

Abstract
Site-specific glycosylation (SSG) of glycoproteins remains a considerable challenge and limits further progress in the areas of proteomics and glycomics. Effective methods require new approaches in sample preparation, detection, and data analysis. While the field has advanced in sample preparation and detection, automated data analysis remains an important goal. A new bioinformatics approach implemented in software called GP Finder automatically distinguishes correct assignments from random matches and compliments experimental techniques that are optimal for glycopeptides, including non-specific proteolysis and high mass resolution LC/MS/MS. SSG for multiple N- and O-glycosylation sites, including extensive glycan heterogeneity, was annotated for single proteins and protein mixtures with a 5% false-discovery rate, generating hundreds of non-random glycopeptide matches and demonstrating the proof-of-concept for a self-consistency scoring algorithm shown to be compliant with the target-decoy approach (TDA). The approach was further applied to a mixture of N-glycoproteins from unprocessed human milk and O-glycoproteins from very-low-density-lipoprotein (vLDL) particles.
Keywords: Glycoproteomics, site-specific glycosylation, tandem mass spectrometry, false-discovery rate, target-decoy approach
Introduction
Characterizing glycoproteins through the determination of site-specific glycosylation (SSG) is one of the key analyses required for understanding glycoprotein functions. SSG describes the glycan heterogeneity for each occupied glycosylation site of a glycoprotein, including N- and O-glycosylation.1 It has gained increased currency in the glycoproteomics2 community, albeit little consensus regarding implementation.3
Determining SSG is complicated by microheterogeneity, which is the large number of glycans occupying a single site. The analysis is also complicated by difficulties associated with the mass spectrometry characterization of glycopeptides. The glycan component is composed of relatively few unique monosaccharides, several with identical masses - a fact that increases the redundancy and similarity of theoretical glycans among competing matches. Because glycopeptides often contain glycans with similar monosaccharide compositions, mass spectrometry (MS) methods, even those capable of obtaining highly accurate masses, are insufficient. Tandem MS is often employed to obtain structural characterization; however, the presence of glycan isomers as well as the difficulty in obtaining comprehensive peptide and glycan fragmentation renders this method wholly insufficient.4 Even if comprehensive fragmentation were available, glycopeptide analysis is still complicated by non-linear glycan structures that often preclude monosaccharide sequencing with de novo5 methods commonly used for peptides. For this reason, chromatographic separation is performed to separate isomeric glycopeptide species. The combination of accurate mass, tandem MS and chromatographic separation provide comprehensive analysis if they can be performed routinely in a rapid, automated manner.
Despite these difficulties, the need for SSG is in high demand in fields as diverse as biomarker discovery and recombinant protein therapeutics (biologics).6 SSG and other post-translational modifications (PTMs) can vary with disease state in the former and in batch-to-batch manufacturing of the latter.7–8 A difference in glycosylation as simple as the presence of fucose can render a biologic ineffective or toxic, depending on the binding receptor of the target entity.9 As patents end for biologics, the so-called biosimilars will be required by regulatory agencies to demonstrate similar PTMs if additional clinical trials demonstrating bioequivalence are to be minimized. Furthermore, industry and academia alike require tools capable of analyzing SSG within complex protein mixtures to understand protein behavior and function.10–11 However, mixture analysis is problematic because the theoretical glycopeptide compositions from different glycoproteins are often quite similar. The recurring challenge in glycoproteomics is achieving distinction amidst similarity.
Common experimental techniques for the analysis of glycopeptides include hydrophilic interaction chromatography (HILIC)12 and generation of diagnostic glycan oxonium ions by tandem MS.13–15 Glycoproteomics has been further enhanced by techniques that address the unique characteristics of the glycoproteome16, such as on-line deglycosylation17 and non-specific proteolysis18. In addition to employing high mass accuracy and high mass resolution with such techniques as FT-ICR MS19, mass spectral techniques have been improved with glycopeptide-centric strategies, such as higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation (HCD-PD-ETD)20. Similar to structural elucidation with nuclear magnetic resonance (NMR), which often requires 1H NMR, 13C NMR, UV/Vis, IR, MS and other complementary techniques, MS-based glycoproteomics is gravitating toward multi faceted approaches using several parallel experiments. Such analyses combine reverse phase (RP) and HILIC chromatography for peptide- and glycan-centric separations as well CID, ECD/ETD, specific and non-specific proteolysis, glycan release (typically PNGase F), and duel polarity ionization.21–23 An example of such an approach is in-gel non-specific proteolysis for elucidating glycoproteins (INPEG)23. Glycoproteomics has also benefitted from novel data analysis approaches such as limiting theoretical possibilities to biologically relevant libraries24–25 and determining N-glycan topology from glycan family information26.
In addition to glycopeptide-tailored instrumentation and sample preparation, several valuable software tools are available to analyze SSG, although they have focused primarily on N-glycosylation.27 Some tools, such as GlycoExtractor28, combine proteomics and glycomics as parallel analyses.29 This strategy often reveals glycan heterogeneity without knowledge of site specificity.30 Other tools are partially or entirely de novo31, requiring only the raw data of detected glycopeptides with no prior knowledge of the protein ID's, such as GlyPID32, a tool that has been used with targeted multi-pass experiments to achieve SSG of complex mixtures. Several tools have taken advantage of proteomics while still retaining the analysis of intact glycopeptides, including GlycoMaster33, GlyDB34, GlycoMiner35, GlycoSpectrumScan36, GlycoPeptideSearch37, ByOnic38, SimGlycan39, Peptoonist40, and GlycoPep Grader41. Peptoonist allows the user to first identify the non-glycosylated peptides with the well-established X!Tandem42 proteomics algorithm and then determine which of the remaining isotopic envelopes, i.e. those not identified as peptides, are potential glycopeptides based on an averagine43 composition for glycopeptides. In addition to the isotopic distribution and accurate mass, tandem MS may be used to identify glycopeptides. Software called GlycoPep Grader performs glycopeptide analysis of tandem MS data starting at the point where the user has generated a list of potential glycopeptide compositions. The on-line software uses an algorithm based on the predictable fragmentation for different categories of glycosylation. Another useful software tool was developed by Joenväärä et al.44 that successfully identified 26 serum glycoproteins from 80 N-glycopeptides.
