

Abstract
The cyclopentenone prostaglandin (CyPG) J2 series, including prostaglandin J2 (PGJ2), Δ12-PGJ2 and 15-deoxy-Δ12, 14 -prostaglandin J2 (15d-PGJ2), are active metabolites of PGD2, exerting multiple effects on neuronal function. However, the physiologic relevance of these effects remains uncertain as brain concentrations of CyPGs have not been precisely determined. In this study, we found that free PGD2 and the J2 series CyPGs (PGJ2, Δ12-PGJ2 and 15d-PGJ2) were increased in post-ischemic rat brain as detected by UPLC-MS/MS with 15d-PGJ2 being the most abundant CyPG. These increases were attenuated by pre-treating with the cyclooxygenase inhibitor piroxicam. Next, effects of chronic exposure to 15d-PGJ2 were examined by treating primary neurons with 15d-PGJ2, CAY10410 (a 15d-PGJ2 analog lacking the cyclopentenone ring structure), or vehicle for 24 h to 96 h. Because we found that the concentration of free 15d-PGJ2 decreased rapidly in cell culture medium, freshly prepared medium containing 15d-PGJ2, CAY10410 or vehicle was changed twice daily to maintain steady extracellular concentrations. Incubation with 2.5 μM 15d-PGJ2, but not CAY10410, increased neuronal cell death without induction of caspase-3 or PARP cleavage, consistent with a primarily necrotic mechanism for 15d-PGJ2-induced cell death which was further supported by TUNEL assay results. Ubiquitinated protein accumulation and aggregation was observed after 96 h 15d-PGJ2 incubation, accompanied by compromised 20S proteasome activity. Unlike another proteasome inhibitor, MG132, 15d-PGJ2 treatment did not activate autophagy or induce aggresome formation. Therefore, the cumulative cytotoxic effects of increased generation of CyPGs after stroke may contribute to delayed post-ischemic neuronal injury.
Introduction
Cyclopentenone prostaglandins (CyPGs) are non-enzymatic metabolites of prostaglandin D2 (PGD2) that covalently modify proteins via adduction of cysteine residues reported to have an array of biological actions in various cell lines (Shibata et al., 2002). In brain, CyPGs have been shown to have both protective (Lin et al., 2006, Pereira et al., 2006) and toxic effects (Rohn et al., 2001, Kondo et al., 2002, Li et al., 2004a, Musiek et al., 2006, Pierre et al., 2009); thus their role in modulating neuronal injury in disease states remains controversial. Exogenous 15d-PGJ2 reduces damage in rodent stroke models via PPARγ-dependent effects (Lin et al., 2006, Pereira et al., 2006). But CyPGs may induce toxicity via PPARγ-independent effects including disruption of the ubiquitin proteasome pathway, resulting in accumulation of ubiquitinated proteins (Ub-proteins) and protein aggregates in neurons (Shibata et al., 2003, Li et al., 2004b, Liu et al., 2011). CyPGs have many well-defined actions in vitro, but whether their concentrations achieve biological significance in vivo remains unclear (Bell-Parikh et al., 2003).
Recent work has shown that modification of proteins by CyPGs could result in the misfolding of target proteins, including UCH-L1 (Koharudin et al., 2010), which may result in loss-of-function. In addition, misfolded proteins are usually prone to aggregation within the cell, which may trigger the unfolded protein response, interfering with the function of the ubiquitin-proteasome system (UPS), resulting in cell injury and death (Ding and Yin, 2008, Nakamura and Lipton, 2009). It has been well documented that misfolded protein accumulation and aggregation are hallmarks of many neurodegenerative diseases including Parkinson’s, Alzheimer’s, and Huntington’s diseases (Gispert-Sanchez and Auburger, 2006, Olanow and McNaught, 2006, Dohm et al., 2008, Irvine et al., 2008). In post-ischemic brain, increased Ub-protein accumulation and aggregation has been reported by various groups (Hu et al., 2000, Hochrainer et al., 2012). Cells exert many protective mechanisms to prevent misfolded protein accumulation and aggregation including: (1) increasing molecular chaperone expression to re-fold proteins, (2) tagging the misfolded proteins with poly-ubiquitin chains (forming poly-ubiquitinated proteins) for recognition and degradation in the UPS, and (3) activating lysosome-based autophagy to clear misfolded proteins or form aggresomes to sequester toxic protein aggregates (Yao, 2010, Zhou et al., 2009). Even though many reports from various groups have shown that CyPGs induce cell toxicity accompanied by the accumulation of Ub-proteins (Shibata et al., 2003, Wang et al., 2006, Liu et al., 2011), the direct effects of CyPGs on proteasome activity, autophagy, and aggresome formation in neurons remain unclear.
