

Abstract
Background and Purpose
Endothelial nitric oxide synthase produces superoxide under physiological conditions leading to hydrogen peroxide (H2O2) -dependent dilations to acetylcholine in isolated mouse cerebral arteries. The purpose of this study was to investigate whether H2O2 was involved in flow-mediated dilation (FMD).
Methods
Cerebral arteries were isolated from 12±2-week-old C57Bl/6 male mice. FMD (0 to 10 μL/min, 2-μL step increase at constant internal pressure) was induced in vessels preconstricted with phenylephrine (30 μmol/L). Simultaneously to diameter acquisition, H2O2 or nitric oxide production was detected by the fluorescent dyes CMH2CFDA or 4,5-diaminofluorescein diacetate, respectively. Results are expressed as mean±SEM of 6 to 8 mice.
Results
FMD (at 10 μL/min, 25±3% of maximal diameter) was prevented (P<0.05) by endothelium removal (6±1%) or endothelial nitric oxide synthase inhibition with N-nitro-L-arginine (11±1%) but not by the specific nitric oxide scavenger 2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl3-oxide (24±3%). Addition of PEG-catalase and silver diethyl dithio-carbamate (superoxide dismutase inhibitor) reduced (P<0.05) FMD to 10±2% and 15±1%, respectively. Simultaneously to FMD, H2O2-associated rise in fluorescence (+133±19 a.u.) was prevented by N-nitro-L-arginine, PEG-catalase, and silver diethyl dithio-carbamate (+55±10, +64±4, and +50±10 a.u., respectively; P<0.05). Inhibition of FMD by PEG-catalase was fully restored by the addition of tetrahydrobiopterin, a cofactor of endothelial nitric oxide synthase (23±3%); this functional reversal in dilation was associated with the simultaneous increase in nitric oxide-associated fluorescence (+418±58 a.u., P<0.05), which was prevented by 2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl3-oxide (+93±26 a.u.). Akt inhibition with triciribine prevented FMD and H2O2-associated rise in fluorescence (3±1% and +23±4% a.u., respectively; P<0.05), but not acetylcholine-induced dilation.
Conclusion
In healthy C57Bl/6 mouse cerebral arteries, Akt-dependent activation of endothelial nitric oxide synthase-derived H2O2 mediates flow-dependent dilation.
Keywords: endothelium, nitric oxide, oxygen radicals, resistance arteries
Shear stress is one of the most important physiological stimuli for vasodilation and is greatly implicated in the regulation of vascular tone and vascular homeostasis by contributing to the maintenance of organ perfusion and vascular integrity.1,2 Several studies have evaluated the implication of endothelial nitric oxide synthase (eNOS) and cyclo-oxygenase (COX) derivatives in flow-mediated dilation (FMD).3–6 However, the implication of each endothelium-dependent relaxing factor (EDRF) varies among vascular beds and pathological states.3,7–11
The release of superoxide (O2•−) has been reported during FMD both in peripheral12,13 and cerebral arteries.14,15 Although an increase in reactive oxygen species (ROS) is traditionally considered as a pathological response, recent investigations show that ROS are implicated in the regulation of vascular function.12,16–18 It is unclear, however, if this concept applies to the physiological regulation of vascular tone and FMD. In a recent study, our group showed that hydrogen peroxide (H2O2) is an EDRF produced by eNOS activity after muscarinic receptor stimulation.19 H2O2 was also reported to induce endothelium-dependent and -independent dilation in healthy rat cerebral arteries18 and identified as an EDRF in FMD of human coronary arteries.1 These results led to the hypothesis that H2O2 could be physiologically implicated in the regulation of cerebral arteries.
The origin of O2•− that leads to the physiological formation of H2O2 is still unsettled. eNOS, however, is able to generate O2•− during enzymatic cycling.20,21 The eNOS-dependent generation of O2•− is nonetheless proposed to be functionally significant only in pathological conditions and related to the limited availability of L-arginine, the substrate of eNOS as well as of its essential cofactor tetrahydrobiopterin (BH4).22,23 In a recent study, Shimokawa’s group suggested that the NOSs system has diverse vasodilator functions depending on the vessel size leading to superoxide production rather than nitric oxide in small vessels.24 There is therefore a need for determining the physiological role of eNOS derived H2O2 in FMD in cerebral arteries.
We hypothesized that eNOS produces physiologically relevant levels of free radicals leading to H2O2-dependent FMD in healthy mouse cerebral arteries.
