

Abstract
We used landscape genetics and statistical models to test how landscape features influence connectivity or create barriers to dispersal for a mountain riparian tree species, Euptelea pleiospermum. Young leaves from 1078 individuals belonging to 36 populations at elevations of 900–2000 m along upper reaches of four rivers were genotyped using eight nuclear microsatellite markers. We found no evidence for the unidirectional dispersal hypothesis in E. pleiospermum within each river. The linear dispersal pattern along each river valley is mostly consistent with the “classical metapopulaton” model. Mountain ridges separating rivers were genetic barriers for this wind-pollinated tree species with anemochorous seeds, whereas river valleys provided important corridors for dispersal. Gene flow among populations along elevational gradients within each river prevails over gene flow among populations at similar elevations but from different rivers. This pattern of gene flow is likely to promote elevational range shifts of plant populations and to hinder local adaptation along elevational gradients. This study provides a paradigm to determine which of the two strategies (migration or adaptation) will be adopted by mountain riparian plants under climate warming.
Introduction
Migration and adaptation are two main responses of mountain plants to climate warming [1]. The predominant direction of gene flow (along elevational gradients or among similar elevations) is of importance in determining which of the two strategies will be adopted [2], [3], [4], [5]. Here, we propose two models of gene flow (Fig. 1) for mountain plants inhabiting restricted linear habitats (e.g. ridge, streamside, valley or adjacent mountains etc.) that transverse elevation gradients. First, gene flow among populations at similar elevations but along adjacent linear habitats is higher than that within the same linear habitat at different elevations (Fig. 1A) because of similar microclimate and flowering times (phenology effect). Support for this model of gene flow has been shown for Poa hiemata [2] and Castanopsis eyrei [3]. Second, gene flow is more likely to occur within each linear habitat than between them (Fig. 1B) because of barriers (e.g. valley or ridge) between linear habitats along elevational gradients (geographic barrier effect). Support for this model has been found in Ainsliaea faurieana [6] and Silene ciliata [5]. The first model of gene flow (Fig. 1A) could promote local adaptation within an elevational zone but also potentially hinder elevational range shifts of plant populations, whereas the second model (Fig. 1B) would render the opposite trend.
Figure 1. Alternative models of gene flow for mountain plants inhabiting linear habitats along elevation gradients.
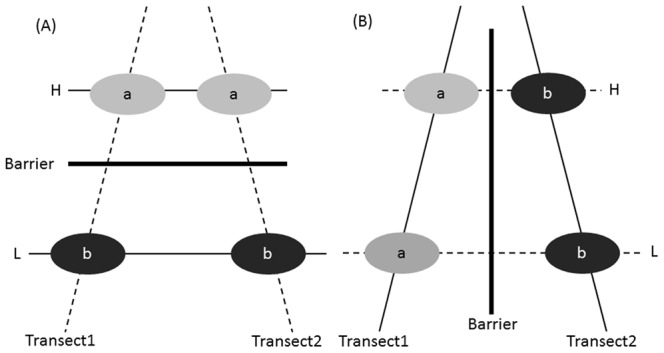
H, high elevation; L, low elevation. Different lower-case letters in different color ellipses represent different genetic groups.
For mountain riparian plants, gene flow is influenced by mountain landscape characteristics, such as elevation, stream flow, river valley and mountain ridge [6], [7], [8]. As dispersal is an important determinant of the level and direction of gene flow, it is essential for understanding how landscape features influence connectivity or create barriers to dispersal [9], [10], [11].
River valleys are long-recognized corridors for riparian or aquatic plants, which are characterized by linear distribution patterns [12], [13]. Previous studies have proposed six hypothetical models about the patterns of dispersal and connectivity between linearly arranged populations [12], [14]. The two extreme models are “spatially extended population” and “regional ensemble” [15] with free and without any ongoing gene flow among populations, respectively. For species characterized predominantly by unidirectional water-mediated dispersal, the linear asymmetrical adjacent flow model and the linear asymmetrical non-adjacent flow model were proposed [16]. The two models predict that downstream populations may harbor higher genetic diversity [17], and a few studies on riparian plants have confirmed this prediction [6], [18], [19]. However, wind- or animal-mediated upstream seed and/or pollen dispersal, which are likely to be promoted by valleys (corridors) along rivers, may weaken or counteract this cumulative effect [8], [12], [20], [21], [22]. Therefore, the two bidirectional dispersal models, the “classical stepping-stone” model and the “classical metapopulation” model, seem to be more common in studied linear populations, especially the latter.
Mountain ridge is the predominant factor causing limited gene flow and genetic differentiation in mountain plants [6], [23]. As streamside in mountainous areas is always at the bottom of a deep valley, linear distribution patterns constrained by highlands on both sides render populations with sharp boundaries and strong natural fragmentation; such spatial structure hampers gene flow among rivers. Therefore, populations at similar elevation along different rivers may be genetically separated by ridges. The linear distribution pattern can, in turn, facilitate gene flow within river valleys, especially for wind-dispersed plants. Therefore, we expected that gene flow of mountain riparian plants would be consistent with the second model (Fig. 1B).
Euptelea pleiospermum Hook. f. et Thoms (Eupteleaceae) is a diploid (2n = 28) [24], deciduous, broad-leaved Tertiary-relict tree species endemic to China, India and Burma [25]. In China, this species distributes across wide altitudinal ranges along streamsides (720–3600 m a.s.l.) [25]. As other riparian plant species, E. pleiospermum is restricted to linear suitable habitats. It is wind-pollinated and flowers in early spring before leaf-out. Adult trees produce numerous indehiscent fruits. The small and light samaras are dispersed first by gravity and secondarily by wind and/or water [26]. For this species, Li [27] has observed water-mediated seed dispersal in a downstream direction by simulation experiments with dyed real and artificial fruits.
