

Abstract
Cartilaginous fishes, divided into Holocephali (chimaeras) and Elasmoblanchii (sharks, rays and skates), occupy a key phylogenetic position among extant vertebrates in reconstructing their evolutionary processes. Their accurate evolutionary time scale is indispensable for better understanding of the relationship between phenotypic and molecular evolution of cartilaginous fishes. However, our current knowledge on the time scale of cartilaginous fish evolution largely relies on estimates using mitochondrial DNA sequences. In this study, making the best use of the still partial, but large-scale sequencing data of cartilaginous fish species, we estimate the divergence times between the major cartilaginous fish lineages employing nuclear genes. By rigorous orthology assessment based on available genomic and transcriptomic sequence resources for cartilaginous fishes, we selected 20 protein-coding genes in the nuclear genome, spanning 2973 amino acid residues. Our analysis based on the Bayesian inference resulted in the mean divergence time of 421 Ma, the late Silurian, for the Holocephali-Elasmobranchii split, and 306 Ma, the late Carboniferous, for the split between sharks and rays/skates. By applying these results and other documented divergence times, we measured the relative evolutionary rate of the Hox A cluster sequences in the cartilaginous fish lineages, which resulted in a lower substitution rate with a factor of at least 2.4 in comparison to tetrapod lineages. The obtained time scale enables mapping phenotypic and molecular changes in a quantitative framework. It is of great interest to corroborate the less derived nature of cartilaginous fish at the molecular level as a genome-wide phenomenon.
Introduction
Chondrichthyans (cartilaginous fishes) occupy a key phylogenetic position among extant vertebrates as one of the early-branching lineages [1]–[3]. Their fossils are considered to be well preserved, largely because of the abundant deposits of dental material [4]. Living chondrichthyans are divided into two subclasses, Holocephali (chimaeras) and Elasmobranchii (sharks, rays and skates) (Figure 1). Based on the fossil records, Holocephali and Elasmobranchii are estimated to have diverged in the lowermost Devonian 410 million years before present (Ma) [5]. The monophyly of each of the two groups was strongly supported by morphological analyses [4], [6], [7], and has been reinforced by recent studies using several genes in the mitochondrial DNA (mtDNA) [8], [9]. More controversially discussed is the relationship between batoids (rays and skates) and sharks inside Elasmobranchii. Earliest morphological studies supported the basal dichotomy between sharks and batoids [10], [11] (Figure 1). However, subsequent extensive analyses of morphological variation suggested that batoids were descendants of one derived shark lineage, which is called the “Hypnosqualea hypothesis” [12], [13]. More recently, the basal dichotomy between batoids and sharks and monophylies of each of these two groups (Figure 1) have been supported by several molecular phylogenetic studies employing genes in mtDNA [9], [14]–[17] and the nucleus-encoded recombination activating gene 1 (RAG1) [18]. Based on this phylogenetic relationship, the monophyletic group containing all sharks is designated ‘Selachimorpha’ (Figure 1). Recently, diverse extinct shark species, assigned to the †Synechodontiformes, were suggested to form a monophyletic group, which is sister to all living sharks [19]. This assignment of Synechodontiformes into a monophyletic group which originated at the basal position of living sharks shifts the previously assumed minimum constraint of 190 Ma as the origin of modern sharks [20] back to the Late Permian around 250 Ma [15], [19].
Figure 1. Relationship of chondrichthyan species.
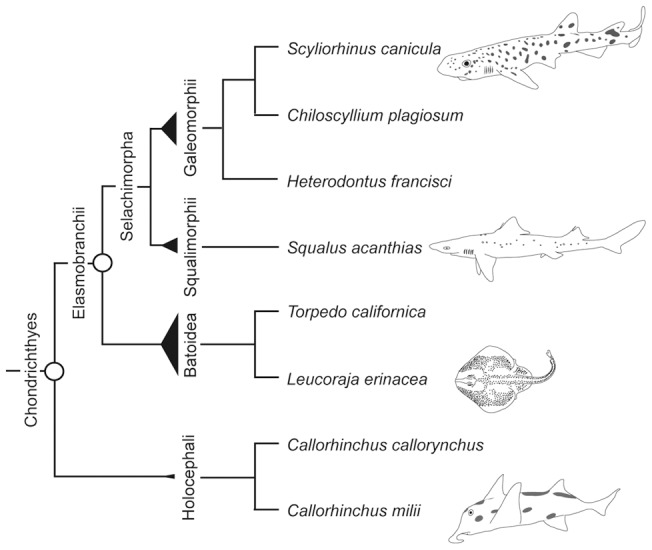
Species tree illustrating the relationship of all chondrichthyan species employed in our analyses, either in the divergence time study or evolutionary rate analysis (see text for alternative views of the phylogenetic relationship). Circles indicate the nodes referred to in the divergence time analysis. Widths of triangles are proportional to the numbers of species for individual groups according to Compagno et al. [69].
Some chondrichthyan species have been subjected to various molecular phylogenetic analyses, and they suggested a decreased rate of molecular evolution in the chondrichthyan lineages [21]–[25]. Martin et al. calculated the molecular evolutionary rate using two mitochondrial genes, cytochrome b (cytb) and cytochrome oxidase I (COI), of 13 shark species and showed their slower rate compared to the mammalian lineage [21]. Later, Martin et al. reinforced that also in nuclear-encoded genes, namely dlx, heat shock protein (HSP) 70 and recombination activating gene (RAG) 1, the rates of molecular evolution for sharks are an order of magnitude slower than those for mammals [24].
For the “Timetree of Life” [26], Heinicke et al. performed a family-level divergence time analysis for the entire Chondrichthyes [20]. They employed nucleotide sequences of the RAG1 gene and mitochondrial 12S and 16S ribosomal RNA (rRNA) genes previously sequenced [14], [18], [27]. Especially among the major chondrichthyan lineages, the analysis employing the RAG1 gene resulted in divergence time estimates of 100 million years older than the estimate with 12S and 16S rRNA genes [20]. Most recently, Inoue et al. inferred divergence times for fourteen divergence points at the family level of chondrichthyans, using whole mtDNA sequences [15]. Summarizing these progresses, further analyses based on the exhaustive use of latest palaeontological data employing more nuclear genes was anticipated.
Robust phylogenetic relationships and accurate divergence times are indispensable for deeper understanding of evolutionary processes. To date, unpretentious representative genes including mitochondrial genes [14], [27] and RAG1 [18], have been frequently selected as markers in molecular phylogenetic analyses. Nowadays, increasing molecular sequence data for diverse organisms and the bioinformatic tools for handling large-scale sequence resources have enhanced the power of so-called ‘phylogenomics’. In this study, we collected nuclear protein-coding sequences and calculated divergence times of two major nodes in the chondrichthyan lineage, the Holocephali-Elasmobranchii and the Batoidea-Selachimorpha splits (Figure 1). Based on our estimates, we measured the relative evolutionary rates of chondrichthyan lineages using available Hox A cluster sequence data.