Materials and Methods
Animals and Tissue Preparation
The procedures and protocols were performed in accordance with our institutional guidelines and the Guide for the Care and Use of Laboratory Animals of Canada. Experiments were conducted on cerebral arteries isolated from 3-month-old male C57BL/6 mice (29±1 g, n=66; Charles River Laboratories, Quebec, Canada) using a method previously described.19 Mice were euthanized by CO2 inhalation and the brain was rapidly removed and placed in ice-cold physiological salt solution (PSS) of the following composition (mmol/L): NaCl 130, KCl 4.7, CaCl2 1.6, MgSO4 1.17, NaHCO3 14.9, KH2PO4 1.18, EDTA 0.026, glucose 10. In all experiments, the PSS was oxygenated by a gas mixture containing 12% O2, 5% CO2, and 83% N2 generating a pO2 of 150±10 mm Hg. Cerebral arteries (anterior, posterior, and posterior communicating; internal diameter of 136±2 μm when pressurized at 60 mm Hg) were carefully isolated, cannulated at both ends, and pressurized at 60 mm Hg on a pressure myograph (Living Systems Instrumentation, Burlington, Vt). The tips of the pipettes used in the study were calibrated and carefully checked to match the vessel inner diameter to minimize resistances.
Reactivity Studies
An equilibration period of 40 minutes was allowed before starting the experiment and the resulting myogenic tone was measured ([Dafter−Dbefore]/Dmax)*100, where Dbefore is the diameter before equilibration, Dafter is the diameter after equilibration time, and Dmax is the maximal diameter obtained in Ca2+-free solution. Similar levels of preconstrictions with phenylephrine (PE; 10 to 30 μmol/L; Table) were obtained before each experiment. FMD was induced using a flow control peristaltic pump (Living Systems Instrumentation) directly connected to the pressured myograph. A single cumulative FMD curve (0 to 10 μL/min, 2-μL step increase at constant internal pressure) was performed on each segment. Two minutes were allowed for each 2-μL step increase. Shear stress was calculated using the following equation: t=4ηQ/πr3 where t is the shear stress (dyn/cm2), η the viscosity (0.8 cp5), Q is the flow rate through the lumen, and r is the inside radius. In this study, the calculated shear stress was comparable to the physiological value of shear stress in arterioles of this size.25
Table.
Myogenic Tone (MT), Contraction With PE (10 or 30 μmol/L), and Maximal Dilation to FMD (10 μL/min) of Cerebral Arteries Isolated From C57Bl/6 Male Mice*
| Groups | MT (%) | PE (%) | FMD (%) | N |
|---|---|---|---|---|
| Control conditions | 8 ± 1 | 40 ± 2 | 25 ± 3 | 13 |
| +L-NNA (10 μmol/L) | 21 ± 4† | 46 ± 5 | 11 ± 1† | 12 |
| +PEG-catalase (50 U/mL) | 15 ± 3† | 43 ± 3 | 16 ± 1† | 12 |
| +PEG-catalase (100 U/mL) | 19 ± 7† | 46 ± 3 | 10 ± 2† | 3 |
| +BH4 (1 mmol/L) | 16 ± 3† | 42 ± 4 | 28 ± 3 | 11 |
| +L-arginine (5 μmol/L) | 12 ± 8 | 49 ± 5 | 24 ± 1 | 6 |
| +PTIO (100 μmol/L) | 8 ± 2 | 40 ± 4 | 26 ± 2 | 8 |
| +ODQ (1 μmol/L) | 25 ± 12† | 49 ± 4 | 4 ± 2† | 4 |
| +DT-3 (25 nmol/L) | 7 ± 2 | 39 ± 6 | 11 ± 1† | 3 |
| +BH4+PTIO | 15 ± 4† | 39 ± 7 | 20 ± 4 | 5 |
| +BH4+PEG-catalase | 18 ± 5† | 45 ± 5 | 26 ± 3 | 6 |
| +BH4+ODQ | 12 ± 5 | 45 ± 4 | 7 ± 2† | 5 |
| +BH4+L-NNA | 17 ± 4† | 44 ± 3 | 11 ± 2† | 4 |
| +BH4+PTIO+PEG-catalase | 21 ± 9† | 47 ± 5 | 9 ± 2† | 5 |
| +Pyruvate (3 mmol/L) | 8 ± 2 | 42 ± 3 | 15 ± 2† | 6 |
| +DETC (1 mmol/L) | 15 ± 2† | 47 ± 3 | 15 ± 1† | 9 |
| +L-NNA+indomethacin (10 μmol/L) | 8 ± 3 | 36 ± 3 | 10 ± 3† | 5 |
| +PEG-catalase+PTIO | 7 ± 2 | 39 ± 4 | 14 ± 3† | 4 |
| +Triciribine (1 μmol/L) | 11 ± 3 | 45 ± 5 | 3 ± 1† | 4 |
| +Apocynin (10 μmol/L) | 12 ± 5 | 45 ± 4 | 33 ± 4 | 4 |
| −Endothelium | 12 ± 5 | 39 ± 9 | 6 ± 1† | 5 |
Note: Data are expressed as mean±SEM.
MT, PE, and FMD are expressed as the percentage of the maximal diameter.
P<0.05 compared with control conditions.