We have previously introduced a software tool named GlycoX45 that yielded comprehensive SSG of single proteins. In contrast to contemporaneous software, GlycoX was equipped to deal with non-specific protease digests, which contrasted to the widely used specific proteases such as trypsin. The use of tryptic digests and non-specific proteases are complementary, each with unique advantages and disadvantages. Tryptic digests yield distinct and well-characterized cleavages that readily yield protein identification. However, because few peptides are glycosylated in nearly all proteins, the vast majority of the peptide products are not glycosylated. Non-glycosylated peptides ionize more effectively than the glycosylated ones, so that one or more glycopeptide enrichment steps are necessary for analysis.46 Tryptic digestion may further produce missed cleavages particularly near the site of glycosylation yielding large glycopeptides that are not amenable using standard tandem MS methods.31,47–48 Conversely, non-specific proteases generate heterogeneous peptide products that split the detection of a particular glycosylation site among a family of related peptides. Identifying the glycopeptide is complicated by the lack of specificity. However, the method is effective at yielding enriched peptide products without extensive purification. In addition, it yields singly glycosylated peptides even when additional glycosylation sites are nearby.
A comprehensive solution for N- and O-glycopeptide analysis is provided to work with both specific and non-specific protease digests.18, 49–55 A new software called GlycoPeptide Finder (GP Finder), is presented, which works independent of the type of enzyme and mass spectrometry platform. Rather than using data-reduction according to a specific enzyme, all possibilities are calculated to accommodate non-specific proteases, such as pronase E, which generates smaller glycopeptides. Glycans are observed with a peptide tag that identifies the protein and the glycosylation site. While the use of non-specific proteases is more challenging for data interpretation because the sizes of the peptide tags are variable, sometimes too short for site determination, the variability can be controlled with the digestion time. The variable peptide lengths around the glycosylation sites correspond to multiple glycopeptides that can be used to provide self-consistency in the assignments.18, 50, 53–54
Methods
Materials and Reagents
Pronase E proteases, cyanogen bromide (CNBr) activated sepharose 4B (S4B) beads, bovine pancreatic ribonuclease, bovine lactoferrin, bovine kappa casein and bovine fetuin were obtained from Sigma Aldrich (St. Louis, MO). Graphitized carbon cartridges were purchased from Grace Davison Discovery Sciences (Deerfield, IL). All chemicals used were either of analytical grade or better.
Protease Digestion and Glycopeptide Cleanup
The site-specific glycosylation analysis workflow of the protein cocktail mixture has been previously described.52 After digestion with pronase, the samples were cleaned up with solid phase extraction (SPE) and collected as two fractions: 20 and 40% acetonitrile. The N-linked glycopeptides appeared primarily in the 20% fraction, whereas the O-linked were in the 40% fraction. For proof of concept we analyzed each fraction separately by data dependent LC/MS/MS; however, the combined data of the two LC/MS/MS runs were processed both separately and simultaneously as if obtained from a single chromatographic experiment. Therefore two fractions need not be collected and analyzed separately for the purposes of the software analysis.
Mass spectrometry analysis
Glycopeptide mixtures were analyzed using an Agilent 1200 series LC system coupled to an Agilent 6520 Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA). The HPLC-Chip/Q-TOF system was equipped with a micro well-plate auto sampler (maintained at 6 °C by a thermostat), a capillary loading pump for sample enrichment, a nano pump as the analytical pump for sample separation, HPLC-Chip Cube, and the Agilent 6520 Q-TOF MS detector. The tandem mass spectra of the glycopeptides were acquired in a data-dependent manner following LC separation on the microfluidic chip. The microfluidic chip consisted of a 9 × 0.075 mm i.d. enrichment column and a 150 × 0.075 mm i.d. analytical column, both packed with 5 μm porous graphitized carbon (PGC) as the stationary phase. Glycopeptides were subjected to collision-induced fragmentation with nitrogen as the collision gas using a series of collision energies that were dependent on the m/z values of the different glycopeptides and peptides. The collision energies correspond to voltages (Vcollision) that were based on the equation: Vcollision = m/z (1.8/100 Da) V − 2.4 V; where the slope and offset of the voltages were set at (1.8/100 Da) and (−2.4), respectively. The preferred charge states were set at 2, 3, >3, and unknown. Detailed experimental set-up and instrumental parameters are provided in the previous publications by Nwosu et al.23, 52
Data Processing
Prior to data analysis, each tandem spectrum was deconvoluted, de-isotoped and adjusted to neutral masses. The mass list of the glycopeptide precursor ions was analyzed with our in-house software, GP Finder, for rapid SSG analysis. The GP Finder is a considerable improvement on the previously developed GlycoX45, offering several new features including glycan libraries with biological filters to reduce false-positive hits, support for protein mixture analysis and tandem MS, faster run-time, and a significantly improved user interface. All glycopeptide assignments were made within a specified tolerance level (≤ 20ppm). Each glycopeptide identity was further verified by tandem MS for detailed structural information. The software and test data are available to download from http://chemgroups.ucdavis.edu/~lebrilla/GPF_docs.zip
Candidate glycopeptide compositions were filtered according the to the presence of diagnostic oxonium ions, accurate mass, and biological rules that are species-specific.25 The tandem MS fragments were filtered by mass and biological rules. The best match for each spectrum was determined by GP Finder according to tandem MS data and self-consistency among the results (Eq. 1 through 4). The algorithms and workflow for GP Finder are provided with further discussion in the Supplemental Materials.
The N- and O-glycosylation sites can be determined in two ways: automatically by the software or manually by the user. Potential N-linked sites are automatically determined by the software by either the consensus sequence NXT or NXS (X is not proline) or the annotated sites that are included in the reviewed sequence entries in the UniProt Knowledgebase. O-linked sites are determined by a number of methods, the most efficient of which is to use annotated UniProt entries. Unfortunately, Q-TOF CID cannot always localize O-glycosylation and the pronase approach may fail on mucin-type O-glycosylation in which there are multiple glycosylation sites separated by only a few residues.56 Although O-linked sites can be specified by the user, their determination is a confirmation rather than a characterization when based solely on UniProt entries.57
Two decoy generation strategies were investigated, both using the actual protein sequences as well as the actual experimental masses. The first strategy, a more traditional method, shuffled the amino acids of the known proteins with a standard (nearly random) Java algorithm, conserving the glycosylation sites and consensus sequences, yet resulting in poor modeling. The second strategy did not shuffle the sequences; instead an 11-Da residue was added as part of each theoretical glycan composition, preserving the true peptide sequences around each site and providing a better model for the random matches to the highly weighted peptide (P) and peptide + HexNAc (PN) peaks. The latter was employed with GP Finder. The decoy data sets were created with 11-Da artificial components to prevent the randomly generated glycan compositions from matching glycans that could actually be present in the sample. The 11-Da residue is small enough that the remaining portion of the glycan composition is comparable in size to the glycan compositions in the target dataset and unique enough that we do not expect it to appear in any compounds of interest. The use of 11 Da also avoids the pitfall of numbers such as 1, 12, 14, 15, 16, and 18 Da, that could be confused with common organic components.