The purpose of the present study is to examine the landscape genetic structure of Euptelea pleiospermum populations located along four main rivers (elevation gradients) within one mountain. Specially, the following questions were addressed: (1) does the recognized water-mediated seed dispersal result in genetic diversity accumulation in downstream populations? If not, which of the other linear models is suitable for populations within rivers? (2) Do river valleys and mountain ridges act as migration corridors and genetic barriers, respectively?
Materials and Methods
Ethics Statement
All necessary permits were obtained for the described field studies. Mr. Jingyuan Yang from the Administration Bureau of the Shennongjia National Nature Reserve issued the permission for each location.
Study Area and Sample Collection
The study area is located in mountain riparian forests of the Shennongjia National Nature Reserve (SNNR) (31°21′20″–31°36′20″ N, 110°03′05″–110°33′50″ E), central China (Fig. 2). Its summit Shennongding (3105.4 m) is the highest peak in central China [28]. SNNR is an important part of the south-central China biodiversity hot-spot and is rich of Tertiary relicts and endemic plants [29], [30]. There are four main river systems in the Shennongjia Mountains: Yandu River and Xiangxi River on the south-facing slope, which are tributaries of the Yangtze River, and Nan River and Du River on the north-facing slope, which are the tributaries of the Han River (Fig. 2). Euptelea pleiospermum is a common tree species in riparian forests at elevations between ∼1200 and ∼1900 m in this area [31].
Figure 2. Distribution of genetic diversity (A, allelic richness (A R) and B, expected heterozygosity (H E)) in 36 Euptelea pleiospermum populations along the four main rivers in the Shennongjia Mountains.

Circles indicate sampling locations, colors represent sample size (N), and circle size is proportional to the values of A R or H E. The white arrows indicate the flow direction of the four rivers (white lines).
Fieldwork was carried out in April and May 2008. Young leaves were collected and immediately dried in a 10 cm ×5 cm plastic bag containing silica gel. This was repeated every ∼100 m from high to low altitude along the four rivers. The sampling range depended on the altitudinal range of E. pleiospermum along each river. In this study, a total of 36 populations were sampled with six to 12 populations for each river (Fig. 2). Because of the sporadic distribution at some altitudes, sample sizes were sometimes relatively low, ranging from six to 61 per population (Table 1 and Fig. 2). A total of 1078 individuals were analyzed in this study.
Table 1. Characteristics of the 36 populations ofEuptelea pleiospermum sampled, including river, population name (Pop.), altitude, latitude (Lat.), longitude (Long.), sample size (N), mean number alleles per locus (A), allelic richness (A R), expected and observed heterozygosity (H E and H O), and inbreeding coefficient (F IS).
| River | Pop. | Altitude (m) | Lat. (N) | Long. (E) | N | A | A R | H E | H O | F IS |
| Yandu | Y1 | 2013 | 31°19.210′ | 110°26.269′ | 33 | 6.00 | 3.65 | 0.558 | 0.386 | 0.267 |
| Y2 | 1962 | 31°19.228′ | 110°26.236′ | 24 | 6.00 | 4.09 | 0.635 | 0.568 | 0.099 | |
| Y3 | 1805 | 31°19.331′ | 110°26.048′ | 6 | 3.88 | 3.88 | 0.606 | 0.521 | 0.245 | |
| Y4 | 1710 | 31°19.550′ | 110°26.031′ | 28 | 5.25 | 3.76 | 0.621 | 0.536 | 0.145 | |
| Y5 | 1640 | 31°19.569′ | 110°25.857′ | 40 | 6.63 | 4.38 | 0.686 | 0.581 | 0.148 | |
| Y6 | 1575 | 31°19.774′ | 110°25.930′ | 61 | 6.75 | 4.33 | 0.657 | 0.518 | 0.165 | |
| Y7 | 1471 | 31°19.960′ | 110°25.512′ | 41 | 6.63 | 4.45 | 0.687 | 0.534 | 0.226 | |
| Y8 | 1320 | 31°20.106′ | 110°25.103′ | 28 | 6.00 | 4.09 | 0.643 | 0.522 | 0.173 | |
| Y9 | 1270 | 31°20.404′ | 110°24.810′ | 11 | 4.63 | 3.80 | 0.558 | 0.455 | 0.169 | |
| Y10 | 1101 | 31°19.541′ | 110°23.851′ | 10 | 4.25 | 3.68 | 0.575 | 0.425 | 0.281 | |
| Y11 | 1005 | 31°19.516′ | 110°23.750′ | 37 | 6.25 | 3.79 | 0.555 | 0.463 | 0.169 | |
| Y12 | 980 | 31°19.351′ | 110°23.615′ | 32 | 6.13 | 4.18 | 0.613 | 0.430 | 0.268 | |
| Xiangxi | X1 | 2003 | 31°24.286′ | 110°19.525′ | 22 | 5.00 | 3.64 | 0.577 | 0.631 | –0.104 |
| X2 | 1910 | 31°24.387′ | 110°19.715′ | 42 | 5.88 | 3.98 | 0.639 | 0.512 | 0.183 | |
| X3 | 1850 | 31°25.095′ | 110°19.914′ | 39 | 6.13 | 3.74 | 0.636 | 0.535 | 0.148 | |