We used (1) PEG-catalase (50 and 100 U/mL); (2) N-nitro-L-arginine (L-NNA; 10 μmol/L), an eNOS inhibitor; (3) pyruvate (3 mmol/L), a H2O2 scavenger26; (4) silver diethyldithiocarbamate (DETC; 1 mmol/L), a superoxide dismutase inhibitor; (5) 2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl 3-oxide (PTIO; 100 μmol/L), a nitric oxide scavenger; (6) indomethacin (10 μmol/L), a nonspecific COX inhibitor; (7) 1H-[1,2,4]-oxadiazole-4,3-aquinoxalin-1-one (ODQ; 10 μmol/L), a soluble guanylate cyclase inhibitor; (8) triciribine (1 μmol/L27), an Akt protein inhibitor; (9) DT-3 (25 nmol/L), a cell-permeant G protein kinase inhibitor28; (10) BH4 (1 mmol/L); (11) apocynin (10 μmol/L), a NAD(P)H oxidase inhibitor; (12) L-arginine (5 mmol/L); and (13) detaNONOate (0.1 μmol/L), a nitric oxide donor. Acetylcholine (ACh; 30 μmol/L) and H2O2 (10 μmol/L) were also used to induce dilation. All drugs were purchased from Sigma-Aldrich Canada Ltd (Oakville, Ontario, Canada), except for triciribine and DT-3, which were purchased from Calbiochem EMD Chemicals Inc (San Diego, Calif). All drugs were directly added to the bath chamber (extraluminal) and the final concentration of ethanol or DMSO never exceeded 0.1%.
Fluorescence Studies
Pressurized cerebral arteries were incubated in oxygenated PSS (37°C) containing either 5 μmol/L of 5- and 6-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate acetyl ester (DCF-DA, a ROS-reacting fluorescent dye; Molecular Probe, Invitrogen, Burlington, Ontario, Canada19,29,30) or 10 μmol/L of 4,5-diaminofluorescein diacetate (DAF-2, a nitric oxide-reacting fluorescent dye; Calbiochem, San Diego, Calif19,31) 30 minutes before the beginning of the experiment, with or without inhibitors. Vessels were then washed with PSS, preconstricted with PE, and dilated with flow (0 to 10 μL/min) or with a single dose of ACh (30 μmol/L) while recording simultaneously the changes in diameter and in fluorescence intensities of fluorescein retained intracellularly after cleavage of the acetate moieties. Fluorescence intensities at 492 to 495 nm (excitation) were measured at 520 nm with an IonOptix Acquire system (IonOptix, Milton, Mass). Before each experiment, basal fluorescence intensity was recorded. Results represent differences between stimulated and basal intensity.
Statistics
n refers to the number of animals used in each protocol. Continuous variables are expressed as mean±SEM. The maximal diameter (Dmax) was determined by changing the PSS to a Ca2+-free PSS.19 Dilations are expressed with the following equation: ([Df−Dmin]/[Dmax−Dmin])*100, where Df is the diameter obtain at each flow value and Dmin is the diameter of PE-induced constriction. One-way analysis of variance were performed to compare the effect of the different inhibitors on FMD curves and the increase in fluorescence intensity at a flow rate of 10 μL/min. Differences were considered to be statistically significant when the probability value was <0.05 (Scheffe’s F test).
Results
Implication of Hydrogen Peroxide and Endothelial Nitric Oxide Synthase in Flow-Mediated Dilation
Flow-mediated dilations of cerebral arteries were reduced by nitric oxide synthase inhibition with L-NNA (10 μmol/L; Figure 1A; Table). The nitric oxide scavenger PTIO (100 μmol/L), however, had no impact on the dilation induced by flow (Figure 1A; Table). The cell-permeable PEG-catalase (50 and 100 U/mL) reduced FMD in a dose-dependent manner (Figure 1; Table). The likely involvement of H2O2 was further confirmed by the inhibitory effect of pyruvate (3 mmol/L), a H2O2 scavenger, and DETC (1 mmol/L), a superoxide dismutase inhibitor (Figure 1B; Table).
Figure 1.
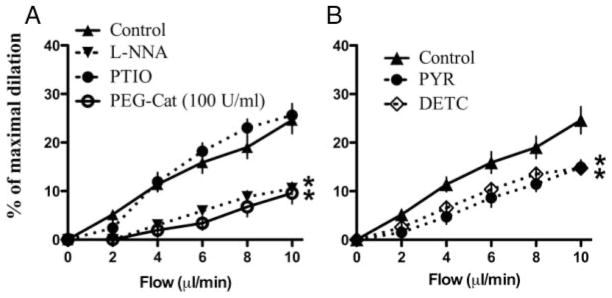
Flow-mediated endothelium-dependent dilations of C57Bl/6 mouse pressurized cerebral arteries: (A) effect of L-NNA (10 μmol/L), PEG-catalase (100 U/mL), and the nitric oxide scavenger PTIO (100 μmol/L). B, Scavenging of H2O2 by pyruvate (3 mmol/L) or superoxide dismutase inactivation by DETC (1 mmol/L). *P<0.05 compared with control (“n” numbers are in the Table).