For the dataset-bias test, the target and decoy libraries compete for top scoring matches to fabricated, incorrect tandem data that have been generated from the actual tandem spectra by adding 11-Da to all peaks in the tandem spectra.58 The adjusted tandem spectra are used for determining whether a set of false tandem spectra will match equivalently to both decoy and target glycopeptide compositions. While it is true that both the false tandem spectra and the decoy glycopeptide compositions use 11-Da, they do not generate matches that are systematically related to 11-Da because the 11-Da component is not included in the composition of the decoy glycopeptides when comparing their in silico fragments to tandem data. Furthermore, the 11-Da component is subtracted once from each experimental precursor mass; whereas, it is added to each of the tandem fragment masses, thus preventing the possibility that a precursor mass and a fragment mass are both adjusted by 11-Da in the same way.
Scores are generated in three distinct phases. First, a Base Score is generated (Eq. 1), followed by a boost in score from self-consistency in the data (Eq. 2) and then subsequent compensation for target-decoy Bias (Eq. 3 and 4). The Base Score is calculated according to the number of fragments observed for each fragment type for a particular theoretical glycopeptide and according to the user-defined weight given to each type of fragment (Eq. 1). The weights applied here were based on the relative importance that we predicted for each fragment type. The Boosted Score for each glycopeptide is calculated in two steps (Eq. 2). The number of unique glycopeptide masses in each peptide family is multiplied by a user-defined weight (we used 1) and then added to the associated Base Score as a temporary adjustment. The average adjusted score for each peptide family from the set of adjusted Base Score values (referred to as the Family Average) is then calculated and added to each associated Base Score (original, non-adjusted value).
A Bias in the target dataset relative to the decoy dataset results from applying self-consistency scoring. Prior to estimating the Bias, GP Finder first calculates the average size of all the decoy peptide families and the average Boosted Score for the entire decoy dataset (referred to as the Average Boosted Decoy Score). Second, a representative score of random self-consistency in the target dataset is obtained by determining the average Boosted Score from all target matches that have a peptide family size equal to the average decoy peptide family size (referred to as the Average Subset Boosted Target Score). The most conservative estimation of the Bias is to subtract the Average Boosted Decoy Score from the Average Subset Boosted Target Score (Eq. 3). The Final Score is calculated by subtracting the Bias from each Boosted Score (Eq. 4).
| Eq. 1 |
where the counts for each fragment type are represented by the following: P=intact peptide, PN=intact peptide + HexNAc, b/y=b and y peptide only fragments, B/Y= b and y glycopeptide fragments with intact peptide, gp=glycopeptides fragmented on glycan and peptide, glycan=glycan-only fragments
| Eq. 2 |
| Eq. 3 |
| Eq. 4 |
The approach performs similarly to specific digestion with QTOF MS/MS, although below what can be done with HCD/ETD combined with high mass accuracy. For this reason, we previously developed a sample preparation method called INPEG23 that enriches for both glycoproteins and glycopeptides without clean-up steps between handling the unprocessed sample and analyzing with LC/MS/MS. INPEG also separates mixtures into less complex subsets that are appropriate for this bioinformatics approach. Furthermore, employing INPEG or some other protein identification strategy is required for GP Finder.23
Although not yet tested, the scoring scheme as implemented here is not likely to work for ETD because it relies heavily on P and PN; however, these parameters can be changed by the user to emphasize peptide backbone fragmentation. To some extent the self-consistency algorithm regains the sensitivity and specificity lost from the nonspecific digest.
Results and Discussion
Experimental and Data Analysis Workflow
An outline of the method is shown in Figure S-1. Digestion of the glycoprotein mixture was followed by solid phase enrichment to yield primarily glycopeptides that were analyzed by nanoflow LC/MS/MS. A large number of spectra were obtained with the vast majority corresponding to glycopeptides (for example, 60% of the nearly 1700 tandem spectra collected for human milk whey and vLDL contained one or more of the diagnostic oxonium ions within a tolerance of m/z ± 0.05). To separate glycopeptide spectra from random, uninformative ones, a number of diagnostic peaks from tandem MS were used, such as the fragments corresponding to the peptide (P) and peptide+HexNAc (PN, or more commonly Y1).
For each sample reported here, a histogram demonstrated the bimodal distribution of scores, which was shown to distinguish the correct matches for previously annotated control samples. The histograms in Figure 1 were populated by the top scores from all tandem spectra of a control mixture. The overlap between the decoy and target score distributions allowed estimation of the false discovery rate (FDR).59
Figure 1.
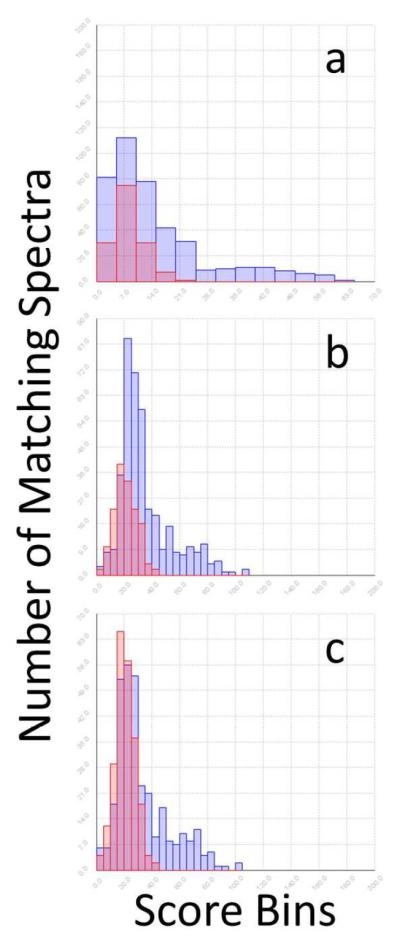
TDA Compliance. Score distributions of the target and decoy N-glycopeptide matches from the mixture of bovine fetuin, lactoferrin, and kappa casein. Data includes both 20 and 40% ACN SPE fractions. a. Without self-consistency. b. With self-consistency. c. With self-consistency and TDA-compliance.
Scoring
An abundance of mass peaks and theoretical matches to all types of fragments requires us to balance the influence of the unique versus common pieces of evidence. Unbalanced weighting of scores leads to either artificially separated target-decoy populations or entirely overlapping ones (Figure S-2). The algorithm effectively uses both common and unique fragments to identify the best matches in a systematic way that often cannot be duplicated by evaluating each spectrum manually.
