(9):5785–5792. doi: 10.4049/jimmunol.179.9.5785. [DOI] [PubMed] [Google Scholar]
- 207.Quinn A, Sercarz EE. T cells with multiple fine specificities are used by non-obese diabetic (NOB) mice in the response to GAD(524-543) Journal of Autoimmunity. 1996;9(3):365–370. doi: 10.1006/jaut.1996.0049. [DOI] [PubMed] [Google Scholar]
- 208.Kaufman DL, Clare-Salzler M, Tian J, et al. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366(6450):69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366(6450):72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 210.Zekzer D, Wong FS, Ayalon O, et al. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. Journal of Clinical Investigation. 1998;101(1):68–73. doi: 10.1172/JCI119878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jaume JC, Parry SL, Madec AM, Sønderstrup G, Baekkeskov S. Suppressive effect of glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing of T cell determinants located within the antibody epitope. Journal of Immunology. 2002;169(2):665–672. doi: 10.4049/jimmunol.169.2.665. [DOI] [PubMed] [Google Scholar]
- 212.Banga JP, Moore JK, Duhindan N, et al. Modulation of antigen presentation by autoreactive B cell clones specific for GAD65 from a type I diabetic patient. Clinical and Experimental Immunology. 2004;135(1):74–84. doi: 10.1111/j.1365-2249.2004.02343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. New England Journal of Medicine. 2009;361(22):2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404(6781):995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 215.Dörner T, Burmester GR. New approaches of B-cell-directed therapy: beyond rituximab. Current Opinion in Rheumatology. 2008;20(3):263–268. doi: 10.1097/BOR.0b013e3282f5e08d. [DOI] [PubMed] [Google Scholar]
- 216.Looney RJ, Anolik J, Sanz I. B cells as therapeutic targets for rheumatic diseases. Current Opinion in Rheumatology. 2004;16(3):180–185. doi: 10.1097/00002281-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 217.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nature Reviews Drug Discovery. 2006;5(7):564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 218.Li Y, Chen F, Putt M, et al. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. Journal of Immunology. 2008;181(5):2961–2972. doi: 10.4049/jimmunol.181.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gatumu MK, Skarstein K, Papandile A, Browning JL, Fava RA, Bolstad AI. Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjögren syndrome in salivary glands of non-obese diabetic mice. Arthritis Research and Therapy. 2009;11(1, article R24) doi: 10.1186/ar2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Molecular Immunology. 2007;44(16):3823–3837. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- 221.Golay J, Zaffaroni L, Vaccari T, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95(12):3900–3908. [PubMed] [Google Scholar]
- 222.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103(7):2738–2743. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 223.Chan HTC, Hughes D, French RR, et al. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Research. 2003;63(17):5480–5489. [PubMed] [Google Scholar]
- 224.Cragg MS, Walshe CA, Ivanov AO, Glennie MJ. The biology of CD20 and its potential as a target for mAb therapy. Current Directions in Autoimmunity. 2005;8:140–174. doi: 10.1159/000082102. [DOI] [PubMed] [Google Scholar]
- 225.Townsend MJ, Monroe JG, Chan AC. B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunological Reviews. 2010;237(1):264–283. doi: 10.1111/j.1600-065X.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 226.Tony HP, Burmester G, Schulze-Koops H, et al. Safety and clinical outcomes of rituximab therapy in patients with different autoimmune diseases: experience from a national registry (GRAID) Arthritis Research and Therapy. 2011;13(3, article R75) doi: 10.1186/ar3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New England Journal of Medicine. 2008;358(7):676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 228.Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JCW. Bone marrow B-lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology. 2007;46(1):29–36. doi: 10.1093/rheumatology/kel148. [DOI] [PubMed] [Google Scholar]
- 229.Cambridge G, Leandro MJ, Teodorescu M, et al. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis and Rheumatism. 2006;54(11):3612–3622. doi: 10.1002/art.22211. [DOI] [PubMed] [Google Scholar]
- 230.Pescovitz MD, Torgerson TR, Ochs HD, et al. Effect of rituximab on human in vivo antibody immune responses. Journal of Allergy and Clinical Immunology. 2011;128(6):1295.e5–1302.e5. doi: 10.1016/j.jaci.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.van der Kolk LE, Baars JW, Prins MH, van Oers MHJ. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood. 2002;100(6):2257–2259. [PubMed] [Google Scholar]
- 232.Gonzalez-Stawinski GV, Yu PB, Love SD, Parker W, Davis RD. Hapten-induced primary and memory humoral responses are inhibited by the infusion of anti-CD20 monoclonal antibody (IDEC-C2B8, Rituximab) Clinical Immunology. 2001;98(2):175–179. doi: 10.1006/clim.2000.4980. [DOI] [PubMed] [Google Scholar]