| X4 | 1715 | 31°24.948′ | 110°20.266′ | 32 | 6.63 | 4.41 | 0.664 | 0.586 | 0.096 | |
| X5 | 1690 | 31°25.057′ | 110°20.338′ | 31 | 6.50 | 4.58 | 0.659 | 0.411 | 0.335 | |
| X6 | 1520 | 31°25.438′ | 110°21.124′ | 33 | 6.25 | 4.18 | 0.611 | 0.564 | 0.072 | |
| X7 | 1478 | 31°25.629′ | 110°21.487′ | 47 | 6.50 | 4.12 | 0.658 | 0.545 | 0.135 | |
| X8 | 1389 | 31°26.015′ | 110°21.534′ | 25 | 5.13 | 4.01 | 0.646 | 0.540 | 0.145 | |
| X9 | 1270 | 31°25.886′ | 110°22.754′ | 22 | 5.50 | 3.96 | 0.651 | 0.483 | 0.271 | |
| X10 | 1190 | 31°26.586′ | 110°23.372′ | 29 | 6.13 | 4.15 | 0.645 | 0.509 | 0.217 | |
| Nan | N1 | 1904 | 31°29.936′ | 110°19.291′ | 36 | 5.88 | 3.71 | 0.622 | 0.604 | 0.027 |
| N2 | 1875 | 31°30.182′ | 110°19.523′ | 31 | 6.13 | 4.28 | 0.671 | 0.589 | 0.126 | |
| N3 | 1725 | 31°32.327′ | 110°20.158′ | 25 | 5.75 | 4.27 | 0.656 | 0.530 | 0.165 | |
| N4 | 1675 | 31°33.736′ | 110°21.564′ | 30 | 5.88 | 4.03 | 0.613 | 0.504 | 0.137 | |
| N5 | 1521 | 31°36.260′ | 110°24.575′ | 37 | 6.63 | 4.13 | 0.611 | 0.520 | 0.133 | |
| N6 | 1410 | 31°36.899′ | 110°25.569′ | 32 | 6.88 | 4.24 | 0.635 | 0.508 | 0.178 | |
| N7 | 1360 | 31°37.079′ | 110°26.047′ | 24 | 5.75 | 3.93 | 0.590 | 0.521 | 0.124 | |
| N8 | 1290 | 31°37.675′ | 110°27.039′ | 15 | 5.25 | 4.08 | 0.629 | 0.558 | 0.153 | |
| Du | D1 | 2070 | 31°28.307′ | 110°09.118′ | 26 | 5.88 | 4.14 | 0.654 | 0.572 | 0.120 |
| D2 | 1940 | 31°28.353′ | 110°08.966′ | 28 | 6.00 | 4.45 | 0.665 | 0.549 | 0.145 | |
| D3 | 1860 | 31°28.134′ | 110°08.860′ | 37 | 6.38 | 4.36 | 0.666 | 0.588 | 0.104 | |
| D4 | 1769 | 31°28.060′ | 110°08.596′ | 32 | 6.25 | 4.22 | 0.667 | 0.633 | 0.052 | |
| D5 | 1682 | 31°27.721′ | 110°08.117′ | 20 | 6.00 | 4.41 | 0.656 | 0.619 | 0.049 | |
| D6 | 1468 | 31°28.473′ | 110°01.316′ | 32 | 6.38 | 4.27 | 0.679 | 0.58 | 0.117 |
Bold values indicate significant departure from Hardy-Weinberg equilibrium.
DNA Extraction and Microsatellite Genotyping
All samples were stored at 4°C pending DNA extraction. Total DNA was extracted from approximately 0.5 g dried leaves using a modified CTAB method [32]. The DNA sample was diluted to 5–10 ng µL−1. Fourteen published primer pairs [33] were tested using 36 individuals (one individual randomly chosen from each population) and eight nuclear microsatellite loci (EP021, EP036, EP059, EP081, EP087, EP091, EP278, and EP294) that detected a suitable level of polymorphism were selected to determine genotypes of the 1078 samples.
The microsatellite genotyping procedure was performed as described by Wei and Jiang [34]. PCR amplifications were carried out in a total volume of 10 µL consisting of 10–50 ng of template DNA, 1 µL 10× reaction buffers, 1.5 mM of MgCl2, 0.2 mM of dNTPs, 0.25 µM of each primer, and 0.5 U of Taq polymerase (Tiangen Biotech Co., LTD., Beijing, China ). PCR reaction was conducted in a PTC-100 thermal cycler (MJ Research, Inc., Cambridge, MA, USA). PCR reactions were performed with the following cycle profile: an initial denaturation at 94°C for 5 min; then 35 cycles of denaturation at 94°C for 50 s, annealing at 56°C for 50 s and extension at 72°C for 1 min; and final extension at 72°C for 10 min. After amplification, identification of the alleles was based on the position of the fragments in relation to a 25-bp marker ladder on a polyacrylamide gel of high resolution with silver stain.
Data Analysis
GENETIX 4.05 [35] was used to calculate the following population diversity indices: mean number of alleles per locus (A), expected (H E) and observed (H O) heterozygosity, and inbreeding coefficient (F IS) across all loci and locus-population combinations. FSTAT 2.9.3.2 [36] was used to estimate allelic richness (A R) for each population and to test for linkage disequilibrium (LD). Departure from Hardy-Weinberg equilibrium (HWE) was tested by Fisher’s exact test in GENEPOP version 4.0 [37]. The significant values for the LD were corrected for multiple comparisons by Bonferroni correction [38]. We used the program MICRO-CHECKER [39] to test for null alleles.
To reveal the pattern of genetic diversity along rivers, regression analyses between genetic diversity parameters (H E and A R) and the position of populations along the course of the river were conducted using the SPSS statistics program (SPSS 15.0 for Windows; SPSS Inc., Chicago, IL, USA).