Results
Genomic and transcriptomic data mining
We employed still limited but fairly large-scale genomic and transcriptomic sequence resources publicly available (see Methods and Figure 2). Through the automated orthology assessment, a total of 203 candidate genes were obtained. After investigation of the corresponding alignment of each gene, 122 candidate genes were excluded from further analyses, because they yielded spurious blast hits corresponding to sequences expanded into abundant copies (>500) in the chimaera genome. These 122 discarded candidates included genes whose mammalian orthologs are known to be involved in pathogen recognition and signal transduction [e.g. the nod-like receptor (NLR) family, the tripartite motif (TRIM) family and proteins containing WD repeats or FYVE zinc-finger domains] or are predicted hypothetical genes.
Figure 2. Work flow of gene family selection for divergence time estimation within chondrichthyans.

See Methods for details of elasmobranch EST assembly and gene prediction on C. milii genomic genomic contigs. Abbreviations: EST, expressed sequence tags; GSS, genome survey sequence.
The remaining 81 candidate genes were further scrutinized regarding their phylogenetic properties, namely their orthologies within individual datasets (see Methods). Here only three candidates (#1, 13 and 18 in Table 1) yielded the maximum-likelihood (ML) tree compatible with the generally accepted species phylogeny. All other candidate genes whose phylogenetic relationship of included species differed from the generally accepted species phylogeny [1] were further analyzed with the statistical framework of the ML method. That is, we calculated the log-likelihood (logL) of the obtained ML tree and that of tree topology in accordance with the generally accepted species phylogeny. The example of the candidate #9, RAN binding protein (RANBP) 1 gene, is illustrated in Figure 3. In this ML tree, teleost fishes, instead of Xenopus laevis, are a sister group of amniotes, which does not agree with the generally accepted phylogenetic relationship [1]. However, the likelihoods of this ML tree (Figure 3) and the generally accepted species phylogeny did not significantly differ (ΔlogL = 1.23±2.35).
Table 1. Overview of nuclear genes used for the divergence time analysis.
| # | Gene name | Protein ID of human ortholog | ΔlogL ± SE | Topology of ML tree | # of sites (aa) | Shape parameter α |
| 1 | Phosphoglycerate kinase 1 (PGK1) | NP_000282.1 | ML | (((((Hs,Md),((Ol,Fr),Dr)),((Sh,Ry),Cm)),Ci),Ds); | 381 | 0.35 |
| 2 | Peptidylprolyl isomerase (cyclophilin)-like 5 isoform | NP_689542.2 | 4.93±7.94 | ((((((((Hs,Md),Gg),Xt),((Sh,Ry),Cm)),(Fr,Ol)),Dr),Ci),Dm); | 225 | 1.30 |
| 3 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 20 | NP_009135.3 | 2.84±2.98 | (((((Ry,Sh),Cm),(((Hs,Md),Gg),Xt)),(((Ol,Fr),Dr),Ci)),Dm); | 171 | 0.67 |
| 4 | Sorting nexin 6 isoform b | NP_689419.2 | 2.22±4.89 | (((((((((Hs,Md),Gg),Xt),Ol),Dr),Fr),((Cm,Ry),Sh)),Ci),Dm); | 144 | 0.44 |
| 5 | ATP/GTP binding protein-like 5 isoform 1 | NP_068603.4 | 0.38±6.55 | (((((Hs,Md),(Xt,Dr)),(Ol,Fr)),((Sh,Ry),Cm)),Ci); | 157 | 0.43 |
| 6 | Testis-specific gene A2 | NP_543136.1 | 2.40±3.19 | ((((Hs,Md),Xt),(((Fr,Ol),Dr),((Ry,Sh),Cm))),Ci); | 159 | 0.60 |
| 7 | Glutamyl-prolyl tRNA synthetase | NP_004437.2 | 3.35±3.90 | (((((((Sh,Ry),Cm),(Ol,Fr)),Xt),Gg),(Hs,Md)),Ci); | 142 | 0.43 |
| 8 | Growth arrest-specific 8 | NP_001472.1 | 1.20±2.72 | (((((((Hs,Md),Xt),Gg),Dr),((Sh,Ry),Cm)),Ci),Dm); | 150 | 0.81 |
| 9 | RAN binding protein 1 | NP_002873.1 | 1.23±2.35 | (((((Hs,(Gg,Md)),((Ol,Fr),Dr)),Xt),((Ry,Sh),Cm)),Ci); | 119 | 0.38 |
| 10 | Ring finger protein 10 | NP_055683.3 | 0.72±1.44 | (((((((Hs,Md),Gg),Xt),((Sh,Ry),Cm)),((Fr,Ol),Dr)),Ci),Dm); | 118 | 0.95 |
| 11 | MDN1, midasin homolog | NP_055426.1 | 3.42±3.76 | ((((((Hs,(Gg,Md)),(Sh,Ry)),Cm),((Ol,Fr),Dr)),Ci),Dm); | 121 | 0.55 |
| 12 | RAB, member RAS oncogene family-like 5 | NP_073614.1 | 0.11±4.21 | (((((Hs,Md),Xt),Gg),((Ol,Dr),((Sh,Ry),Cm))),Ci); | 108 | 2.05 |
| 13 | IWS1 homolog | NP_060439.1 | ML | (((((((Hs,Md),Xt),Gg),(Fr,Ol)),((Cm,Sh),Ry)),Ci),Dm); | 117 | 1.21 |
| 14 | Trinucleotide repeat containing 5 | NP_006577.2 | 1.30±2.09 | ((((((Hs,Md),Gg),((Ol,Fr),Dr)),Xt),((Sh,Ry),Cm)),Dm); | 107 | 0.77 |
| 15 | M-phase phosphoprotein 10 | NP_005782.1 | 2.19±2.30 | (((((((Hs,Md),Gg),Xt),(Dr,Fr)),(Sh,Ry)),Cm),Dm); | 98 | 0.88 |
| 16 | WD40 Protein Ciao 1 protein | NP_004795.1 | 1.26±2.04 | ((((((Hs,Md),(Xt,Gg)),(Fr,Dr)),((Sh,Ry),Cm)),Ci),Dm); | 98 | 0.53 |
| 17 | Ceroid-lipofuscinosis, neuronal 5 (CLN5) | NP_006484.1 | 0.60±1.18 | ((((Hs,Md),(Xt,Gg)),(Ol,Dr)),((Sh,Ry),Cm)); | 130 | 0.38 |
| 18 | CCR4-NOT transcription complex | NP_055331.1 | ML | (((((Hs,Md),Xt),((Fr,Ol),Dr)),((Cm,Sh),Ry)),Dm); | 126 | 0.57 |
| 19 | Splicing factor 3a, subunit 1 (SF3A1) | NP_005868.1 | 4.85±6.19 | ((((((((Hs,Md),Gg),Xt),Fr),Sh),((Cm,Ry),Ol)),Dr),Dm); | 171 | 0.17 |
| 20 | Thyroid hormone receptor interactor 12 (TRIP12) | NP_004229.1 | 0.63±4.44 | ((((Hs,(Md,Gg)),(((Ol,Fr),Dr),((Cm,Sh),Ry))),Xt),Ci); | 131 | 0.55 |
The log-likelihood difference (ΔlogL) between the ML tree and the topology based on the generally accepted species phylogeny is shown with standard error. The topology of the ML tree is shown in newick format. The number of amino acid sites used for tree inference is shown for each gene, as well as their corresponding shape parameter α for gamma distribution. Abbreviations: aa, amino acid; ML, maximum likelihood tree; Dm, Drosophila melanogaster; Ds, Drosophila simulans; Ci, Ciona intestinalis; Cm, chimaeras; Sh, sharks; Ry, rays/skates; Fr, Takifugu rubripes; Ol, Oryzias latipes; Dr, Danio rerio; Hs, Homo sapiens; Md, Monodelphis domestica; Gg, Gallus gallus; Xt, Xenopus tropicalis.