To confirm these pharmacological data, H2O2 production was assessed in pressurized vessels after incorporation of the fluorescent ROS-reactive dye, DCF-DA.19,29,30 FMD was associated with an increase in fluorescence intensity: ROS-dependent signals (Figure 2A, C) were abolished by L-NNA, PEG-catalase, DETC, and pyruvate, demonstrating the specificity of the dye for H2O2 in our experimental conditions. Apocynin, a NAD(P)H oxidase inhibitor, neither reduced FMD (Table) nor DCF-DA fluorescence (Figure 2C).
Figure 2.

Representative changes in fluorescence intensities induced by flow (4, 6, and 8 μL/min) in pressurized mouse cerebral arteries. Arteries were loaded with (A) DCF-DA (5 μmol/L) before (control) and after incubation with PEG-catalase and (B) DAF-2 (10 μmol/L) before (control) and after addition of BH4). C, Average increases in fluorescence intensities of arteries loaded with DCF-DA or (D) DAF-2 during flow-mediated dilation (10 μL/min; n=3 to 5 per group). *P<0.05 compared with control; †P<0.05 compared with BH4.
Nitric oxide production was assessed after incorporation of the fluorescent nitric oxide-reactive dye, DAF-2.19 FMD was not associated with an increase in fluorescence intensity, which was unaffected by the addition of PEG-catalase and DETC (Figure 2B, D).
In contrast, addition of the nitric oxide donor detaNONO-ate (0.1 μmol/L) induced a potent dilation of 73±5% of Dmax and increased DAF-associated fluorescence by 959±47 a.u. (n=4). These responses were reduced (P<0.05) by addition of PTIO (46±6% and 88±24 a.u., respectively).
Effects of Tetrahydrobiopterin on Endothelial Nitric Oxide Synthase Activity and Nitric Oxide Production
Addition of BH4 (1 mmol/L) did not alter FMD (Figure 3A; Table) but led to the production of nitric oxide as revealed by the appearance of a strong DAF-2-associated fluorescence that was prevented by PTIO and L-NNA (Figure 2B, D). Although PTIO or PEG-catalase alone did not alter FMD in the presence of BH4 (Table), the dilation to flow was prevented by combining PTIO and PEG-catalase (Table). This was associated with a reduction in both nitric oxide- and H2O2-associated fluorescence from 418±58 and 571±94 a.u. to 45±4 and 58±20 a.u., respectively (P<0.05).
Figure 3.

Effects of BH4 (1 mmol/L), a nitric oxide synthase cofactor; DT-3, a protein kinase G inhibitor (25 nmol/L), and ODQ, a soluble guanylate cyclase inhibitor (1 μmol/L), on (A–B) flow-mediated dilation and (C) changes in fluorescence intensities induced by flow (10 μL/min) in pressurized mouse cerebral arteries loaded with DCF-DA (5 μmol/L; “n” numbers are reported in the Table). *P<0.05 compared with control.
In contrast to BH4, L-arginine (5 mmol/L) neither affected FMD (10 μL/min; Table) nor nitric oxide-associated fluorescence (26±4 a.u.).
Vasodilator Mechanism of Action of Hydrogen Peroxide
ODQ (1 μmol/L), an inhibitor of the sGC, reduced the dilation triggered by flow as efficiently as endothelial denudation (Figures 3B and 4; Table). ODQ, however, did not prevent H2O2-associated rise in fluorescence (Figure 3C). In the presence of BH4, ODQ still reduced FMD (Figure 3A). Cell-permeant G protein kinase inhibition with DT-3 reduced FMD, but not the H2O2-associated rise in fluorescence (Figure 3B, C).
Figure 4.
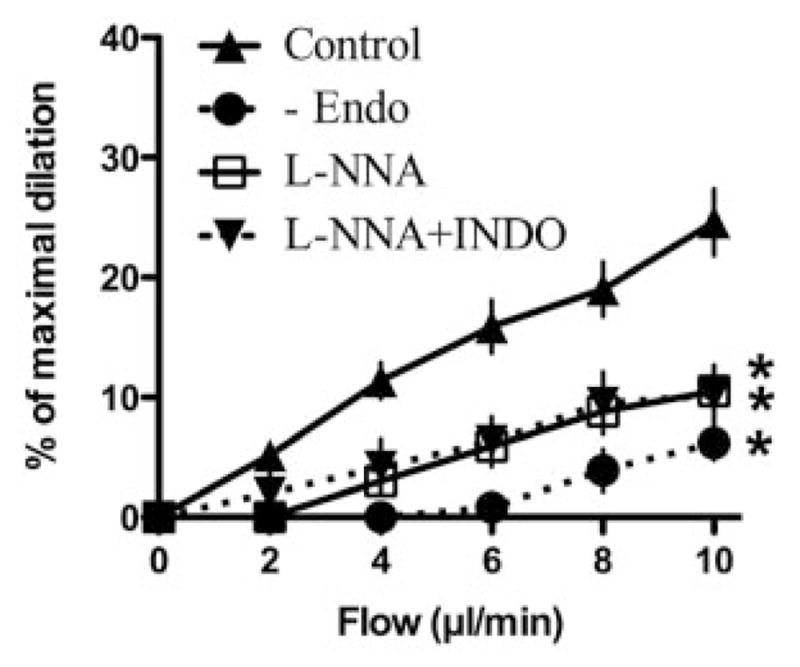
Effect of removal of the endothelium (-Endo), L-NNA (10 μmol/L), and addition of indomethacin (10 μmol/L) on flow-mediated dilations of pressurized cerebral arteries isolated from C57Bl/6 mice (“n” numbers are reported in the Table). *P<0.05 compared with control.