Prior to examining the test data, we predicted the relative importance of each type of fragment that could be generated from an intact glycopeptide precursor ion.60 Predictions were based on widely observed diagnostic fragments that are characteristic within a broad range of collision energies.38, 44, 61–63
The P and PN fragments are frequently observed for glycopeptides and are particularly rare for incorrect matches because of the unique amino acid composition required for the intact peptide. The b and y peptide fragments are somewhat unique and therefore also rare for incorrect matches with exception of some overlap between NXS and NXT consensus sequences, common among both target and decoy matches. Unfortunately, the extent of peptide fragmentation with collision induced dissociation (CID) is attenuated for glycopeptides because the glycosidic bonds are labile and the fragmentation process is stochastic. Another important set of diagnostic peaks are the glycopeptide fragments containing a fragmented glycan and an intact peptide (B and Y ions); however, a few false matches to these peaks are somewhat common. While testing this scoring scheme, we monitored data that had been previously annotated. We found that even in the rare cases when a random false match had both P and PN, if the assigned fragments did not distinguish them (Figure S-3), the self-consistency did.
Unfortunately, the parameters of mass deconvolution can favor some of these fragment types over others. For this reason, we employed commercial mass deconvolution software prior to input into GP Finder that assumes peptide molecular composition and identifies all the peptide fragments at the expense of a few glycopeptide fragments (Figure S-4 and S-5). The glycan-only fragments were readily identified and useful for filtering out poor matches; however, similar glycosylation among theoretical matches is common, making glycan-only fragments poor differentiators between two or more seemingly good matches (except when a pair of peaks is detected for NeuAc and NeuAc-H2O, eliminating all non-sialylated possibilities). Some fragment types are ignored to reduce random matches, including internal peptides and certain rearrangements (Figure S-6 and S-7), leaving some peaks unassigned.
The extent of random matches is modeled by a decoy dataset that competes with target data for the highest scoring match to each tandem spectrum. If the scoring method truly favors correct assignments and if sufficient data points are collected, the histogram of all the top scores for the target data should include two distributions - matches with considerable evidence (presumably correct) and random matches (presumably incorrect).59 The decoy distribution should model the random matches and overlap with the lower-scoring population of target matches. By calculating the percent overlap between the decoy and the high-scoring target population, the false discovery rate (FDR) is estimated.
Target-Decoy Approach and Self-Consistency
A dataset-bias test provides evidence that the decoy dataset correctly models the target data.58 As shown in Figure S-8, the decoy and target datasets generated a similar shape and frequency of matches to incorrect tandem spectra. While a successful test is not proof of correct modeling, a failed test is proof of incorrect modeling.
The decoy dataset was included with the target data during the analysis of real tandem data and competed with the target data (Figure 1a). The decoy distribution modeled the shape of the low scoring target matches well; however, the heights were not the same. Based on the dataset-bias test, we expected the intensities to be similar. The inconsistency indicated either that the decoy data did not correctly model the incorrect matches in the target data or that the low scoring target data included some correct assignments.
We extracted potentially correct target matches out of the low scoring distribution by applying a self-consistency scoring algorithm (Figure 1b). The self-consistency algorithm identified families50, 52 of peptide heterogeneity. Unlike tryptic digestion that provides only glycan-based self-consistency, as described by Goldberg et al.38, pronase digestion generates peptide-based self-consistency. Each glycan is expected to be detected on multiple glycopeptides with different peptide tag lengths around the same glycosylation site. The self-consistency described here provides more than an occasional opportunistic boost in score, as described by Gupta et al.64 for the common double-pass filters that are used for boosting scores of neighboring peptides detected from a given protein; in contrast, the self-consistency in pronase digestion is consistently observed50, 52 and is empirically indicative of correct assignments (Table S-1). We tested several variations of these steps, as shown in Figure S-2.
Recent candid discussion in the field64–65 benefited our analysis by drawing our attention to the requirements for rigorous TDA-compliant analysis. The generally accepted premise of TDA is that the “matches to decoy peptide sequences and false matches to sequences from the original database follow the same distribution.”66 The timely discussion showed us that the random self-consistency in the target data will necessarily gain an advantage over the random self-consistency in the decoy data. Assuming the target data contains at least some correct matches to the tandem spectra, the target data will naturally have more high-scoring correct matches than the decoy. The boost in score for the incorrect (random) target matches as a result of self-consistency with correct target matches is therefore not accounted for in the decoy data. While some random incorrect target matches gain a boost in score and are included in the results, an even more abundant population of correct matches is boosted under these conditions. Furthermore, the average boost in score that is caused by random matches in the target data can be determined and removed as shown in Figure 1c where the decoy and low-scoring target distributions are similar in shape and height after employing the TDA-compliant self-consistency algorithm.
The self-consistency scoring presents an unknown factor: the ratio of correct to incorrect matches that have been boosted within the target data. We have accounted for this bias by calculating the difference between the random score boost for the target and decoy data. The algorithm is discussed in detail in the Methods section and is provided in the Supplementary Materials.
Figure 2 shows the self-consistency among three spectra for N-glycopeptides with the same glycosylation site. The top two fragmentation spectra are related by having the same glycan composition but different peptide, for which both spectra show the presence of P, Hex:HexNAc, and several related Y glycopeptide fragments. The bottom two fragmentation spectra have a common peptide but different yet related glycans. The resultant fragmentation data both exhibit the presence of the PN+HexNAc and b3 peptide ions. All three spectra contain fragments of PN and 2HexNAc. The components of the scores are provided in Table 1. Additional spectra showing SSG, including O-linked examples, are provided in Figure S-5 and S-9.
Figure 2.

Self-consistent Data. Tandem mass spectra of site-specific N-glycopeptide analysis at site 495 of bovine lactoferrin showing putative glycan structures. Glycan self-consistency is shown in a (m/z 1068.9363) and b (m/z 1089.9696). Peptide self-consistency is shown in b and c (m/z 988.4189). Blue squares: HexNAc, Gray circles: Hex, Green circles: Man. * Denotes precursor.