Figure 3. Phylogenetic tree ofRAN binding protein 1 genes.
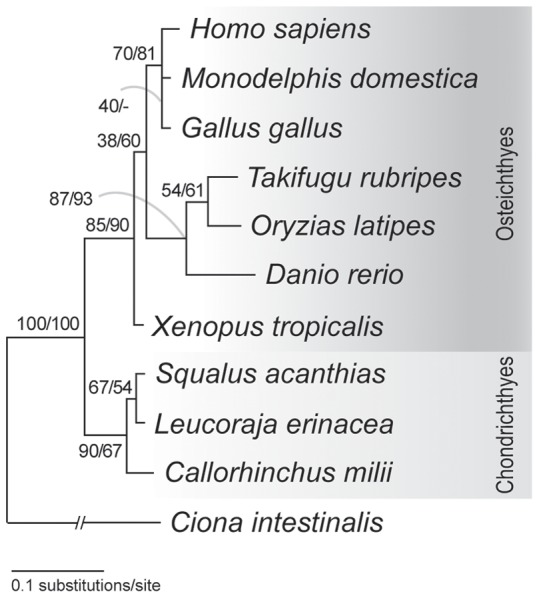
This gene is listed as candidate #9 in Table 1. The tree was reconstructed with the maximum-likelihood (ML) method (see Methods). Bootstrap values were calculated with 100 resamplings. Support values at nodes indicate, in order, probabilities in the ML and the neighbor-joining (NJ) analysis. 119 amino acid sites were included for tree inference (shape parameter for gamma distribution α = 0.38). Note that the topology of this ML tree is not consistent with the generally accepted species phylogeny, but the log-likelihood of the tree topology consistent with the species phylogeny was not significantly lower than that of the ML tree (Table 1). For this reason, this gene was included in the final dataset.
After this assessment, 20 candidate genes remained in the final dataset with each of them containing from 98 [candidate #15: M-phase phosphoprotein (MPP) 10] to 381 [candidate #1: phosphoglycerate kinase (PGK) 1] amino acids in the gene-by-gene alignments (Table 1). The total number of amino acids in the final concatenated dataset was 2973.
Divergence time estimation employing nuclear protein-coding genes
Holocephali-Elasmobranchii split
With the concatenated dataset, we first inferred the divergence time between Holocephali and Elasmobranchii (Figure 1). We employed fossil-based time constraints, which had two options for the split between Batoidea and Selachimorpha (node 11) (Table S2). In fact, the difference between the two options for the relatively young node did not influence the results of the time inference for the older node, namely the Holocephali-Elasmobranchii split. Our MCMCTREE analysis resulted in the estimated divergence time of about 421 Ma as posterior mean in late Silurian and a 95% confidence interval (CI) of 410–441 Ma, for the Holocephali-Elasmobranchii split (node 10 in Table 2 and Figure 4). In Figure 4, above the 95% CI time bars in the graph, the marginal densities are shown in light grey. Notably, the median divergence time (depicted above the 95% CI time bars) for node 10 is with a value of 419 Ma slightly younger than the mean value, which however aims in the direction of the previous assumed time points of this divergence [5], [15] (Figure 4).
Table 2. Estimated divergence times.
| Node # | Constraint set I | Constraint set II | ||
| Time | CI | Time | CI | |
| 1 | 566 | 536–582 | 566 | 536–582 |
| 2 | 547 | 520–576 | 547 | 520–576 |
| 3 | 454 | 437–464 | 454 | 437–464 |
| 4 | 418 | 416–421 | 418 | 416–421 |
| 5 | 343 | 331–351 | 343 | 331–351 |
| 6 | 319 | 312–330 | 319 | 312–330 |
| 7 | 148 | 124–171 | 148 | 124–171 |
| 8 | 157 | 149–163 | 157 | 149–163 |
| 9 | 112 | 95–139 | 112 | 95–138 |
| 10 | 421 | 410–441 | 420 | 410–440 |
| 11 | 306 | 252–387 | 261 | 193–373 |
Divergence time estimates (posterior mean) and their 95% confidence intervals (CIs) in Ma (million years from present) for eleven nodes indicated in Figure 4. See Table S2 for details of the time constraints. Different minimum constraints of node #11 resulted in different divergence time estimates.
Figure 4. Estimated timetree of vertebrates.

Timetree produced by MCMCTREE in PAML 4.4 [62] implementing the relaxed molecular clock method. A total of 19 time constraints (see Table S2) used for the calculation are shown as arrowheads at the eleven nodes. 2973 amino acid sites were analyzed derived from a total of 20 nuclear genes. Horizontal bars indicate 95% confidence intervals (CI) of the divergence time estimates. All estimates and 95% CIs are listed in Table 1. The marginal densities obtained in TRACER 1.5 are shown in light grey above the bars. Rates given by MCMCTREE are shown above the individual branches.
Batoidea-Selachimorpha split
Our second analysis target, the split between Batoidea and Selachimorpha in the Elasmobranchii lineage (node 11; also see Figure 1), was estimated at 306 Ma in the late Carboniferous period, with a 95% CI of 252–387 Ma based on constraint set I (Table 2 and Figure 4). On the other hand, based on constraint set II, this divergence was estimated at 261 Ma in the late Permian with a 95% CI of 193–373 Ma (Table 2). These 95% CIs are remarkably large presumably because of the missing constraint for any younger node. However, the estimated divergence time of 306 Ma (based on constraint set I), which is already biased through our lower constraint of 250 Ma [19], is consistent with palaeontological records. The median of the calculated marginal densities again reveals a younger divergence time of 300 Ma, which slightly aims at the direction of the previous assumed time points of divergence [15], [19].