To provide direct evidence for the dispersal pattern along each river, contemporary and historical migration rates between populations along each river were calculated by using the programs BayesAss version 1.3 [40] and MIGRATE version 3.2.2.windows [41], respectively. BayesAss was run using a Markov chain Monte Carlo (MCMC) length of 3,000,000 with a burn-in period of 1,000,000 (initial conditions of Δp, Δm, and ΔF = 0.15). MIGRATE uses Bayesian inference with long chains (500,000 steps sampled, 5,000 steps recorded) and 1,000 burn-in per chain. We then compared mean values of downstream and upstream migration rates along each river valley, and significance was tested with a paired-samples t-test.
Pairwise population differentiation was estimated at two scales: among populations and among rivers. F ST [42] and D EST [43] were calculated using FSTAT 2.9.3.2 [36] and SMOGD 1.2.5 [44], respectively. Significance tests for F ST were based on 1000 randomizations. We compare genetic differentiation among populations from different rivers and that among populations within the same river (inter-F ST vs intra-F ST and inter-D EST vs intra-D EST, respectively), and significance was tested by performing an independent-samples t-test.
To analyze the level and partitioning of genetic variation among geographical groups, among populations within geographical groups, and within populations, analysis of molecular variance (AMOVA) [45] was performed with 1000 permutations in ARLEQUIN version 3.1 [46]. There were two kinds of geographical groups: populations along each river (Yandu, Xiangxi, Nan, and Du) and populations in each altitudinal zone (12, from 900 m to 2000 m).
To test whether the individuals from the same river valley or from similar altitudes can be clustered into genetically homogeneous groups, nonspatial and spatially sensitive Bayesian clustering were implemented by using STRUCTURE version 2.3.3 [47], [48], [49], [50] and TESS version 2.3.1 [51]. Previous STRUCTURE analysis employed a Markov chain Monte Carlo (MCMC) approach to cluster individuals into K panmictic groups without a priori assignment of individuals to sampling locations and by minimizing deviations from Hardy-Weinberg equilibrium and linkage equilibrium. The LOCPRIOR model, recently developed by Hubisz et al. [52], allowed us to infer weak population structure with the assistance of sample group information. To guide an empirical estimate of the number of genetic populations, we performed five replicates of each simulation from K = 1 to K = N P +3 (N P, the number of sampling locations), with a burn-in period of 100,000, and MCMC iterations of 1,000,000, based on the LOCPRIOR model and the admixture model with correlated allele frequencies. We applied Evanno et al.’s [53] ad hoc statistic, ΔK, to calculate the uppermost hierarchical level of structure (K). For the spatially sensitive TESS analysis (admixture model: BYM; spatial interaction parameter: 0.6), we set 10 runs per K max (maximal number of clusters), for K max ranging from two to six, with 300,000 total sweeps and a burn-in of 100,000.We used the program DISTRUCT version 1.1 [54] to create bar charts for the output data derived from STRUCTURE and TESS analyses.
To test for isolation by distance (IBD) [55], the correlation between genetic distances (F ST/(1–F ST)) and altitudinal distances (logarithmically transformed) among populations was executed using the Mantel test implemented in GenAlEx 6.41 [56]. We also performed partial Mantel test to check for the effect of altitudinal distance after accounting for geographical distance by using FSTAT 2.9.3.2 [36]. The IBD tests were performed at two spatial scales: each of the four rivers and the whole Shennongjia Mountains. Latitude and longitude coordinates for each population were used to calculate pairwise geographical distances between populations.
Results
Microsatellite Polymorphism, Hardy-Weinberg Equilibrium and Linkage Disequilibrium
A total of 102 alleles at eight nuclear microsatellite loci were revealed across 1078 individuals of 36 sampled populations of E. pleiospermum. At the species level, all eight nuclear microsatellite markers were highly polymorphic, having 6–29 alleles per locus with an average of 12.8. At the intra-population level, mean number of alleles per locus (A) ranged from 3.88 to 6.88, and allelic richness (A R) varied from 3.64 to 4.58 (Table 1 and Fig. 2A). The expected heterozygosity (H E) ranged from 0.555 to 0.687 (Fig. 2B), and observed heterozygosity (H O) ranged from 0.386 to 0.633 (Table 1). The level of inbreeding for each population ranged from –0.104 to 0.335 and significant inbreeding coefficients (F IS) were detected in 34 of 36 populations (Table 1).
The test for HWE found that 135 of 288 locus-population combinations were significant, including 32 at locus EP021, 21 at EP036, three at EP059, 17 at EP081, 22 at EP087, nine at EP091, eight at EP278 and 23 at EP294. However, no locus displayed consistent deviation from HWE across all populations. Furthermore, MICROCHECKER revealed potential null alleles in five of the 36 populations, with three at locus EP021 and two at EP297. This suggested that departure from HWE is potentially resulted from both technical (e.g. null allele) and biological (e.g. inbreeding) reasons. The test for genotypic disequilibrium found that only one (EP036×EP294) of 28 locus pairs showed significant genotypic disequilibrium in 12 populations. Because no consistent genotypic disequilibrium was found between any locus pair across all populations, all loci were used for further analyses.
Linear Population Model along Rivers
Linear regression analyses did not reveal any significant negative relationship between genetic diversity and the position of populations along the course of the rivers (Table 2 and Fig. 2), suggesting that there was no accumulation of genetic diversity in downstream populations. In accordance with this result, we found no significant difference between downstream and upstream migration rates, although the former was a little higher than the latter along all the four rivers (Table 3, Table S1 and Table S2).