Table 1.
| Scores |
Fragment Counts |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glycopeptide | Protein | Final | Base | Fam. Aver. | Bias | Fam. Size | P | PN | b/y p | B/Y | gp | glycan |
| IV495NQ 4 Hexose 5 HexNAc | TRFL | 67 | 43 | 29 | 5 | 12 | 1 | 1 | 0 | 6 | 0 | 5 |
| GLIV495N 4 Hexose 5 HexNAc | TRFL | 74 | 50 | 29 | 5 | 12 | 1 | 1 | 1 | 6 | 0 | 8 |
| GLIV495N 4 Hexose 4 HexNAc | TRFL | 60 | 35 | 30 | 5 | 14 | 0 | 1 | 1 | 7 | 0 | 5 |
| SGEP152TS154TPT Hex HexNAc 2 NeuAc | CASK | 86 | 49 | 40 | 3 | 22 | 1 | 1 | 3 | 3 | 0 | 8 |
| GEP152TS154TPT Hex HexNAc 2 NeuAc | CASK | 95 | 58 | 40 | 3 | 22 | 1 | 1 | 4 | 4 | 0 | 10 |
| GEP152TS154TPT Hex HexNAc NeuAc | CASK | 69 | 43 | 29 | 3 | 10 | 1 | 1 | 3 | 2 | 0 | 5 |
O-glycosylation can be challenging to match confidently, even though it consistently generates more extensive peptide fragmentation than N-glycosylation. The challenge is due to the lack of a consensus sequence coupled with the high occurrence of serine and threonine residues around O-glyosylation sites. For instance, the candidate glycopeptides for the spectra shown in Figure S-10 could be glycosylated on either of the two glycosylation sites on the same peptide, making these results site-directed as opposed to site-specific. Site-directed results for these spectra distinguish glycan heterogeneity at sites 152 and 154 from the surrounding sites: 142, 157, 163, and 186, as shown in Table 1. Without self-consistency the scores for sites 152 and 154 are equal, even though there is some ambiguity with site 152 because the data lacks the y3 and y4 peptide ions that may be more likely to be observed with glycosylation on site 152 (coincidentally, the y4 peptide ion is observed in a potentially isomeric compound that eluted seven minutes earlier than the data shown in Figure S-10). Furthermore, the peptide heterogeneity shows proteolytic cleavage at several sites C-terminal to 154 yet does not show cleavage of residues 153 or 154 C-terminal to 152. However, we cannot yet make conclusions from the lack of data in a particular spectrum or peptide family. Nonetheless the two possibilities are slightly different using self-consistency scoring. It is also unclear at this point how much bias is imposed on site 154 from its simultaneous proximity to both sites 152 and 157. For this reason, as well as the fact that few false matches are available for plotting populations of random false matches per spectrum, the results are not claimed to be inherently correct, rather they are a pragmatic way of comparing and presenting the evidence.
Some degree of qualitative legitimacy is demonstrated by analyzing tandem MS data with an intentionally low or high mass tolerance for the method, such as 5 ppm and 1 Da. The result is that the decoy and target distributions are not separated; however, the distributions are in fact separated with an appropriate mass tolerance for the method, such as 80 ppm. The correct mass tolerance window is an effective sweet-spot that distinguishes correct self-consistency scoring from random self-consistency.
Analysis of single protein with one site of glycosylation
We analyzed a glycoprotein with well-characterized glycosylation: bovine pancreatic ribonuclease (RNaseB). As part of the in silico digest for the analysis of RNaseB, we also included the sequences from a three-protein mixture to probe the actual FDR. That the method is superior to manual analysis is illustrated in this application. Manual analysis, previously performed on this data by an experienced analyst, yielded 26 glycopeptides, all of which were also selected by GP Finder as the top possibilities for their respective spectra (Table S-2). GP Finder identified 18 additional RNaseB glycopeptides that scored within the 5% FDR. Although the number of data points precludes a solid statistical analysis, the separation of the decoy and target matches was characteristic of data that we consider to be high quality (Figure S-11). Manual analysis revealed that the true FDR at the threshold indicated by the 5% target-decoy overlap was 17%. The true 5% FDR threshold was a few points higher than the value calculated with TDA, encompassing 46 rather than 58 matches. The discrepancy was not surprising because the dataset was small, with less certainty regarding the true shape of the decoy distribution in the decaying region of the histogram. The discrepancy may also be caused by our pragmatic method for calculating the target-decoy bias, for which the average random match will be corrected, while some subset of matches will either be over- or under-corrected.
Analysis of three-protein mixture each with multiple sites of glycosylation
A protein mixture composed of bovine fetuin, lactoferrin and kappa casein was created to serve as a substantially more difficult problem with 18 sites of N- and O-glycosylation. The height of the decoy distribution modeled the low-scoring target distribution considerably better after employing the TDA-compliant self-consistency algorithm, providing greater confidence in the assignments (Figure 1c). The total number of target N-glycopeptide matches above the 5% FDR was 106 without self-consistency, 133 with self-consistency and 78 with the TDA-compliant self-consistency analysis (Table S-3 and S-4). Some matches were ambiguous because they included multiple top score possibilities for a single tandem mass spectrum and generally lacked sufficient peptide fragmentation to differentiate the possibilities. The inclusion of the RNaseB sequence (not actually present in the sample) generated one false match that scored just over the actual 5% FDR threshold. Although only peptide self-consistency was used to determine each glycoform, glycan self-consistency emerged from the results, as did heterogeneity of glycan types, such as the complex/hybrid and high mannose glycoforms observed on bovine lactoferrin site 495 (Figure 2 and S-5).
The O-linked matches behaved similarly to the N-linked ones, with increased separation between the correct and incorrect assignments after applying the self-consistency algorithm. The total number of target O-glycopeptides above the 5% FDR was 92 without self-consistency, 115 with self-consistency and 61 with the TDA-compliant self-consistency analysis (Table S-3 and S-4). Additional validity is shown by comparing the results with an analysis considering only one of the O-linked glycoproteins present in the sample. While the empirically determined 5% FDR did not change between the two analyses, the size of the high-scoring population did change from 36 to 61 when both O-glycoproteins were included in the analysis (Figure S-12).
Another source of ambiguity was the overlap among spectra assigned to both N- and O-linked glycopeptides. After comparing the separately analyzed N and O matches (generated from the same tandem MS data), 5% of the TDA-compliant results were false as a result of the overlap and required manual interpretation (Figure S-3).
A second technique for calculating FDR corroborates the results obtained with the target-decoy approach and provides additional discovery rate statistics. By entering the heights of the histogram bars for the target dataset from Figure 1C and S-12 B into a commercial peak deconvolution software (PeakFit67), the underlying distributions of the target dataset were calculated (Figure S-13) and used to estimate the FDR, false-negative rate (FNR), and accuracy.59, 68 The respective values for the N-linked analysis are 8.7 %, <0.0 %, and 98.4 %. The values for the O-linked analysis are 10.4 %, 14.9 %, and 97.0 %.