Molecular evolutionary rates in chondrichthyan lineages
Our application of the calculated divergence time of 306 Ma, as well as previously estimated divergence times to the Hox A cluster sequences (see Methods), revealed much lower absolute rates in the cartilaginous fish lineages in comparison to tetrapod species (Table 3). The final comparison showed an increasing evolutionary rate in Hox A genes from H. francisci to X. tropicalis (sorted from lower rate to higher rate: Hf, Sc, Le, Gg, Hs, Ac, Xt). As depicted, H. francisci showed the lowest evolutionary rate of the three chondrichthyans included in our study, namely 0.004×10−8 substitutions/site/year. L. erinacea as batoid representative showed the highest rate in this group with 0.009×10−8 substitutions/site/year, but highly similar to the rate of S. canicula (0.008×10−8 substitutions/site/year). The human Hox A genes revealed averagely an evolutionary rate of 0.028×10−8 substitutions/site/year. This induces a 7.0 times to 3.1 times decreased rate for chondrichthyans in comparison to human, looking at species employed in our analysis (Table 3). Considering the four tetrapod representatives, G. gallus showed the lowest rate of sequence divergence, however only with a 1.3 times lower rate compared to the human, and 1.4 times decreased rate in comparison to A. carolinensis (Table 3). By contrast with L. erinacea, chicken showed a 2.4 times higher evolutionary rate. This comparison, of a chondrichthyan representing the highest evolutionary rate, and a tetrapod representing the lowest evolutionary rate, reveals the largest difference between two species in our analysis, when rates are sorted according to values.
Table 3. Distances and evolutionary rates for chondrichthyan and tetrapod representatives.
| Species pair | Hf, Sc | Le, Sc | Hf, Le | Hs, Gg | Hs, Ac | Hs, Xt | ||||||
| # of aa sites | 3121 | 2891 | 3128 | 2341 | 2344 | 2748 | ||||||
| Divergence time (Ma) | 203 | 306 | 306 | 312 | 312 | 330 | ||||||
| Distance | Hf-Cm | 0.052 | Le-Cm | 0.073 | Hf-Cm | 0.054 | Hs-Cm | 0.223 | Hs-Cm | 0.205 | Hs-Cm | 0.212 |
| Sc-Cm | 0.063 | Sc-Cm | 0.068 | Le-Cm | 0.068 | Gg-Cm | 0.203 | Ac-Cm | 0.218 | Xt-Cm | 0.222 | |
| Hf-Sc | 0.027 | Le-Sc | 0.049 | Hf-Le | 0.038 | Hs-Gg | 0.155 | Hs-Ac | 0.183 | Hs-Xt | 0.200 | |
| O-Hf | 0.008 | O-Le | 0.027 | O-Hf | 0.012 | O-Hs | 0.087 | O-Hs | 0.085 | O-Hs | 0.095 | |
| O-Sc | 0.019 | O-Sc | 0.022 | O-Le | 0.027 | O-Gg | 0.068 | O-Ac | 0.098 | O-Xt | 0.105 | |
| Rate (×10−8) | O-Hf | 0.004 | O-Le | 0.009 | O-Hf | 0.004 | O-Hs | 0.028 | O-Hs | 0.027 | O-Hs | 0.029 |
| O-Sc | 0.009 | O-Sc | 0.007 | O-Le | 0.009 | O-Gg | 0.022 | O-Ac | 0.031 | O-Xt | 0.032 | |
Distances (number of substitutions per site) were calculated by codeml for different pairs of species using Callorhinchus milii (Cm) as outgroup. Applying divergence times estimated in this study (306 Ma) as well as previous studies (203 Ma, 312 Ma and 330 Ma), evolutionary rates were calculated for three chondrichthyans and four tetrapod species. Abbreviations: aa, amino acid; Ma, million years ago; Hf, Heterodontus francisci; Sc, Scyliorhinus canicula; Le, Leucoraja erinacea; Gg, Gallus gallus; Hs, Homo sapiens; Ac, Anolis carolinensis; Xt, Xenopus tropicalis; O, Last common ancestor of the two selected species.
We also applied the whole Hox A cluster nucleotide sequences including non – coding regions (see Methods). This analysis showed less difference in the overall substitution rates, but the same order of species was observed, when rates are sorted according to values. H. francisci resulted in the lowest rate, followed by S. canicula and L. erinacea. G. gallus, again, showed the lowest rate of substitution in comparison between tetrapod species (data not shown).
Discussion
Marker gene selection and orthology assessment
More and more abundant molecular sequence data has enabled so-called ‘phylogenomics’ reconstructing more precisely the evolutionary history of many species by combining a number of independent nuclear loci ([28], [29]; also see [24] for review). The study by Li et al. on ray-finned fish phylogeny was one of the first demonstrations of in silico selection of phylogenetic markers [30]. This approach has been adopted in phylogenetic analyses involving a variety of taxa [31]–[34]. Similarly, in this study, we organized an in silico pipeline to handle available transcriptomic and genomic sequence data and chose nuclear marker genes to establish a solid time scale for cartilaginous fish evolution.
For each selected candidate gene, orthology was carefully assessed during phylogenetic analyses (see Methods). We excluded several candidates in which the tree topology based on the generally accepted phylogenetic relationships is rejected with significant statistical confidence. The 20 genes employed in our analysis were accepted in the likelihood analyses to support the currently accepted species phylogeny, although sometimes not as the ML tree (Figure 3). As divergence time estimation should be based on dataset including only orthologs, paralogy which can be detected by the inconsistency of the tree topology with the accepted species tree can mislead divergence time estimate. In this sense, our careful assessment should have consolidated our orthologous sequence dataset.
Batoidea–Selachimorpha split in the late Carboniferious period
The earliest fossils assigned to Chondrichthyes are assigned to the Silurian (444–416 Ma), and later chondrichthyan fossils become more widespread in the Devonian (416–359 Ma) [5]. Based on the fossil evidence, Holocephali and Elasmobranchii are estimated to have diverged 410 Ma [5]. For our estimation of the Holocephali-Elasmobranchii split, likewise with Inoue et al. [15], we adopted the time constraint for the split between Osteichthyes and Chondrichthyes (Table S2) based on Benton et al. [35], to stabilize the adjacent younger node of the Holocephali-Elasmobranchii split. Thus, our analysis employing nuclear sequence data yielded a similar divergence time estimate of 421 Ma (410–441Ma).
Moreover, for the estimation of the Batoidea-Selachimorpha divergence time, a study by Klug et al. attracted our attention. In this study, the †Synechodontiformes were identified as monophyletic group, sister to all extant sharks [19]. Klug et al. discuss that this assignment makes it necessary to enlarge the concept of neoselachian systematics to include this completely extinct group, which is considered to represent stem-group neoselachians [19]. The earliest fossils of †Synechodontiformes were assigned to the Early Permian (295 Ma) [36], and Klug et al. concluded that the origin of neoselachians can be as young as the Late Permian about 250 Ma [19]. Applying this above-mentioned record of 250 Ma for the Batoidea-Selachimorpha divergence as a minimum time constraint, presuming Batoidea as sister group to Selachimorpha, our calculation resulted in a mean divergence time of 306 Ma (252–387 Ma). In comparison with Heinicke et al. indicating a much older divergence of 393 Ma [20], our estimate would need to assume a shorter ghost range (306–250 Ma) based on the fossil records for early divergences within Neoselachians.