Endothelial Regulation of Flow-Mediated Dilation
FMD was strongly reduced by L-NNA with no additive effects of indomethacin, suggesting no involvement of COX derivatives during FMD of mouse cerebral arteries (Figure 4). FMD and its associated increase in H2O2 fluorescence were abolished by Akt inhibition with triciribine (Figure 5A, C; Table). In contrast, triciribine only limited ACh-induced endothelium-dependent dilation (30 μmol/L) and its associated increase in H2O2 fluorescence (Figure 5B, D).
Figure 5.

Effect of removal of the endothelium (-Endo) and triciribine, an inhibitor of Akt (TB; 1 μmol/L) on (A) FMD (10 μL/min) and (B) endothelium-dependent ACh-induced (30 μmol/L) dilation of pressurized cerebral arteries isolated from C57Bl/6 mice. Effect of TB on H2O2-associated rise in fluorescence induced by (C) FMD (10 μL/min) and (D) ACh (30 μmol/L; “n” numbers are in the Table). *P<0.05 compared with control; †P<0.05 compared with FMD.
Impact of Endothelial Nitric Oxide Synthase Activity on Myogenic Tone
L-NNA, PEG-catalase, DETC, and ODQ increased myogenic tone, whereas Akt inhibition and PTIO had no effect (Table). The rise in myogenic tone induced by L-NNA was prevented by COX-1/2 inhibition, suggesting that addition of L-NNA reveals a COX-derived contracting factor (Table).
Exogenous Hydrogen Peroxide Dilation
Exogenous H2O2 (10 μmol/L, n=4) induced a large dilation (65±4%). Addition of L-NNA or BH4 did not affect this response (60±6% and 52±7%, respectively). Addition of ODQ, however, prevented H2O2-induced dilation (6±2%).
Discussion
The main finding of the present study is that FMD is mediated by H2O2 derived from eNOS activity in pressurized cerebral arteries isolated from young and healthy C57Bl/6 mice. FMD was sensitive to eNOS inhibition and H2O2 scavengers and associated with an increase in H2O2-associated fluorescence. In addition, Akt-dependent activation of eNOS is mandatory for FMD, unlike for ACh-induced dilation. These results support the concept that eNOS-derived H2O2 is a physiologically relevant signaling molecule in healthy mice.
Endothelium removal abolished FMD in cerebral artery consistent with previous reports.5,7,13,32 Inhibition of eNOS by L-NNA limited the dilation similarly to H2O2 scavengers, whereas PTIO, a nitric oxide scavenger, had no impact. These results strongly suggest that H2O2 originates from stimulated eNOS activity and is responsible for FMD in mouse cerebral arteries. This hypothesis is supported by the absence of H2O2-associated fluorescence in cerebral arteries incubated in the presence of L-NNA and PEG-catalase and by the demonstration that FMD is not associated with an increase in nitric oxide-associated fluorescence. These data are in line with recent work showing that H2O2 is an important signaling molecule in cerebral14 and peripheral33 arteries.
We19 and others1,34 previously reported that H2O2 is an EDRF derived from eNOS activity after Ach-induced dilation of mouse arteries. It is known that eNOS is able to generate O2•− during enzymatic cycling.20,21 The eNOS-dependent generation of O2•− is proposed, however, to be only functionally significant in pathological conditions and related to the limited availability of eNOS cofactors.22,23 In this study, the addition of BH4, but not L-arginine, induced an increase in nitric oxide-associated fluorescence, but neither BH4 nor L-arginine had significant effect on the extent of FMD. This increase in nitric oxide-associated fluorescence intensity was blocked by PTIO and L-NNA, confirming that eNOS produces nitric oxide in the presence of an excess of BH4. The level of BH4 is significantly lower in the brain than in other mouse organs as previously reported by Heales’ group35; this could be a reason for the preferential production of H2O2, but it remains to be demonstrated. The NAD(P)H oxidase is known to be a source of superoxide and H2O2 during endothelium-dependent dilation of rat cerebral arteries.14 In our hands, apocynin failed to reduce FMD as well as the increase in H2O2-associate fluorescence, suggesting no involvement in the activity of the NAD(P)H oxidase in the H2O2-dependent dilation of mouse cerebral arteries.