Analysis of unknown protein mixtures
We applied the method to a mixture of glycoproteins from human very-low-density-lipoprotein (vLDL) nanoparticles. Tryptic analysis identified two O-glycoproteins, apolipoprotein C-III (APOC3, P02656) and apolipoprotein E (APOE, P02649), as well as apolipoprotein B (APOB, P04114), an N-glycoprotein. GP Finder identified 11 glycopeptides with high confidence (completely outside the decoy distribution) for APOC3 and APOE (SSG maps provided in Figure 3 and score histograms in Figure S-14), for which the TDA-compliant self-consistency scoring draws a clearer distinction between the 11 high-scoring matches and the low scoring matches. Both O-glycoproteins were glycosylated on the sites that were annotated in the UniProt flat file. The confident assignments were associated with excellent tandem spectra that were also verified manually (Figure S-15).
Figure 3.
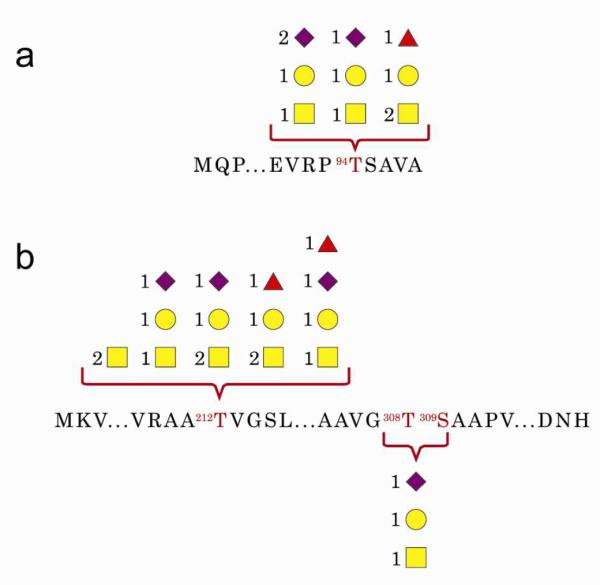
SSG Map of O-Glycoproteins. SSG of a mixture of proteins extracted from vLDL nanoparticles for two O-glycoproteins. a. APOC3 b. APOE. Yellow squares: HexNAc, Yellow circles: Hex, Purple diamonds: NeuAc. Red triangles: dHex.
SSG was also analyzed for a refined pool of electrophoretically separated glycoproteins from raw human milk using the recently published INPEG method (Figure S-16). The analysis included concurrent digests from parallel gel bands with non-specific proteases and trypsin, providing separate glycopeptide-rich and tryptic peptide-rich samples for each band of refined human milk glycoproteins. Following digestion of the refined milk pool, the sample was separated and detected on-line with LC/MS/MS (Figure 4), reducing ion-suppression and revealing site-specific heterogeneity (SSG maps provided in Figure S-17 and S-18). Two glycoproteins were identified from one band, lactoferrin (TRFL_HUMAN, P02788) and polymeric immunoglobulin receptor (PIGR_HUMAN, P01833), resulting in 22 and 58 glycopeptide assignments at a 5% FDR (histogram provided in Figure S-19) covering 3 and 6 glycosylation sites, respectively.
Figure 4.

Separation of Glycopeptides. On-line separation and detection of glycopeptides from human lactoferrin and polymeric immunoglobulin receptor, two glycoproteins isolated in a gel band after SDS-PAGE of raw human milk.
Conclusion
An ideal scoring scheme generates separate populations of correct and incorrect matches after plotting all top scores in a histogram according to the frequency of scores within certain score bins. Diagnostic fragment types are awarded more significance than others. Two control samples demonstrate that the weighting factors applied here favor correct matches over incorrect ones for the described experimental conditions.
The method relies on the assumption that the decoy dataset correctly models the random false matches in the target data.69 Although we probe the validity of this assumption by analyzing previously annotated datasets, performing dataset-bias tests and plotting the score distributions, we have relied on the FDR more as an effective means to filter out poor target matches than as a precise calculator for FDR. The validity of the FDR approximation is strengthened by our observation that poor tandem MS data does not produce target scores that are distinguishable from random decoy scores.
The final result is an automated and confident annotation of the site-specific or site-directed heterogeneity of every glycoprotein in the control mixture (Table S-4). While the self-consistency of peptides around each site is both expected and useful, the algorithm does not require a non-specific protease and could potentially benefit from data reduction with specific proteases. This analysis is a step towards automated glycoproteomics, taking advantage of the power behind protein identification with shot-gun proteomics66, 70 and the specificity of a glycopeptide-optimized mixture analysis method. The method has been demonstrated on unknown glycoprotein mixtures from vLDL nanoparticles and human milk and has identified glycopeptides with score distributions that distinguish confident from random assignments.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Agilent Technologies for their contributions. Financial support was provided by the National Institutes of Health (RO1GM049077 for C. B. Lebrilla) and the Korean Converging Research Center Program through the Ministry of Education, Science and Technology (2011K000968 for H. J. An).