Possible sources of further improvement
It could be practical to discuss what might higher the resolution of the study. One simplistic idea is to increase the sequence information. At the moment, there is no truly genome-wide resource for any chondrichthyan species – the C. milii genome was only highly partially sequenced, and the resulting assembly does not cover many universal genes [37]. It can be augmented with deep transcriptomic sequencing. Apart from the amount of original sources, phylogenetic marker gene selection can also result in a remarkable difference. Our orthology assessment, primarily based on the Blast bit scores and secondarily on ML tree inferences, played a crucial role in removing any noisy data caused by possibly non-orthologous gene set for divergence time estimation. However, our rigorous criteria led to a relatively small number of phylogenetic marker genes (Table 1). While being aware of its risk to include possible cases with hidden paralogy, relaxing the selection criteria regarding orthology may result in a large increase in the number of genes in divergence time estimation. Another possible source of improvement lies in taxon sampling. Flexible choice of species in ingroup or outgroup may lead to reservation of more sites in the alignment used in divergence time estimation.
On the other hand, inclusion of more fossil records could also largely improve the results. In this study, divergence time constraints based on fossil records were narrow enough for non-chondrichthyan lineages. Apparently, fossil records in the chondrichthyan lineages are currently scarce and remain to be augmented by future effort.
Decreased molecular evolutionary rates in chondrichthyan lineages
The decreased rate of molecular evolution for a number of chondrichthyan species is already suggested since some chondrichthyan species have been subjected to various molecular analyses [21]–[25]. Mulley et al. suggested that the genomes of cartilaginous fish generally are more highly conserved than those of tetrapods or teleost fish, whereas for instance the investigation by Ravi et al. showed that teleosts show higher rate of chromosomal rearrangements and that protein-coding sequences in teleost fish genomes are evolving faster than in mammals [22], [38]. Based on the slow evolution observed in the individual genes of our study (Table 3), we support the hypothesis by Mulley et al. that the entire genomes of chondrichthyans may be evolving more slowly. On the other hand, the loss of the Hox C cluster was implicated in the elasmobranch lineage [39], and this is a remarkable drastic change that has never been observed in any other jawed vertebrate lineage. This contrast, seen between different features of the genomes, demands a caution in discussing rates of molecular evolution.
A question whether rate of morphological evolution is associated with that of molecular evolution has been repeatedly discussed regarding so-called ‘living fossils’, such as tuatara [40], [41] and coelacanth [42], [43]. To date, embryonic development was investigated in detail for some chondrichthyan species including the small spotted catshark Scyliorhinus canicula [44], the chimaera Callorhinchus milii [45] and the clearnose skate, Raja eglanteria [46]. Including more representatives of currently missing lineages, it is expected to get thorough and quantitative framework of morphological evolution. Our analysis of molecular evolutionary rate focusing on Hox A cluster resulted in at least 2.4-fold decrease in the chondrichthyan lineage in comparison to the tetrapod lineage, which remains to be confirmed at a genomic scale.
Conclusions
Large-scale data of nuclear gene sequences for chondrichthyan species were collected. We employed the amino acid sequence alignment with 2973 unambiguously aligned amino acid sites of 20 protein-coding genes in Bayesian-based divergence time analyses. Assuming an origin of sharks in the late Permian no less than 250 Ma and the batoids as sister clade to sharks within Neoselachii, result in a mean divergence time of 306 Ma for the Selachimorpha-Batoidea split.
Moreover, our analysis revealed a lower rate of molecular evolution for chondrichthyan lineages by a factor of at least 2.4.
Methods
Bioinformatic analysis pipeline
The procedure for this step is outlined in Figure 2. Expressed sequence tags (ESTs) of four elasmobranch species, Squalus acanthias [47], Leucoraja erinacea [47], Chiloscyllium plagiosum and Torpedo californica were downloaded from NCBI and assembled with the program Phrap [48], [49] with the default parameters. All resulting contigs and singletons were comprehensively translated into peptide sequences with all possible six open reading frames. The genome sequences of Callorhinchus milii were subjected to ab initio gene prediction with the program GenScan [50]. Using each peptide sequence of the predicted C. milii genes plus all annotated peptide sequences for chimaeras as a query, Blastp searches were performed towards three peptide sequence databases for 1) sharks, 2) rays/skates and 3) human (as outgroup). The human sequences were retrieved from NCBI RefSeq. The shark and ray/skate sequence collections included annotated peptide sequences retrieved from NCBI GenBank. As the preliminary orthology assessment step, the bit scores of the Blastp searches [51] were evaluated so that C. milii query sequences whose bit score in the search towards human (S co) is smaller than those in the search towards both sharks and rays/skates (S cs and S cr). As a result, 203 genes were passed onto downstream analyses.
Phylogenetic analysis and taxon sampling
For each of the 203 selected candidate genes which passed the preliminary orthology assessment step (Figure 2), homologous sequences of bilaterians to each C. milii sequences selected in the preliminary orthology assessment step were retrieved from NCBI GenBank and Ensembl to build molecular phylogenetic trees. First a multiple alignment of amino acid sequences was constructed using the alignment editor XCed in which the alignment algorithm MAFFT is implemented [52]. The 5′ and 3′ ends of the alignment that contained large stretches of missing data, compared with the chondrichthyan species, were truncated to include the most efficient number of unambiguously aligned sites and resulted in alignments between 98 aa [for M-phase phosphoprotein 10 (MPP10) gene] and 381 aa [for phosphoglycerate kinase 1 (PGK1) gene].
Preliminary neighbor joining (NJ) trees [53] were inferred on XCed. The candidate genes in which a rough orthology could be confirmed were further analyzed in more detail, by inferring their trees by the maximum-likelihood (ML) method [54] using PHYML version 2.4.4 [55], assuming the JTT+I+Γ4 model. Generally, to avoid wrong assignment of orthology we started with a more extensive dataset including a large number of bilaterian sequences. In several rounds of NJ and ML analyses, we basically retained orthologs of 12 core species for the final data set with few exceptions (Table S1). This data set included four representatives of tetrapods (Homo sapiens, Monodelphis domestica, Gallus gallus and Xenopus tropicalis), three teleost fish species (Oryzias latipes, Takifugu rubripes and Danio rerio) and three selected chondrichthyans, namely one representative of each lineage (Holocephali, Batoidea and Selachimorpha). Ciona intestinalis and Drosophila melanogaster were used as outgroup.