Our data, those of Rosenblum,15 and of Sobey’s group14 demonstrate that free radicals are involved in the physiological regulation of the cerebrovascular tone. Free radicals, however, are commonly associated with endothelial dysfunction and cellular damages triggered by oxidative stress.34 Many physiological regulatory effects are mediated by H2O2 and other ROS that are chemically derived from superoxide.36 NAD(P)H, for example, is essential for the regeneration of GSH from GSSG, reactivation of H2O2-inactivated catalase, and regeneration of thioredoxin.37 It is therefore essential to maintain adequate levels of NAD(P)H as well as the normal function of these enzymes. Hence, controlled production of free radicals has a physiological meaning, whereas unregulated oxidative stress is deleterious.
The fact that PTIO addition blocked the rise in nitric oxide-associated fluorescence after the addition of det-aNONOate demonstrates the specificity of DAF-2 dye for nitric oxide as previously discussed.19 DCF-DA is known to recognize all ROS species at high concentrations such as O2−, OH−, and H2O2 with a greater specificity for ONOO−.38 DCF-DA fluorescence signal was, however, abolished by PEG-catalase, pyruvate, and DETC, but not PTIO (data not showed), demonstrating greater selectivity of the dye for H2O2 than nitric oxide or its derived reactive nitrogen species. Hence, in these healthy arteries, and in our experimental conditions, peroxynitrites are not produced.
The mechanism of eNOS activation by flow is not fully understood. It has been proposed that Akt-dependent phosphorylation of eNOS is essential for nitric oxide production.14 Our study demonstrates that FMD is abolished by Akt inhibition and this was associated with a reduction in H2O2-associated fluorescence. Thus, shear stress-dependent activation of Akt is responsible for eNOS activation. To support this hypothesis, we used another Akt inhibitor (LY294002). LY294002, however, is also a PI3-K inhibitor and reduced the precontraction induced by PE (data not shown), preventing its use. In contrast to flow, muscarinic receptor activation directly promotes eNOS activation without requiring Akt-dependent activation. Importantly, neither the dilation nor H2O2-associated fluorescence was abolished by Akt inhibition after muscarinic receptor stimulation. Akt inhibition reduced, nonetheless, ACh-dependent dilation, suggesting that there is a basal level of Akt activity influencing eNOS cycling.
Addition of ODQ to block the sGC abrogated FMD without affecting H2O2-associated fluorescence. Furthermore, exogenous H2O2-induced dilation was unaffected by L-NNA or BH4, but prevented by ODQ, which is in agreement with our previous report.19 To further validate the involvement of the cGMP pathway in FMD, we used a cell-permeant G protein kinase inhibitor. FMD dilation, but not H2O2 production, was abolished by DT-3, confirming the implication of cGMP in H2O2-dependent FMD. In the presence of BH4, FMD was also abolished by ODQ, confirming that H2O2 shares with nitric oxide a similar vasodilator pathway.18,39
FMD was not completely blocked by L-NNA with no additive effects of indomethacin, suggesting the involvement of another EDRF. Previous studies1,7 suggested the role of an EDRF during FMD. This factor may account for the residual response obtained in the presence of L-NNA. It is also interesting that COX inhibition had no effect on FMD. This is another difference with ACh-induced endothelium dependent dilation of cerebral arteries in which prostacyclin significantly contributes to this response.19 Like in our previous report,19 myogenic tone was increased by L-NNA but reduced by combining indomethacin, suggesting that in the absence of nitric oxide, a COX-derived contracting factor is produced or its effect revealed. We observed the same response in renal arteries,40 which suggests that both constrictors and dilators coexist to regulate tone. In age and in the presence of risk factor for cardiovascular diseases, the expression of these contracting factors dominate as a consequence of a reduced nitric oxide synthase activity.41–43
In conclusion, our results suggest that flow triggers Akt-dependent eNOS activation leading to a dilation essentially induced by H2O2 in pressurized cerebral arteries isolated from healthy and young C57Bl/6 mice. Like nitric oxide, H2O2 activates the sGC. Our results highlight the multifaceted function of eNOS in its mechanisms of regulation of vascular function.
Acknowledgments
Sources of Funding
This work has been supported in part by the Foundation of Montreal Heart Institute, the Heart and Stroke Foundation of Quebec, and the Canadian Institute for Health Research (MOP87388). A.D. holds the Frederick Banting and Charles Best Canada Graduate Scholarships–Doctoral Award in association with the Canadian Institute for Health Research.
Footnotes
Disclosures
None.