Table 2. Correlation between genetic diversity (A R, Allelic richness and H E, expected heterozygosity) and the position of populations along the course of the river at different spatial scales.
| Position | A R | H E | ||
| r | p | r | p | |
| Shennongjia Mountain | 0.001 | 0.995 | −0.188 | 0.272 |
| Yandu River | 0.052 | 0.871 | −0.197 | 0.540 |
| Xiangxi River | 0.295 | 0.408 | 0.477 | 0.163 |
| Nan River | 0.127 | 0.764 | −0.467 | 0.244 |
| Du River | 0.122 | 0.818 | 0.646 | 0.166 |
Table 3. Comparison of downstream and upstream migration rates ofEuptelea pleiospermum along each of the four rivers (Mean ± S.E.).
| Contemporary migration rate | Historical migration rate | ||||||
| Downstream | Upstream | p | Downstream | Upstream | p | ||
| Yandu River | 0.023±0.006 | 0.016±0.005 | 0.461 | 24.05±3.33 | 17.68±1.51 | 0.107 | |
| Xiangxi River | 0.015±0.007 | 0.010±0.005 | 0.563 | 21.35±2.56 | 20.91±2.59 | 0.913 | |
| Nan River | 0.029±0.010 | 0.015±0.009 | 0.313 | 22.80±2.21 | 18.14±1.82 | 0.127 | |
| Du River | 0.038±0.017 | 0.023±0.006 | 0.351 | 25.52±4.80 | 24.36±4.45 | 0.868 | |
Contemporary and historical migration rates were calculated by using the programs BayesAss version 1.3 MIGRATE version 3.2.2., respectively.
Moderate levels of pairwise differentiation (F ST and D EST) were found between populations within each river valley. Significance test revealed that most values of F ST were significantly different from zero (Table S3).
Mantel test detected significant IBD within the Yandu River and Du River (Table 4). However, after controlling for geographical distance, the partial Mantel test detected no significant relationships between genetic and altitudinal distances within any of the four rivers (Table 4).
Table 4. Results of Mantel tests and partial Mantel test of pairwise genetic distance (F ST/(1–F ST)) and the natural logarithm of altitudinal distance for Euptelea pleiospermum at different spatial scales.
| Mantel test | Partial Mantel test | |||
| Altitudinal distance | r | p | r | p |
| Shennongjia Mountain | 0.149 | 0.000 | 0.142 | 0.000 |
| Yandu River | 0.517 | 0.000 | 0.175 | 0.163 |
| Xiangxi River | 0.118 | 0.439 | 0.090 | 0.560 |
| Nan River | 0.271 | 0.163 | −0.029 | 0.886 |
| Du River | 0.596 | 0.019 | 0.389 | 0.169 |
Roles of Mountain Ridges and River Valley
When individuals from the same river were seen as one population, all pairwise F ST values between rivers were significantly different from zero, and the pairwise D EST values also showed similar levels of genetic differentiation (Table 5).
Table 5. Matrix of pairwise differentiation (D EST, above diagonal and F ST, below diagonal) among rivers.
| Yandu River | Xiangxi River | Nan River | Du River | |
| Yandu River | – | 0.016 | 0.018 | 0.016 |
| Xiangxi River | 0.013 | – | 0.019 | 0.030 |
| Nan River | 0.026 | 0.015 | – | 0.027 |
| Du River | 0.012 | 0.015 | 0.026 | – |
All values of F ST are significant based on permutation tests following a sequential Bonferroni correction.
We found moderate levels of genetic differentiation among most pair of populations from different rivers (Table S3). Furthermore, average F ST values were higher for inter-river population pairs than for intra-river population pairs (F ST inter: 0.069±0.001; F ST intra: 0.061±0.003, P = 0.007), and D EST values showed the same trend (D EST inter: 0.091±0.002; D EST intra: 0.082±0.004, P = 0.032) (See Table S3 for details).
The results of the AMOVA were shown at Table 6. There was significant differentiation among different rivers (0.99%; P<0.001). However, no significant differentiation was found among populations located at different altitudinal zones (0.20%; P = 0.206).
Table 6. Effects of mountain ridges (A) and river valleys (B) on differences among 36Euptelea pleiospermum populations as determined by analysis of molecular variance (AMOVA).
| Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation | p |
| (A) | |||||
| Among rivers | 3 | 85.401 | 0.028 | 0.99 | <0.001 |
| Among populations within river valleys | 32 | 411.263 | 0.173 | 6.20 | <0.001 |
| Within populations | 2120 | 4598.853 | 2.594 | 92.81 | <0.001 |
| Total | 2155 | 5995.517 | 2.795 | ||
| (B) | |||||
| Among altitudinal zones | 11 | 170.286 | 0.005 | 0.20 | 0.206 |
| Among populations within altitudinal zones | 24 | 326.378 | 0.189 | 6.79 | <0.001 |
| Within populations | 2120 | 5498.853 | 2.594 | 93.02 | <0.001 |
| Total | 2155 | 5995.517 | 2.788 |
With the four rivers as sample group information, STRUCTURE analysis revealed a clear peak of Evanno et al.’s [53] ad hoc statistic ΔK at K = 4. The bar plot assuming K = 4 showed that individuals from different rivers were clustered into distinct groups (Fig. 3A). However, the same analysis did not clearly detect genetic structure when the 12 altitudinal zones were used in the LOCPRIOR model (data not shown). Results from TESS paralleled those obtained from STRUCTURE, although there are some minor differences (Fig. 3).
Figure 3. Population structure ofEuptelea pleiospermum estimated by the programs STRUCTURE 2.3.4 (A) and TESS 2.3.1 (B).