In each step of refinement, we needed to be careful in use of these nuclear genes not to overlook ‘hidden paralogy’ [56], [57]. For instance, secondary losses or delayed identifications of gene family members after the 2R-WGDs [58] could lead to a confusing pattern, in which paralogous sequences appear to be orthologous [59]. For candidate genes in which a pattern of the teleost-specific genome duplication (TSGD) was observed [60], the more divergent subtype of each teleost fish species was excluded for further analyses. If the orthology of a candidate could not be confirmed, these sequences were not incorporated in the following phylogenetic analyses. Generally, resultant tree topologies were assessed in light of the tree topology supported by Kikugawa et al. [1].
For all groups in which an orthology could be confirmed but differences of relationships between groups emerged, we finally calculated the log-likelihoods (logL) of two trees. First, applying the currently accepted species phylogeny and second, assuming the phylogeny maintained in the ML analysis, using TREE-PUZZLE version 4.2 [61]. We compared the determined likelihoods of the two trees and considered their difference (ΔlogL). If the standard error (SE) of the second-best calculated likelihood was larger than the ΔlogL of the two trees, these provided candidate genes were included in further analysis for divergence time estimation (ΔlogL/SE<1, accepted; ΔlogL/SE>1, rejected).
Divergence time inference
For the divergence time analysis, a Bayesian-based method implemented in the MCMCTREE program in the PAML 4.4 package [62] was used, implying a relaxed molecular clock that take into account rate variation across lineages (clock = 2) [63]. The unambiguously assigned alignment sites of each candidate were concatenated to one input file. The assumed tree topology, accepted for all single genes in the likelihood analysis, was applied and each node outside the chondrichthyan lineage, except Chordata, was constrained by a soft minimum and maximum calibration point (Table S2) based on the fossil record [35]. For the Selachimorpha/Batoidea split, in one analysis, a calibration point of 190 Ma was adopted according to a previous study [15] and in another, we adopted a lower constraint of 250 Ma based on a recently reported fossil record [19]. Inside the chondrichthyan lineage only a lower hard time constraint was applied. Using CODEML implemented in the PAML package, the substitution rate and the gamma shape parameter alpha was estimated using the JTT model [64]. Two priors were set for the final MCMCTREE analysis, the overall substitution rate (rgene_gamma) at G (1, 5.2) and the rate-drift parameter (sigma2_gamma) at G (1, 5.6) for our dataset containing 2973 amino acids (20 nuclear genes).
One time unit was used as 100 million years, because the node ages should fall between 0.01 and 10 [62]. Markov chain Monte Carlo (MCMC) approximation with a burn-in period of 50,000 cycles was obtained, and every 20 cycles were taken to create a total of 200,000 samples. Two replicates with different random seed numbers were performed to work out possible failure of the Markov chains to converge to their stationary distribution [15]. After completion of the analysis the output was plotted in TRACER 1.5 (available at http://beast.bio.ed.ac.uk/Tracer) to re-examine their convergence and to obtain the marginal densities of the calculated mean (Figure 4).
Quantification of evolutionary rates
To infer evolutionary rates of three chondrichthyan representatives [Scyliorhinus canicula (Sc), Leucoraja erinacea (Le) and Heterodontus francisci (Hf)] in comparison to tetrapods [human (Hs), chicken (Gg), Anolis carolinensis (Ac) and Xenopus tropicalis (Xt)] we downloaded all available amino acid sequences of Hox A protein-coding genes from GenBank [65] or Ensembl [66] for the above-mentioned species as well as for the elephant shark [Callorhinchus milii (Cm)] (accession numbers: Table S3). All available Hox A genes (HoxA1-A7, A9-A11 and A13) were concatenated into one sequence file for each species and we created six amino acid alignment datasets using the alignment editor XCed. The numbers of unambiguously aligned amino acid sites in Hox A protein-coding alignments are found in Table 3. These sites were extracted into a new input file and the distances (number of substitutions per site) between each pair of species were calculated running CODEML of the PAML 4.4 package [62], applying the JTT model [65]. A loose bound for the root was set at 420 Ma (in dataset 1–3), and at 450 Ma (in dataset 4–6). To further calculate the distance of each ingroup species (A, B) to a hypothetical ancestor (O) a relative rate test was performed with an outgroup (C) as below.
To infer absolute rates of evolution, the calculated distances (K OA and K OB) were divided through our estimated divergence time (306 Ma for shark-ray, dataset 2 and 3), as well as previously estimated divergence times (203 Ma [15] for Hf-Sc in dataset 1, 312 Ma [35] for Hs-Gg and Hs-Ac in dataset 4 and 5, and 330 Ma [35] for Hs-Xt, respectively) and thus we performed a quantification of divergence rates by pairwise comparison.
In a supplementary investigation, we employed the complete Hox A cluster nucleotide sequences to calculate pairwise distances and evolutionary rates of species described above. We downloaded sequence data of whole Hox A clusters from Ensembl [66]: Hs (GhRC37, chromosome 7, base position 27128722 to 27250000), Gg (WASHUC2.1, chromosome 2, base position 32508052 to 32636817), Ac (AnoCar2.0, Scaffold GL343275.1, base position 1364049 to 1613271, reverse complement (r.c.)) and Xenopus tropicalis (assembly 4.2 by the Joint Genome Institute, Scaffold GL172692.1, base position 1377582 to 1529191, r.c.) and GenBank [65]: Sc (FQ032658.1, r.c.), Le (FJ944024.1, r.c.), Hf (AF224262.1, r.c.; AF479755.1, r.c.) and Cm (FJ824598.1). All Hox A cluster sequences were aligned using mVista [67]. The multiple alignment was transferred to MEGA 5 and a complete deletion of gaps and missing data was set. This condition finally resulted in 8081 nucleotide sites, for which pairwise distances were computed [68] with the p-distance method.
Supporting Information
List of species included in this analysis.
(PDF)
List of time constraints. Upper (U) and lower (L) time constraints in million years from present (Ma) applied for nodes in estimating divergence times in the chondrichthyan lineage.
(PDF)
Accession numbers of Hox A proteins employed in the evolutionary rate analysis. The sequences with the given accession numbers were retrieved from the NCBI or Ensembl database.
(PDF)
Acknowledgments
We thank John Mulley, Ziheng Yang, Jun Inoue, Shaohua Fan, Tereza Manousaki, and Jürgen Kriwet for valuable discussion and insightful comments. Our gratitude extends to two anonymous reviewers who gave us constructive comments about the manuscript.