References
- 1.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 2.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol. 2007;292:H93–H100. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 3.Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Lüscher TF. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. doi: 10.1161/01.cir.91.5.1314. [DOI] [PubMed] [Google Scholar]
- 4.Koller A, Sun D, Huang A, Kaley G. Corelease of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. Am J Physiol. 1994;267:H326–H332. doi: 10.1152/ajpheart.1994.267.1.H326. [DOI] [PubMed] [Google Scholar]
- 5.Shipley RD, Kim SJ, Muller-Delp JM. Time course of flow-induced vasodilation in skeletal muscle: contributions of dilator and constrictor mechanisms. Am J Physiol. 2005;288:H1499–H1507. doi: 10.1152/ajpheart.00489.2004. [DOI] [PubMed] [Google Scholar]
- 6.Fujii K, Heistad DD, Faraci FM. Flow-mediated dilatation of the basilar artery in vivo. Circ Res. 1991;69:697–705. doi: 10.1161/01.res.69.3.697. [DOI] [PubMed] [Google Scholar]
- 7.Miura H, Wachtel RE, Liu Y, Loberiza FR, Jr, Saito T, Miura M, Gutterman DD. Flow-induced dilation of human coronary arterioles: important role of Ca(2+)-activated K(+) channels. Circulation. 2001;103:1992–1998. doi: 10.1161/01.cir.103.15.1992. [DOI] [PubMed] [Google Scholar]
- 8.Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 9.Koller A, Sun D, Kaley G. Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ Res. 1993;72:1276–1284. doi: 10.1161/01.res.72.6.1276. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Delp JM, Spier SA, Ramsey MW, Delp MD. Aging impairs endothelium-dependent vasodilation in rat skeletal muscle arterioles. Am J Physiol. 2002;283:H1662–H1672. doi: 10.1152/ajpheart.00004.2002. [DOI] [PubMed] [Google Scholar]
- 11.Sun D, Liu H, Yan C, Jacobson A, Ojaimi C, Huang A, Kaley G. COX-2 contributes to the maintenance of flow-induced dilation in arterioles of eNOS-knockout mice. Am J Physiol. 2006;291:H1429–H1435. doi: 10.1152/ajpheart.01130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laurindo FR, de Pedro MA, Barbeiro HV, Pileggi F, Carvalho MH, Augusto O, da Luz PL. Vascular free radical release. Ex vivo and in vivo evidence for a flow-dependent endothelial mechanism. Circ Res. 1994;74:700–709. doi: 10.1161/01.res.74.4.700. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolos AC, Gutterman DD. Mitochondrial sources of generation play a key role in flow- H2O2 mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 14.Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cerebr Blood Flow Metab. 2006;26:836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- 15.Rosenblum WI. Hydroxyl radical mediates the endothelium-dependent relaxation produced by bradykinin in mouse cerebral arterioles. Circ Res. 1987;61:601–603. doi: 10.1161/01.res.61.4.601. [DOI] [PubMed] [Google Scholar]
- 16.Rush JW, Ford RJ. Nitric oxide, oxidative stress and vascular endothelium in health and hypertension. Clin Hemorheol Microcirc. 2007;37:185–192. [PubMed] [Google Scholar]
- 17.Ungvari Z, Wolin MS, Csiszar A. Mechanosensitive production of reactive oxygen species in endothelial and smooth muscle cells: role in microvascular remodeling? Antioxid Redox Signal. 2006;8:1121–1129. doi: 10.1089/ars.2006.8.1121. [DOI] [PubMed] [Google Scholar]
- 18.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Stroke. 1997;28:2290–2295. doi: 10.1161/01.str.28.11.2290. [DOI] [PubMed] [Google Scholar]
- 19.Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Porasuphatana S, Tsai P, Rosen GM. The generation of free radicals by nitric oxide synthase. Comp Biochem Phys Part C. 2003;134:281–289. doi: 10.1016/s1532-0456(02)00271-5. [DOI] [PubMed] [Google Scholar]
- 21.Stroes E, Hijmering M, van Zandvoort M, Wever R, Rabelink TJ, Faassen EE. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998;438:161–164. doi: 10.1016/s0014-5793(98)01292-7. [DOI] [PubMed] [Google Scholar]
- 22.Alp NJ, Chanon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1–9. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 23.Katusic ZS, d’Uscio LV. Tetrahydrobiopterin. Mediator of endothelial protection. Arteroscler Thromb Vasc Biol. 2004;24:397–398. doi: 10.1161/01.ATV.0000121569.76931.0b. [DOI] [PubMed] [Google Scholar]