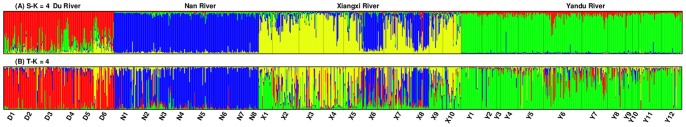
Each individual is represented by a thin vertical line, which is partitioned into four colored segments that represent the individual’s estimated membership fractions in the four clusters. Black vertical lines separate individuals of different populations. Labels below the plot provide population codes, which are the same as in Table 1. Labels above the plot (Du River, Nan River, Xiangxi River and Yandu River) are sampling information.
Both Mantel and partial Mantel tests found significant correlations between F ST-based genetic distances and altitudinal distances within the whole Shennongjia Mountains (Table 4).
Discussion
Linear Population Model within Rivers
We found no evidence for the unidirectional dispersal hypothesis in E. pleiospermum (Tables 2 and 3, Fig. 2), although water-mediated seed dispersal in a downstream direction has been observed [27]. In other words, upstream dispersal of pollen and seed mediated by wind are efficient mechanisms to eliminate the downstream accumulation of genetic diversity caused by water-mediated seed dispersal. Besides upstream dispersal, Honnay et al. [22] recently pointed out that higher seed recruitment opportunities in upstream habitats due to density dependence of recruitment is another explanation. However, this seems not the case for E. pleiospermum in the study area, because the pattern of genetic diversity along the Yandu River supports the central-marginal hypothesis [57].
The “regional ensemble” and “classical stepping-stone” models were rejected because IBD was not observed within any of the four rivers (Table 4), indicating that gene flow along each river was across a wide elevational range. The “spatially extended population”, a single genetically uniform panmictic unit, was also rejected because most of the genetic differentiation values among populations were significant. As inferred above, the bidirectional long-distance dispersal within each river valley and the significant genetic differentiation among populations indicated that the linear dispersal pattern for E. pleiospermum is most consistent with the “classical metapopulaton” model. This model was also confirmed in another riparian tree species, Populus nigra [20]; anemochory is the most important dispersal mode for this species as well.
Contrasting Roles of Mountain Ridges and River Valleys
Our results suggested that mountain ridges and river valleys played contrasting roles in shaping the genetic structure of E. pleiospermum at the landscape scale. Mountain ridges between rivers can be recognized as genetic barriers, while river valleys act as corridors for gene flow. In other words, gene flow of E. pleiospermum is consistent with the second model (Fig. 1B).
Four results lead to the conclusion that mountain ridges act as genetic barriers. First, individuals from each river were clustered into distinct groups (Fig. 3). Furthermore, the 12 altitudinal zones were not clustered into different groups by STRUCTURE analysis and this result suggested that populations from different rivers at similar elevations could not be clustered into the same group. Second, the F ST and D EST values for inter-river comparisons were higher than intra-river comparisons. This has been found in another mountain riparian plant, Ainsliaea faurieana [6]. Third, we found significant IBD in the whole Shennongjia Mountains but lack of IBD within all four rivers (Table 4). Fourth, when each river was seen as a population, we detected significant genetic differentiation among them (Table 5). This was confirmed by the results of AMOVA, which showed that there was significant differentiation among rivers, whereas no significant differentiation was found among altitudinal zones (Table 6). Mountain ridges have been found as genetic barriers for another wind dispersed tree species, Betula maximowicziana [23].
However, unambiguous evidence for gene flow among rivers is that the X6 (1520 m) and X8 (1389 m) populations from Xiangxi River were clustered into the Nan River group, as revealed by STRUCTURE and TESS analyses (Fig. 3). This can be partly explained by the ex situ conservation performed with plant materials collected from the Nan River. There is one national highway connecting the two rivers, and there was an ex situ conservation site at ∼1400 m along the Xiangxi River in the past. Furthermore, it has been reported that roads can serve as dispersal corridors for plants [58], [59]. However, it is not yet clear why X1 (2003 m) and X9 (1270 m) populations were clustered into the Yandu River group (Fig. 3B).
Our results also support that linear riparian zones in deep valleys provide important corridors for gene flow. First, as mentioned above, lack of IBD in each river indicated that gene flow along the river valley was not restricted (Table 4). This has previously been reported for riparian and aquatic plants [6], [14]. Second, as mentation above, the F ST and D EST values for inter-river comparisons were higher as compared to intra-river comparisons. Third, the Bayesian clustering approach divided individuals from the four rivers into four distinct groups (Fig. 3), indirectly indicating that gene flow within each river was relatively high.
Conclusions and Implications
We found no accumulation of genetic diversity at the downstream populations of E. pleiospermum within each of the four rivers. This was supported by the estimates of contemporary and historical migration rates, which revealed that the downstream migration rate was not significantly higher than that in upstream direction. The linear dispersal pattern along each river valley was mostly consistent with the “classical metapopulaton” model. Mountain ridges among rivers were genetic barriers for this wind-pollinated tree species with anemochorous seeds, whereas river valleys provided important corridors for dispersal. In summary, the streamside population of E. pleiospermum is consistent with the second model of gene flow (Fig. 1B) for mountain plants restricted to linear habitats along elevational gradients. Here, it should be mentioned that the variation of sample size, especially small number of sample in some of the populations (less than 20), may influence in our conclusions.
Gene flow among E. pleiospermum populations along elevational gradients within each river prevailed over gene flow among populations at similar elevations but from different rivers. This pattern of gene flow could promote elevational range shifts of plant populations but potentially hinder local adaptation along elevational gradients. Therefore, range shift along elevations is the most likely strategy for E. pleiospermum in the face of climate warming. As mountain riparian zones are crucial refuges for tertiary-relict plants [60], [61], [62], [63], this study provides a paradigm to determine which of the two strategies (migration or adaptation) will be adopted by these plants under climate warming.
Supporting Information
Contemporary migration rates between Euptelea pleiospermum populations along each river.