References
- 1.Kolarich D, Jensen PH, Altmann F, Packer NH. Determination of site-specific glycan heterogeneity on glycoproteins. Nat. Protoc. 2012;7:1285–98. doi: 10.1038/nprot.2012.062. [DOI] [PubMed] [Google Scholar]
- 2.Pan S, Chen R, Aebersold R, Brentnall TA. Mass spectrometry based glycoproteomics--from a proteomics perspective. Mol. Cell. Proteomics. 2011;10:R110, 003251. doi: 10.1074/mcp.R110.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doerr A. Glycoproteomics. Nat. Methods. 2012;9:36–36. [Google Scholar]
- 4.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007;849:115–28. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 5.Hughes C, Ma B, Lajoie GA. De novo sequencing methods in proteomics. Methods Mol. Biol. 2010;604:105–21. doi: 10.1007/978-1-60761-444-9_8. [DOI] [PubMed] [Google Scholar]
- 6.Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006;24:1241–52. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 7.Butler M. Optimisation of the cellular metabolism of glycosylation for recombinant proteins produced by Mammalian cell systems. Cytotechnology. 2006;50:57–76. doi: 10.1007/s10616-005-4537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, et al. Simultaneous analysis of glycosylated and sialylated prostate-specific antigen revealing differential distribution of glycosylated prostate-specific antigen isoforms in prostate cancer tissues. Anal. Chem. 2011;83:240–5. doi: 10.1021/ac102319g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peipp M, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112:2390–9. doi: 10.1182/blood-2008-03-144600. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins N, Parekh RB, James DC. Getting the glycosylation right: implications for the biotechnology industry. Nat. Biotechnol. 1996;14:975–81. doi: 10.1038/nbt0896-975. [DOI] [PubMed] [Google Scholar]
- 11.Dove A. The bittersweet promise of glycobiology. Nat. Biotechnol. 2001;19:913–7. doi: 10.1038/nbt1001-913. [DOI] [PubMed] [Google Scholar]
- 12.Alpert AJ. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. 1990;499:177–96. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan B, Addona TA, Carr SA. Selective detection of glycopeptides on ion trap mass spectrometers. Anal. Chem. 2004;76:3112–8. doi: 10.1021/ac035427d. [DOI] [PubMed] [Google Scholar]
- 14.Demelbauer UM, Zehl M, Plematl A, Allmaier G, Rizzi A. Determination of glycopeptide structures by multistage mass spectrometry with low-energy collision-induced dissociation: comparison of electrospray ionization quadrupole ion trap and matrix-assisted laser desorption/ionization quadrupole ion trap reflectron time-of-flight approaches. Rapid Commun. Mass Spectrom. 2004;18:1575–82. doi: 10.1002/rcm.1521. [DOI] [PubMed] [Google Scholar]
- 15.Dalpathado DS, Desaire H. Glycopeptide analysis by mass spectrometry. Analyst. 2008;133:731–8. doi: 10.1039/b713816d. [DOI] [PubMed] [Google Scholar]
- 16.Sun B, et al. Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Mol Cell Proteomics. 2007;6:141–9. doi: 10.1074/mcp.T600046-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Qu Y, et al. Integrated sample pretreatment system for N-linked glycosylation site profiling with combination of hydrophilic interaction chromatography and PNGase F immobilized enzymatic reactor via a strong cation exchange precolumn. Anal. Chem. 2011;83:7457–63. doi: 10.1021/ac201665e. [DOI] [PubMed] [Google Scholar]
- 18.Dodds ED, Seipert RR, Clowers BH, German JB, Lebrilla CB. Analytical performance of immobilized pronase for glycopeptide footprinting and implications for surpassing reductionist glycoproteomics. J. Proteome Res. 2009;8:502–12. doi: 10.1021/pr800708h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Emmett MR, Marshall AG. Liquid chromatography electrospray ionization Fourier transform ion cyclotron resonance mass spectrometric characterization of N-linked glycans and glycopeptides. Anal. Chem. 2010;82:6542–8. doi: 10.1021/ac1008833. [DOI] [PubMed] [Google Scholar]
- 20.Saba J, Dutta S, Hemenway E, Viner R. Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int J Proteomics. 2012;2012:560391. doi: 10.1155/2012/560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leymarie N, Zaia J. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal. Chem. 2012;84:3040–8. doi: 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deguchi K, et al. Complementary structural information of positive- and negative-ion MSn spectra of glycopeptides with neutral and sialylated N-glycans. Rapid Commun. Mass Spectrom. 2006;20:741–6. doi: 10.1002/rcm.2368. [DOI] [PubMed] [Google Scholar]
- 23.Nwosu CC, et al. In-gel nonspecific proteolysis for elucidating glycoproteins: a method for targeted protein-specific glycosylation analysis in complex protein mixtures. Anal. Chem. 2013;85:956–63. doi: 10.1021/ac302574f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Go EP, et al. GlycoPep DB: a tool for glycopeptide analysis using a “Smart Search”. Anal. Chem. 2007;79:1708–13. doi: 10.1021/ac061548c. [DOI] [PubMed] [Google Scholar]
- 25.Kronewitter SR, et al. The development of retrosynthetic glycan libraries to profile and classify the human serum N-linked glycome. Proteomics. 2009;9:2986–94. doi: 10.1002/pmic.200800760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldberg D, Bern M, North SJ, Haslam SM, Dell A. Glycan family analysis for deducing N-glycan topology from single MS. Bioinformatics. 2009;25:365–71. doi: 10.1093/bioinformatics/btn636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dallas DC, Martin WF, Hua S, German JB. Automated glycopeptide analysis--review of current state and future directions. Brief Bioinform. 2012 doi: 10.1093/bib/bbs045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Artemenko NV, Campbell MP, Rudd PM. GlycoExtractor: a web-based interface for high throughput processing of HPLC-glycan data. J. Proteome Res. 2010;9:2037–41. doi: 10.1021/pr901213u. [DOI] [PubMed] [Google Scholar]
- 29.Tang H, Mechref Y, Novotny MV. Automated interpretation of MS/MS spectra of oligosaccharides. Bioinformatics. 2005;21(Suppl 1):i431–9. doi: 10.1093/bioinformatics/bti1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geyer H, Geyer R. Strategies for analysis of glycoprotein glycosylation. Biochim. Biophys. Acta. 2006;1764:1853–69. doi: 10.1016/j.bbapap.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Peltoniemi H, Joenvaara S, Renkonen R. De novo glycan structure search with the CID MS/MS spectra of native N-glycopeptides. Glycobiology. 2009;19:707–14. doi: 10.1093/glycob/cwp034. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, et al. Mapping site-specific protein N-glycosylations through liquid chromatography/mass spectrometry and targeted tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2010;24:965–72. doi: 10.1002/rcm.4474. [DOI] [PubMed] [Google Scholar]
- 33.Xin L, Shan P. GlycoMaster — Software for Glycopeptide Identification with Combined ETD and CID/HCD Spectra. J. Biomol. Tech. 2011;22:S51–S52. [Google Scholar]