- 24.Takaki A, Morikawa K, Tsutsui M, Murayama Y, Tekes E, Yamagishi H, Ohashi J, Yada T, Yanagihara N, Shimokawa H. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. doi: 10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papaioannou TG, Karatzis EN, Vavuranakis M, Lekakis JP, Stefanadis C. Assessment of vascular wall shear stress an implications for athero-sclerotic disease. Int J Cardiol. 2006;113:12–18. doi: 10.1016/j.ijcard.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 26.Mallet RT, Sun J, Knott EM, Sharma AB, Olivencia-Yurvati AH. Metabolic cardioprotection by pyruvate: recent progress. Exp Biol Med. 2005;230:435–443. doi: 10.1177/153537020523000701. [DOI] [PubMed] [Google Scholar]
- 27.Shein NA, Tsenter J, Alexandrovich AG, Horowitz M, Shohami E. Akt phosphorylation is required for heat acclimation-induced neuroprotection. J Neurochem. 2007;103:1523–1529. doi: 10.1111/j.1471-4159.2007.04862.x. [DOI] [PubMed] [Google Scholar]
- 28.Dostmann WRG, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Iα inhibit NO-induced cerebral dilation. Proc Natl Acad Sci U S A. 2000;97:14772–14777. doi: 10.1073/pnas.97.26.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatoum OA, Otterson MF, Kopelman D, Miura H, Sukhotnik I, Larsen BT, Selle RM, Moulder JE, Gutterman DD. Radiation induces endothelial dysfunction in murine intestinal arterioles via enhanced production of reactive oxygen species. Arterioscler Thromb Vasc Biol. 2006;26:287–294. doi: 10.1161/01.ATV.0000198399.40584.8c. [DOI] [PubMed] [Google Scholar]
- 30.Chaytor AT, Edwards DH, Bakker LM, Griffith TM. Distinct hyperpolarizing and relaxant roles for gap junctions and endothelium-derived H2O2 in NO-independent relaxations of rabbit arteries. Proc Natl Acad Sci U S A. 2003;100:15212–15217. doi: 10.1073/pnas.2435030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sylvester FA, Stepp DW, Frisbee JC, Lombard JH. High-salt diet depresses acetylcholine reactivity proximal to NOS activation in cerebral arteries. Am J Physiol. 2002;283:H353–H363. doi: 10.1152/ajpheart.00127.2002. [DOI] [PubMed] [Google Scholar]
- 32.Bergaya S, Hilgers RH, Meneton P, Dong Y, Bloch-Faure M, Inagami T, Alhenc-Gelas F, Lévy BI, Boulanger CM. Flow-dependent dilation mediated by endogenous kinins requires angiotensin AT2 receptors. Circ Res. 2004;94:1623–1629. doi: 10.1161/01.RES.0000131497.73744.1a. [DOI] [PubMed] [Google Scholar]
- 33.Matoba T, Shimokawa H. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Pharmacol Sci. 2003;92:1–6. doi: 10.1254/jphs.92.1. [DOI] [PubMed] [Google Scholar]
- 34.Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 35.Lam AAJ, Hyland K, Heales SJR. Tetrahydrobiopterin availability, nitric oxide metabolism and glutathione status in the hph-1 mouse; implications for the pathogenesis and treatment of tetrahydrobiopterin deficiency states. J Inherit Metab Dis. 2007;30:256–262. doi: 10.1007/s10545-006-0502-x. [DOI] [PubMed] [Google Scholar]
- 36.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 37.Ying W. NAD-/NADH and NADP-/NADPH in cellular functions and cell death: regulation and biological consequences. Antiox Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez J, Specian V, Maloney R, Jourd’heuil D, Feelisch M. Performance of diamino fluorophores for the localization of sources and targets of nitric oxide. Free Radic Biol Med. 2005;38:356–368. doi: 10.1016/j.freeradbiomed.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 39.Iesaki T, Gupte SA, Kaminski PM, Wolin MS. Inhibition of guanylate cyclase stimulation by NO and bovine arterial relaxation to peroxynitrite and H2O2. Am J Physiol. 1999;46:H978–H985. doi: 10.1152/ajpheart.1999.277.3.H978. [DOI] [PubMed] [Google Scholar]
- 40.Gendron ME, Thorin E. A change in the redox environment and thromboxane A2 production precede endothelial dysfunction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H2508–H2515. doi: 10.1152/ajpheart.00352.2007. [DOI] [PubMed] [Google Scholar]
- 41.Auch-Schwelk W, Katusic ZS, Vanhoutte PM. Thromboxane A2 receptor antagonists inhibit endothelium-dependent contractions. Hypertension. 1990;15:699–703. doi: 10.1161/01.hyp.15.6.699. [DOI] [PubMed] [Google Scholar]
- 42.Yang D, Feletou M, Levens N, Zhang JN, Vanhoutte PM. A diffusible substance(s) mediates endothelium-dependent contractions in the aorta of SHR. Hypertension. 2003;41:143–148. doi: 10.1161/01.hyp.0000047651.45322.16. [DOI] [PubMed] [Google Scholar]
- 43.Quyyumi AA, Dakak N, Andrews NP, Husain S, Arora S, Gilligan DM, Panza JA, Cannon RO., III Nitric oxide activity in human coronary circulation. Impact of risk factors for coronary atherosclerosis. J Clin Invest. 1995;95:1747–1755. doi: 10.1172/JCI117852. [DOI] [PMC free article] [PubMed] [Google Scholar]