(DOC)
Historical migration rates between Euptelea pleiospermum populations along each river.
(DOC)
Pair-wise population D EST (above the diagonal) and F ST (below the diagonal).
(XLS)
Acknowledgments
We thank Dong He and Qiang Li for their assistance in sampling and lab work. We are indebted to Scott B. Franklin for reading and commenting on earlier draft of this manuscript. We also thank Dr. João Pinto and four anonymous reviewers for valuable comments on previous version of this manuscript.
Funding Statement
The study was supported by the National Natural Science Foundation of China (Nos. 31070465 and 31100344). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jump AS, Peñuelas J (2005) Running to stand still: adaptation and the response of plants to rapid climate change. Ecol Lett 8: 1010–1020. [DOI] [PubMed] [Google Scholar]
- 2. Byars SG, Parsons Y, Hoffmann AA (2009) Effect of altitude on the genetic structure of an alpine grass, Poa hiemata . Ann Bot 103: 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi MM, Michalski SG, Chen XY, Durka W (2011) Isolation by elevation: genetic structure at neutral and putatively non-neutral loci in a dominant tree of subtropical forests, Castanopsis eyrei . PLoS ONE 6(6): e21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hahn T, Kettle CJ, Ghazoul J, Frei ER, Matter P, et al. (2012) Patterns of genetic variation across altitude in three plant species of semi-dry grasslands. PLoS ONE 7(8): e41608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. García-Fernández A, Segarra-Moragues JG, Widmer A, Escudero A, Irondo JM (2012) Unravelling genetics at the top: mountain islands or isolated belts? Ann Bot 110: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mitsui Y, Isagi Y, Setoguchi H (2010) Multiple spatial scale patterns of genetic diversity in riparian populations of Ainsliaea faurieana (Asteraceae) on Yakushima Island, Japan. Am J Bot 97: 101–110. [DOI] [PubMed] [Google Scholar]
- 7. Prentis PJ, Mather PB (2008) Fine-scale patterns of genetic variation indicate non-equilibrium gene frequency divergence in the stream lily, Helmholtzia glaberrima . Freshwater Biol 53: 973–980. [Google Scholar]
- 8. Hu LJ, Uchiyama K, Shen HL, Ide Y (2010) Multiple-scaled spatial genetic structure of Fraxinus mandshurica over a riparian-mountain landscape in Northeast China. Conserv Genet 11: 77–87. [Google Scholar]
- 9. Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18: 189–197. [Google Scholar]
- 10. Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, et al. (2007) Putting the ‘landscape’ in landscape genetics. Heredity 98: 128–142. [DOI] [PubMed] [Google Scholar]
- 11. Storfer A, Murphy MA, Spear SF, Holderegger R, Waits LP (2010) Landscape genetics: where we are now? Mol Ecol 19: 3496–3514. [DOI] [PubMed] [Google Scholar]
- 12. Tero N, Aspi J, Siikamäki P, Jäkäläniemi A, Tuomi J (2003) Genetic structure and gene flow in a metapopulation of an endangered plant species, Silene tatarica . Mol Ecol 12: 2073–2085. [DOI] [PubMed] [Google Scholar]
- 13. Fér T, Hroudová Z (2008) Detecting dispersal of Nuphar lutea in river corridors using microsatellite markers. Freshwater Biol 53: 1409–1422. [Google Scholar]
- 14. Pollux BJA, Luteijn A, van Groenendael JM, Ouborg NJ (2009) Gene flow and genetic structure of the aquatic macrophyte Sparganium emersum in a linear unidirectional river. Freshwater Biol 54: 64–76. [Google Scholar]
- 15. Freckleton RP, Watkinson AR (2002) Large-scale spatial dynamics of plants: metapopulations, regional ensembles and patchy populations. J Ecol 90: 419–434. [Google Scholar]
- 16. Markwith SH, Scanlon MJ (2007) Multiscale analysis of Hymenocallis coronaria (Amaryllidaceae) genetic diversity, genetic structure, and gene movement under the influence of unidirectional stream flow. Am J Bot 94: 151–160. [DOI] [PubMed] [Google Scholar]
- 17. Ritland K (1989) Genetic differentiation, diversity, and inbreeding in the mountain monkeyflower (Mimulus caespitosus) of the Washington Cascades. Can J Bot 67: 2017–2024. [Google Scholar]
- 18. Liu Y, Wang Y, Huang H (2006) High interpopulation genetic differentiation and unidirectional linear migration patterns in Myricaria laxiflora (Tamaricaceae), an endemic riparian plant in the Three Gorges Valley of the Yangtze River. Am J Bot 93: 206–215. [DOI] [PubMed] [Google Scholar]
- 19. Kikuchi S, Suzuki W, Sashimura N (2009) Gene flow in an endangered willow Salix hukaoana (Salicaceae) in natural and fragmented riparian landscapes. Conserv Genet 12: 79–89. [Google Scholar]
- 20. Imbert E, Lefèvre F (2003) Dispersal and gene flow of Populus nigra (Salicaceae) along a dynamic river system. J Ecol 91: 447–456. [Google Scholar]
- 21. Prentis PJ, Vesey A, Meyers NM, Mather PB (2004) Genetic structuring of the stream lily Helmholtzia glaberrima (Philydraceae) within Toolona Creek, south-eastern Queensland. Aust J Bot 52: 201–207. [Google Scholar]
- 22. Honnay O, Jacquemyn H, Nackaerts K, Breyne P, Van Looy K (2010) Patterns of population genetic diversity in riparian and aquatic plant species along rivers. J Biogeogr 37: 1730–1739. [Google Scholar]
- 23. Tsuda Y, Sawada H, Ohsawa T, Nakao K, Nishikawa H, et al. (2010) Landscape genetic structure of Betula maximowicziana in the Chichibu mountain range, central Japan. Tree Genet Genomes 6: 377–387. [Google Scholar]
- 24. Pan KY, Lu AM, Wen J (1991) Chromosome number and development of gametophytes in Euptelea pleiospermum (Eupteleaceae). Acta Phytotaxon Sin 29: 439–444. [Google Scholar]
- 25.Fu LK, Jin JM (1992) China plant red data book: rare and endangered plants. Beijing: Science Press. 680–681 p.