Funding Statement
This work was supported by the Young Scholar Fund of the University of Konstanz and the research grant (KU2669/1-1) from the Deutsche Forschungsgemeinschaft (DFG) to SK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kikugawa K, Katoh K, Kuraku S, Sakurai H, Ishida O, et al. (2004) Basal jawed vertebrate phylogeny inferred from multiple nuclear DNA-coded genes. BMC Biol 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janvier P (1996) Early vertebrates. Oxford: Oxford University Press. [Google Scholar]
- 3.Nelson JM (1994) Fishes of the world. New York: Wiley. [Google Scholar]
- 4. Maisey JG (1984) Higher Elasmobranch Phylogeny and Biostratigraphy. Zool J Linn Soc-Lond 82: 33–54. [Google Scholar]
- 5.Coates MI, Sequeira SEK (2001) Early sharks and primitive gnathostome interrelationships. In: Ahlberg PE, editor. Major Events in Early Vertebrate Evolution Palaeontology, phylogeny, genetics and development. London: Taylor & Francis. 241–262. [Google Scholar]
- 6.Grogan ED, Lund R (2004) The origin and relationships of early chondrichthyes. In: Carrier JC, Musick JA, Heithaus MR, editors. Biology of sharks and their relatives. Boca Raton: CRC Press. 3–32. [Google Scholar]
- 7. Maisey JG (1986) Heads and tails: A chordate phylogeny. Cladistics 2: 201–256. [DOI] [PubMed] [Google Scholar]
- 8. Arnason U, Gullberg A, Janke A (2001) Molecular phylogenetics of gnathostomous (jawed) fishes: old bones, new cartilage. Zool Scr 30: 249–255. [Google Scholar]
- 9. Mallatt J, Winchell CJ (2007) Ribosomal RNA genes and deuterostome phylogeny revisited: more cyclostomes, elasmobranchs, reptiles, and a brittle star. Mol Phylogenet Evol 43: 1005–1022. [DOI] [PubMed] [Google Scholar]
- 10.Bigelow HB, Schroeder WC (1948) Sharks. In: Tee-Van J, Breder CM, Hildebrand SF, Parr AE, Schroeder WC, editors. Fishes of the western North Atlantic Part 1. New Haven: Sears Foundation for Marine Research, Yale University. 59–546. [Google Scholar]
- 11.Bigelow HB, Schroeder WC (1953) Sawfishes, guitarfishes, skates and rays. In: Tee-Van J, Breder CM, Hildebrand SF, Parr AE, Schroeder WC, editors. Fishes of the western North Atlantic Part 2. New Haven: Sears Foundation for Marine Research, Yale University. [Google Scholar]
- 12.Shirai S (1992) Squalean phylogeny: a new framework of “squaloid” sharks and related taxa. Sapporo: Hokkaido University Press. [Google Scholar]
- 13.de Carvalho MR (1996) Higher-level elasmobranch phylogeny, basal squaleans, and paraphyly. In: Stiassny MLJ, Parenti LR, Johnson GD, editors. Interrelationships of fishes. San Diego: Academic Press. 35–84. [Google Scholar]
- 14. Douady CJ, Dosay M, Shivji MS, Stanhope MJ (2003) Molecular phylogenetic evidence refuting the hypothesis of Batoidea (rays and skates) as derived sharks. Mol Phylogenet Evol 26: 215–221. [DOI] [PubMed] [Google Scholar]
- 15. Inoue JG, Miya M, Lam K, Tay BH, Danks JA, et al. (2010) Evolutionary origin and phylogeny of the modern holocephalans (Chondrichthyes: Chimaeriformes): a mitogenomic perspective. Mol Biol Evol 27: 2576–2586. [DOI] [PubMed] [Google Scholar]
- 16. Velez-Zuazo X, Agnarsson I (2011) Shark tales: a molecular species-level phylogeny of sharks (Selachimorpha, Chondrichthyes). Mol Phylogenet Evol 58: 207–217. [DOI] [PubMed] [Google Scholar]
- 17. Winchell CJ, Martin AP, Mallatt J (2004) Phylogeny of elasmobranchs based on LSU and SSU ribosomal RNA genes. Mol Phylogenet Evol 31: 214–224. [DOI] [PubMed] [Google Scholar]
- 18.Maisey JG, Naylor GJP, Ward DJ (2004) Mesozoic elasmobranchs, neoselachian phylogeny and the rise of modern elasmobranch diversity. In: Arratia G, Tintori A, editors. Mesozoic fishes 3– systematics, paleoenvironments and biodiversity. München: Verlag Dr. Friedrich Pfeil. 17–56. [Google Scholar]
- 19. Klug S (2010) Monophyly, phylogeny and systematic position of the dagger Synechodontiformes (Chondrichthyes, Neoselachii). Zool Scr 39: 37–49. [Google Scholar]
- 20.Heinicke M, Naylor G, Hedges S (2009) Cartilaginous fishes (Chondrichthyes). In: Kumar S, Hedges S, editors. The Timetree of Life. New York: Oxford University Press. 320–327. [Google Scholar]
- 21. Martin AP, Naylor GJ, Palumbi SR (1992) Rates of mitochondrial DNA evolution in sharks are slow compared with mammals. Nature 357: 153–155. [DOI] [PubMed] [Google Scholar]
- 22. Mulley JF, Zhong YF, Holland PW (2009) Comparative genomics of chondrichthyan Hoxa clusters. BMC Evol Biol 9: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J, Lee AP, Kodzius R, Brenner S, Venkatesh B (2009) Large number of ultraconserved elements were already present in the jawed vertebrate ancestor. Mol Biol Evol 26: 487–490. [DOI] [PubMed] [Google Scholar]
- 24. Martin AP (1999) Substitution rates of organelle and nuclear genes in sharks: implicating metabolic rate (again). Mol Biol Evol 16: 996–1002. [DOI] [PubMed] [Google Scholar]
- 25. Martin AP, Palumbi SR (1993) Body Size, Metabolic-Rate, Generation Time, and the Molecular Clock. Proc Natl Acad Sci U S A 90: 4087–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hedges SB, Kumar S (2009) The Timetree of Life. New York: Oxford University Press. [Google Scholar]
- 27. Iglesias SP, Lecointre G, Sellos DY (2005) Extensive paraphylies within sharks of the order Carcharhiniformes inferred from nuclear and mitochondrial genes. Mol Phylogenet Evol 34: 569–583. [DOI] [PubMed] [Google Scholar]
- 28. Delsuc F, Brinkmann H, Philippe H (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6: 361–375. [DOI] [PubMed] [Google Scholar]
- 29. Philippe H, Delsuc F, Brinkmann H, Lartillot N (2005) Phylogenomics. Annu Rev Ecol Evol Syst 36: 541–562. [Google Scholar]
- 30. Li C, Orti G, Zhang G, Lu G (2007) A practical approach to phylogenomics: the phylogeny of ray-finned fish (Actinopterygii) as a case study. BMC Evol Biol 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Townsend TM, Alegre RE, Kelley ST, Wiens JJ, Reeder TW (2008) Rapid development of multiple nuclear loci for phylogenetic analysis using genomic resources: an example from squamate reptiles. Mol Phylogenet Evol 47: 129–142. [DOI] [PubMed] [Google Scholar]
- 32. Shen XX, Liang D, Wen JZ, Zhang P (2011) Multiple genome alignments facilitate development of NPCL markers: a case study of tetrapod phylogeny focusing on the position of turtles. Mol Biol Evol 28: 3237–3252. [DOI] [PubMed] [Google Scholar]