- 34.Ren JM, Rejtar T, Li L, Karger BL. N-Glycan structure annotation of glycopeptides using a linearized glycan structure database (GlyDB) J. Proteome Res. 2007;6:3162–73. doi: 10.1021/pr070111y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozohanics O, et al. GlycoMiner: a new software tool to elucidate glycopeptide composition. Rapid Commun. Mass Spectrom. 2008;22:3245–54. doi: 10.1002/rcm.3731. [DOI] [PubMed] [Google Scholar]
- 36.Deshpande N, Jensen PH, Packer NH, Kolarich D. GlycoSpectrumScan: fishing glycopeptides from MS spectra of protease digests of human colostrum sIgA. J. Proteome Res. 2010;9:1063–75. doi: 10.1021/pr900956x. [DOI] [PubMed] [Google Scholar]
- 37.Pompach P, Chandler KB, Lan R, Edwards N, Goldman R. Semi-Automated Identification of N-Glycopeptides by Hydrophilic Interaction Chromatography, nano-Reverse-Phase LC-MS/MS, and Glycan Database Search. J. Proteome Res. 2012;11:1728–40. doi: 10.1021/pr201183w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bern M, Cai Y, Goldberg D. Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 2007;79:1393–400. doi: 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- 39.Apte A, Meitei NS. Bioinformatics in glycomics: glycan characterization with mass spectrometric data using SimGlycan. Methods in Molecular Biology. 2010;600:269–81. doi: 10.1007/978-1-60761-454-8_19. [DOI] [PubMed] [Google Scholar]
- 40.Goldberg D, et al. Automated N-glycopeptide identification using a combination of single- and tandem-MS. J. Proteome Res. 2007;6:3995–4005. doi: 10.1021/pr070239f. [DOI] [PubMed] [Google Scholar]
- 41.Woodin CL, et al. GlycoPep grader: a web-based utility for assigning the composition of N-linked glycopeptides. Anal. Chem. 2012;84:4821–9. doi: 10.1021/ac300393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craig R, Cortens JP, Beavis RC. The use of proteotypic peptide libraries for protein identification. Rapid Commun. Mass Spectrom. 2005;19:1844–50. doi: 10.1002/rcm.1992. [DOI] [PubMed] [Google Scholar]
- 43.Senko MW, Beu SC, McLafferty FW. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 44.Joenvaara S, Ritamo I, Peltoniemi H, Renkonen R. N-glycoproteomics - an automated workflow approach. Glycobiology. 2008;18:339–49. doi: 10.1093/glycob/cwn013. [DOI] [PubMed] [Google Scholar]
- 45.An HJ, Tillinghast JS, Woodruff DL, Rocke DM, Lebrilla CB. A new computer program (GlycoX) to determine simultaneously the glycosylation sites and oligosaccharide heterogeneity of glycoproteins. J. Proteome Res. 2006;5:2800–8. doi: 10.1021/pr0602949. [DOI] [PubMed] [Google Scholar]
- 46.Kaji H, Yamauchi Y, Takahashi N, Isobe T. Mass spectrometric identification of N-linked glycopeptides using lectin-mediated affinity capture and glycosylation site-specific stable isotope tagging. Nat. Protoc. 2006;1:3019–27. doi: 10.1038/nprot.2006.444. [DOI] [PubMed] [Google Scholar]
- 47.Grass J, Pabst M, Chang M, Wozny M, Altmann F. Analysis of recombinant human follicle-stimulating hormone (FSH) by mass spectrometric approaches. Anal. Bioanal. Chem. 2011;400:2427–38. doi: 10.1007/s00216-011-4923-5. [DOI] [PubMed] [Google Scholar]
- 48.Chen R, et al. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res. 2009;8:651–61. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- 49.Hua S, An HJ. Glycoscience aids in biomarker discovery. BMB Rep. 2012;45:323–30. doi: 10.5483/bmbrep.2012.45.6.132. [DOI] [PubMed] [Google Scholar]
- 50.Hua S, et al. Site-specific protein glycosylation analysis with glycan isomer differentiation. Anal. Bioanal. Chem. 2012;403:1291–1302. doi: 10.1007/s00216-011-5109-x. [DOI] [PubMed] [Google Scholar]
- 51.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-Glycosylation Sites and Site Heterogeneity in Glycoproteins. Anal. Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 52.Nwosu CC, et al. Simultaneous and extensive site-specific N- and O-glycosylation analysis in protein mixtures. J. Proteome Res. 2011;10:2612–24. doi: 10.1021/pr2001429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seipert RR, Dodds ED, Lebrilla CB. Exploiting Differential Dissociation Chemistries of O-Linked Glycopeptide Ions for the Localization of Mucin-Type Protein Glycosylation. J. Proteome Res. 2008;8:493–501. doi: 10.1021/pr8007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clowers BH, Dodds ED, Seipert RR, Lebrilla CB. Site determination of protein glycosylation based on digestion with immobilized nonspecific proteases and Fourier transform ion cyclotron resonance mass spectrometry. J. Proteome Res. 2007;6:4032–40. doi: 10.1021/pr070317z. [DOI] [PubMed] [Google Scholar]
- 55.Froehlich JW, et al. Nano-LC–MS/MS of Glycopeptides Produced by Nonspecific Proteolysis Enables Rapid and Extensive Site-Specific Glycosylation Determination. Anal. Chem. 2011;83:5541–5547. doi: 10.1021/ac2003888. [DOI] [PubMed] [Google Scholar]
- 56.Christiansen MN, Kolarich D, Nevalainen H, Packer NH, Jensen PH. Challenges of determining O-glycopeptide heterogeneity: a fungal glucanase model system. Anal. Chem. 2010;82:3500–9. doi: 10.1021/ac901717n. [DOI] [PubMed] [Google Scholar]
- 57.Williams TI, et al. Investigations with O-linked protein glycosylations by matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Mass Spectrom. 2008;43:1215–23. doi: 10.1002/jms.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 60.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007;6:654–61. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritchie MA, Gill AC, Deery MJ, Lilley K. Precursor ion scanning for detection and structural characterization of heterogeneous glycopeptide mixtures. J. Am. Soc. Mass Spectrom. 2002;13:1065–77. doi: 10.1016/S1044-0305(02)00421-X. [DOI] [PubMed] [Google Scholar]
- 62.Harazono A, et al. Simultaneous glycosylation analysis of human serum glycoproteins by high-performance liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;869:20–30. doi: 10.1016/j.jchromb.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 63.Harazono A, Kawasaki N, Kawanishi T, Hayakawa T. Site-specific glycosylation analysis of human apolipoprotein B100 using LC/ESI MS/MS. Glycobiology. 2005;15:447–62. doi: 10.1093/glycob/cwi033. [DOI] [PubMed] [Google Scholar]
- 64.Gupta N, Bandeira N, Keich U, Pevzner P. Target-Decoy Approach and False Discovery Rate: When Things May Go Wrong. J Am Soc Mass Spectr. 2011;22:1111–1120. doi: 10.1007/s13361-011-0139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bern M, Kil YJ. Comment on “Unbiased statistical analysis for multi-stage proteomic search strategies”. J. Proteome Res. 2011;10:2123–7. doi: 10.1021/pr101143m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods. 2007;4:787–97. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 67.Computer Software Reviews. J Am Chem Soc. 1992;114:7961–7962. [Google Scholar]
- 68.Brosch M, Choudhary J. Scoring and validation of tandem MS peptide identification methods. Methods Mol Biol. 2010;604:43–53. doi: 10.1007/978-1-60761-444-9_4. [DOI] [PubMed] [Google Scholar]
- 69.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 2008;7:3354–63. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eng JK, McCormack AL, Yates JRI. An approach to correlate MS/MS data to amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.