- 26. Wei XZ, Liao JX, Jiang MX (2010) Effects of pericarp, storage conditions, seed weight, substrate moisture content, light, GA3 and KNO3 on germination of Euptelea pleiospermum . Seed Sci Technol 38: 1–13. [Google Scholar]
- 27.Li JX (2008) Seed dispersal and temporal-spatial pattern of rare and endangered species Euptelea pleiospermum in Shennongjia region, Hubei, China. Master thesis, Graduate University of Chinese Academy of Sciences, Beijing.
- 28.Zhu ZQ, Song CS (1999) Scientific survey of Shennongjia Nature Reserve. Beijing: China Forestry Publishing House.
- 29. Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403: 853–858. [DOI] [PubMed] [Google Scholar]
- 30. Ying TS (2001) Species diversity and distribution pattern of seed plants in China. Biodivers Sci 9: 393–398. [Google Scholar]
- 31. Wei XZ, Huang HD, Jiang MX, Yang JY (2008) Quantitative characteristics and spatial distribution patterns of Euptelea pleiospermum populations in riparian zones of the Shennongjia area, central China. J Plant Ecol (Chinese version) 32: 825–837. [Google Scholar]
- 32. Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15. [Google Scholar]
- 33. Zhang J, Yao X, Wei X, Chen L, Jiang M (2008) Development and characterization of 14 polymorphic microsatellite loci in the endangered tree Euptelea pleiospermum (Eupteleaceae). Mol Ecol Resour 8: 314–316. [DOI] [PubMed] [Google Scholar]
- 34. Wei X, Jiang M (2012) Limited genetic impacts of habitat fragmentation in an “old rare” relict tree, Euptelea pleiospermum (Eupteleaceae). Plant Ecol 213: 909–917. [Google Scholar]
- 35.Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996–2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier, France.
- 36.Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices, (version 2.9.3.2). Available: http://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 2005 Aug 23. Updated from Goudet (1995).
- 37. Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8: 103–106. [DOI] [PubMed] [Google Scholar]
- 38. Rice WR (1989) Analyzing tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- 39. van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538. [Google Scholar]
- 40. Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beerli P (2009) How to use migrate or why are markov chain monte carlo programs difficult to use? In: Bertorelle G, Bruford MW, Hauffe HC, Rizzoli A, Cernesi C, editors. Population genetics for animal conservation, volume 17 of Conservation Biology. Cambridge: Cambridge University Press. 42–79.
- 42. Weir B, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- 43. Jost L (2008) G ST and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026. [DOI] [PubMed] [Google Scholar]
- 44. Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10: 556–557. [DOI] [PubMed] [Google Scholar]
- 45. Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinforma Online 1: 47–50. [PMC free article] [PubMed] [Google Scholar]
- 47. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pritchard JK, Wen X, Falush D (2009) Documentation for STRUCTURE software: version 2.3. Available: http://pritch.bsd.uchicago.edu/structure.html. Accessed 2012 Jul.
- 49. Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7: 747–756. [Google Scholar]
- 52. Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- 54. Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138. [Google Scholar]
- 55. Wright S (1943) Isolation by distance. Genetics 28: 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wei X, Jiang M (2012) Contrasting relationships between species diversity and genetic diversity in natural and disturbed forest tree communities. New Phytol 193(3): 779–786. [DOI] [PubMed] [Google Scholar]
- 58. Balkenhol N, Waits LP (2009) Molecular road ecology: exploring the potential of genetics for investigating transportation impacts on wildlife. Mol Ecol 18: 4151–4164. [DOI] [PubMed] [Google Scholar]
- 59. Holderegger R, Di Giulio M (2010) The genetic effects of roads: A review of empirical evidence. Basic Appl Ecol 11: 522–531. [Google Scholar]
- 60. Hampe A, Arroyo J (2002) Recruitment and regeneration in populations of an endangered South Iberian Tertiary relict tree. Biol Conserv 107: 263–271. [Google Scholar]
- 61. Suzuki W, Osumi K, Masaki T, Takahashi K, Daimaru H, et al. (2002) Disturbance regimes and community structures of a riparian and an adjacent terrace stand in the Kanumazawa Riparian Research Forest, northern Japan. Forest Ecol Manag 157: 285–301. [Google Scholar]
- 62. Mejías JA, Arroyo J, Marañón T (2007) Ecology and biogeography of plant communities associated with the post Plio-Pleistocene relict Rhododendron ponticum subsp. baeticum in southern Spain. J Biogeogr 34: 456–472. [Google Scholar]
- 63. Wei XZ, Jiang MX, Huang HD, Yang JY, Yu J (2010) Relationships between environment and mountain riparian plant communities associated with two rare tertiary-relict tree species, Euptelea pleiospermum (Eupteleaceae) and Cercidiphyllum japonicum (Cercidiphyllaceae). Flora 205: 841–852. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contemporary migration rates between Euptelea pleiospermum populations along each river.
(DOC)
Historical migration rates between Euptelea pleiospermum populations along each river.
(DOC)
Pair-wise population D EST (above the diagonal) and F ST (below the diagonal).
(XLS)