- 233.Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(10):4658–4663. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Anderson KC, Bates MP, Slaughenhoupt BL. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63(6):1424–1433. [PubMed] [Google Scholar]
- 235.Popa C, Leandro MJ, Cambridge G, Edwards JCW. Repeated B lymphocyte depletion with rituximab in rheumatoid arthritis over 7 yrs. Rheumatology. 2007;46(4):626–630. doi: 10.1093/rheumatology/kel393. [DOI] [PubMed] [Google Scholar]
- 236.Jones RB, Ferraro AJ, Chaudhry AN, et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and Rheumatism. 2009;60(7):2156–2168. doi: 10.1002/art.24637. [DOI] [PubMed] [Google Scholar]
- 237.van Vollenhoven RF, Emery P, Bingham CO, et al. Longterm safety of patients receiving rituximab in rheumatoid arthritis clinical trials. Journal of Rheumatology. 2010;37(3):558–567. doi: 10.3899/jrheum.090856. [DOI] [PubMed] [Google Scholar]
- 238.de la Torre I, Leandro MJ, Edwards JCW, Cambridge G. Baseline serum immunoglobulin levels in patients with rheumatoid arthritis: relationships with clinical parameters and with B-cell dynamics following rituximab. Clinical and Experimental Rheumatology. 2012;30(4):554–560. [PubMed] [Google Scholar]
- 239.Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. Journal of Investigative Dermatology. 2008;128(12):2850–2858. doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- 240.Stasi R, Del Poeta G, Stipa E, et al. Response to B-cell-depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood. 2007;110(8):2924–2930. doi: 10.1182/blood-2007-02-068999. [DOI] [PubMed] [Google Scholar]
- 241.Hamaguchi Y, Uchida J, Cain DW, et al. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. Journal of Immunology. 2005;174(7):4389–4399. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 242.Müller C, Murawski N, Wiesen MHJ, et al. The role of sex and weight on rituximab clearance and serum elimination half-life in elderly patients with DLBCL. Blood. 2012;119(14):3276–3284. doi: 10.1182/blood-2011-09-380949. [DOI] [PubMed] [Google Scholar]
- 243.Hollander N. Immunotherapy for B-cell lymphoma: current status and prospective advances. doi: 10.3389/fimmu.2012.00003. Frontiers in Immunology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus A systematic review of off-label use in 188 cases. Lupus. 2009;18(9):767–776. doi: 10.1177/0961203309106174. [DOI] [PubMed] [Google Scholar]
- 245.Diaz-Lagares C, Perez-Alverez R, Garcia-Hernandez FJ, et al. Rates of and risk factors for severe infections in patients with systemic autoimmune diseases receiving biological agents off-label. Arthritis Research & Therapy. 2011;13, article R112 doi: 10.1186/ar3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zocher M, Baeuerle PA, Dreier T, Iglesias A. Specific depletion of autoreactive B lymphocytes by a recombinant fusion protein in vitro in vivo. International Immunology. 2003;15(7):789–796. doi: 10.1093/intimm/dxg076. [DOI] [PubMed] [Google Scholar]
- 247.Tchorbanov AI, Voynova EN, Mihaylova NM, et al. Selective silencing of DNA-specific B lymphocytes delays lupus activity in MRL/lpr mice. European Journal of Immunology. 2007;37(12):3587–3596. doi: 10.1002/eji.200737143. [DOI] [PubMed] [Google Scholar]
- 248.Mihaylova N, Voynova E, Tchorbanov A, et al. Selective silencing of disease-associated B-lymphocytes by chimeric molecules targeting their FcγIIb receptor. International Immunology. 2008;20(2):165–175. doi: 10.1093/intimm/dxm133. [DOI] [PubMed] [Google Scholar]
- 249.Dimitrova I, Gesheva V, Nikolova K, et al. Target silencing of disease-associated B-lymphocytes by chimeric molecules in SCID model of pristane-induced autoimmunity. Lupus. 2010;19(11):1261–1271. doi: 10.1177/0961203310371153. [DOI] [PubMed] [Google Scholar]
- 250.Józsi M, Prechl J, Bajtay Z, Erdei A. Complement receptor type 1 (CD35) mediates inhibitory signals in human B lymphocytes. Journal of Immunology. 2002;168(6):2782–2788. doi: 10.4049/jimmunol.168.6.2782. [DOI] [PubMed] [Google Scholar]
- 251.Iking-Konert C, Stocks S, Weinsberg F, et al. First clinical trials of a new heteropolymer technology agent in normal healthy volunteers and patients with systemic lupus erythematosus: safety and proof of principle of the antigen-heteropolymer ETI-104. Annals of the Rheumatic Diseases. 2004;63(9):1104–1112. doi: 10.1136/ard.2003.016691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Blank M, Manosroi J, Tomer Y, et al. Suppression of experimental systemic lupus erythematosus (SLE) with specific anti-idiotypic antibody-saporin conjugate. Clinical and Experimental Immunology. 1994;98(3):434–441. doi: 10.1111/j.1365-2249.1994.tb05509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang X, Zhang A, Liu Y, et al. Anti-idiotypic antibody specific to GAD65 autoantibody prevents type 1 diabetes in the NOD mouse. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0032515.32515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bollmann FM. Rheumatic autoimmune diseases: proposed elimination of autoreactive B-cells with magnetic nanoparticle-linked antigens. Medical Hypotheses. 2012;78(4):479–481. doi: 10.1016/j.mehy.2012.01.010. [DOI] [PubMed] [Google Scholar]