- 33. Aschliman NC, Nishida M, Miya M, Inoue JG, Rosana KM, et al. (2012) Body plan convergence in the evolution of skates and rays (Chondrichthyes: Batoidea). Mol Phylogenet Evol 63: 28–42. [DOI] [PubMed] [Google Scholar]
- 34. Li C, Matthes-Rosana KA, Garcia M, Naylor GJ (2012) Phylogenetics of Chondrichthyes and the problem of rooting phylogenies with distant outgroups. Mol Phylogenet Evol 63: 365–373. [DOI] [PubMed] [Google Scholar]
- 35.Benton M, Donoghue P, Asher R (2009) Calibrating and constraining molecular clocks. In: Kumar S, Hedges S, editors. The Timetree of Life. Oxford: Oxford University Press. 35–86. [Google Scholar]
- 36. Ivanov A (2005) Early Permian chondrichthyans of the Middle and South Urals. Revista Brasileira de Paleontologia 8: 127–138. [Google Scholar]
- 37. Dessimoz C, Zoller S, Manousaki T, Qiu H, Meyer A, et al. (2011) Comparative genomics approach to detecting split-coding regions in a low-coverage genome: lessons from the chimaera Callorhinchus milii (Holocephali, Chondrichthyes). Brief Bioinform 12: 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ravi V, Venkatesh B (2008) Rapidly evolving fish genomes and teleost diversity. Curr Opin Genet Dev 18: 544–550. [DOI] [PubMed] [Google Scholar]
- 39. King BL, Gillis JA, Carlisle HR, Dahn RD (2011) A natural deletion of the HoxC cluster in elasmobranch fishes. Science 334: 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hay JM, Subramanian S, Millar CD, Mohandesan E, Lambert DM (2008) Rapid molecular evolution in a living fossil. Trends Genet 24: 106–109. [DOI] [PubMed] [Google Scholar]
- 41. Miller HC, Moore JA, Allendorf FW, Daugherty CH (2009) The evolutionary rate of tuatara revisited. Trends Genet 25: 13–15. [DOI] [PubMed] [Google Scholar]
- 42. Amemiya CT, Powers TP, Prohaska SJ, Grimwood J, Schmutz J, et al. (2010) Complete HOX cluster characterization of the coelacanth provides further evidence for slow evolution of its genome. Proc Natl Acad Sci U S A 107: 3622–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Casane D, Laurenti P (2013) Why coelacanths are not ‘living fossils’: A review of molecular and morphological data. Bioessays 35: 332–338. [DOI] [PubMed] [Google Scholar]
- 44. Ballard WW, Mellinger J, Lechenault H (1993) A Series of normal stages for development of Scyliorhinus canicula, the lesser spotted dogfish (Chondrichthyes, Scyliorhinidae). J Exp Zool 267: 318–336. [Google Scholar]
- 45. Didier DA, Leclair EE, Vanbuskirk DR (1998) Embryonic staging and external features of development of the chimaeroid fish, Callorhinchus milli (Holocephali, Callorhinchidae). J Morphol 236: 25–47. [DOI] [PubMed] [Google Scholar]
- 46. Luer CA, Walsh CJ, Bodine AB, Wyffels JT (2007) Normal embryonic development in the clearnose skate, Raja eglanteria, with experimental observations on artificial insemination. Environ Biol Fish 80: 239–255. [Google Scholar]
- 47. Parton A, Bayne CJ, Barnes DW (2003) Analysis and functional annotation of expressed sequence tags from in vitro cell lines of elasmobranchs: Spiny dogfish shark (Squalus acanthias) and little skate (Leucoraja erinacea). Comp Biochem Physiol Part D Genomics Proteomics 5: 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordon D (2003) Viewing and editing assembled sequences using Consed. Curr Protoc Bioinformatics Unit 11.2. doi: 10.1002/0471250953.bi1102s02 [DOI] [PubMed]
- 49. Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8: 195–202. [DOI] [PubMed] [Google Scholar]
- 50. Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268: 78–94. [DOI] [PubMed] [Google Scholar]
- 51. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saitou N, Nei M (1987) The neighbor-joining method – a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425. [DOI] [PubMed] [Google Scholar]
- 54. Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17: 368–376. [DOI] [PubMed] [Google Scholar]
- 55. Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704. [DOI] [PubMed] [Google Scholar]
- 56. Daubin V, Gouy M, Perriere G (2001) Bacterial molecular phylogeny using supertree approach. Genome Inform 12: 155–164. [PubMed] [Google Scholar]
- 57. Gribaldo S, Philippe H (2002) Ancient phylogenetic relationships. Theor Popul Biol 61: 391–408. [DOI] [PubMed] [Google Scholar]
- 58. Dehal P, Boore JL (2005) Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol 3: e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kuraku S (2010) Palaeophylogenomics of the vertebrate ancestor – impact of hidden paralogy on hagfish and lamprey gene phylogeny. Integr Comp Biol 50: 124–129. [DOI] [PubMed] [Google Scholar]
- 60. Kuraku S, Meyer A (2009) The evolution and maintenance of Hox gene clusters in vertebrates and the teleost-specific genome duplication. Int J Dev Biol 53: 765–773. [DOI] [PubMed] [Google Scholar]
- 61. Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18: 502–504. [DOI] [PubMed] [Google Scholar]
- 62. Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- 63. Rannala B, Yang Z (2007) Inferring speciation times under an episodic molecular clock. Syst Biol 56: 453–466. [DOI] [PubMed] [Google Scholar]
- 64. Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8: 275–282. [DOI] [PubMed] [Google Scholar]
- 65. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2011) GenBank. Nucleic Acids Res 39: D32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Flicek P, Amode MR, Barrell D, Beal K, Brent S, et al. (2011) Ensembl 2011. Nucleic Acids Res 39: D800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.VISTA Genome Browser. Available: http://genome.lbl.gov/vista/index.shtml Accessed 2013 June 1.
- 68. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 28: 2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Compagno LJV, Didier DA, Burgess GH (2005) Classification of Chondrichthyan Fish. In: Fowler SL editor. Sharks, Rays, and Chimaeras, the Status of the Chondrichthyan Fishes. Gland: IUCN/SSC Shark Specialist Group. 4–11. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of species included in this analysis.
(PDF)
List of time constraints. Upper (U) and lower (L) time constraints in million years from present (Ma) applied for nodes in estimating divergence times in the chondrichthyan lineage.
(PDF)
Accession numbers of Hox A proteins employed in the evolutionary rate analysis. The sequences with the given accession numbers were retrieved from the NCBI or Ensembl database.
(PDF)