In the current study, concentrations of free CyPGs including three J2-series CyPGs: 15d-PGJ2, PGJ2 and Δ12-PGJ2, were measured in rat brain 24 h after temporary focal ischemia via highly specific ultra performance liquid chromatography (UPLC) -MS/MS. Since CyPGs are highly reactive, the actual concentration of free CyPGs may decrease rapidly in culture medium and within the cell due to adduction of proteins and other molecules. Thus, we adopted an in vitro treatment paradigm providing twice daily changes of cell culture medium containing fresh 15d-PGJ2 upon primary neurons. This paradigm was used to study the toxicity of 15d-PGJ2 in vitro, the effects of 15d-PGJ2 on proteasome activity, accumulation of ubiquitinated proteins, and the formation of protein aggregates.
Materials and Methods
Animal studies were performed in accordance with the ethical standards in the Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Reagents and Antibodies
15d-PGJ2 and 9,10-dihydro-15-deoxy-Δ12,14 -prostaglandin J2 (CAY 10410) were purchased from Cayman Chemical (Ann Arbor, MI). Antibody sources were as follows: Mouse monoclonal anti-mono- and poly- ubiquitinated proteins antibody (clone FK2), and anti-poly-ubiquitinated proteins antibody (clone FK1) were from Enzo Life Sciences (Plymouth Meeting, PA). Monoclonal anti-ubiquitin antibody was from Covance (Berkeley, CA); Anti-PARP, anti-Caspase-3, anti-cleaved Caspase-3, and anti-LC3B antibodies were from Cell Signaling (Boston, MA); Anti-UCH-L1 antibody was from Sigma-Aldrich (St. Louis, MO); anti-NeuN antibody was from Millipore (Temecula, CA) and anti-MAP-2 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA); Cy3-conjugated and Alexafluor 488-conjugated secondary antibodies were from Jackson Immunoresearch Lab (West Grove, PA). The ProteoStat aggresome detection kit was purchased from Enzo Life Sciences. Ultra performance liquid chromatography organic solvents and water were from VWR (West Chester, PA). Anti-β-actin antibody and all other chemicals were from Sigma-Aldrich (St. Louis, MO). The Click-iT TUNEL Alexa Fluor image assay kit was purchased from Invitrogen (Grand Island, NY).
Middle cerebral artery occlusion (MCAO)
Animal studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee following The Guide for the Care and Use of Laboratory Animals. Anesthetic induction was achieved using inhaled 4% isoflurane in 50% nitrous oxide, balance oxygen, then lowered to 1.5% throughout surgery. Core body temperature was maintained at 37 +/− 0.5 °C through the use of a water-filled heating pad. MCAO was induced in adult male Sprague Dawley (Charles Rivers Laboratories, Wilmington, MA) rats, 250 – 275 g, by making a midline incision at the trachea, retracting soft tissues and advancing a nylon suture into the MCA for a duration of 90 min as previously described (Ahmad et al., 2009). Animals were monitored continuously during anesthetic induction and surgery, and for 30 min. following reperfusion, with daily monitoring thereafter. Lidocaine (1%) was applied to the wound after suture removal and wound closure. One hour prior to MCAO rats were treated with vehicle (2% methyl cellulose, n = 6) or piroxicam (30 mg/kg in vehicle, n = 6) via oral gavage. After 24h reperfusion, rats were anesthetized with isoflurane, followed by decapitation. Core infarct and penumbral brain tissues were rapidly dissected and frozen until CyPG measurement. Core and penumbra (n = 4) from naive rats were used as control.
Measurement of PGD2 and CyPGs in rat brain
15d-PGJ2, PGD2, PGJ2/Δ12 PGJ2, and 15-deoxy PGD2 (15d- PGD2) were measured by UPLC-MS/MS, as previously described with some modifications (Miller et al., 2009). Samples were homogenized in 0.12 M potassium phosphate buffer (5mM MgCl2 and 0.113 mM BHT) and centrifuged. The supernatant was removed and 7.5 ng each of deuterated 15d-PGJ2 and PGD2 were used as the internal standard. Samples were loaded onto Oasis HLB solid phase extraction cartridges (Waters, OAsis, Milford, MA) and washed with 1 ml volumes of 5% methanol before elution with 100% methanol. Extracts were dried under nitrogen and reconstituted in 125 μL of 80:20 methanol: de-ionized water. Analytes were separated via reverse phase UPLC with an Acquity UPLC BEH C18 1.7μM, 2.1 × 100 mm column. MS analysis was performed via a Thermo TSQ Quantum Ultra triple quadrupole mass spectrometer (ThermoFisher Scientific). Quantitation by selected reaction monitoring (SRM) analysis on PGD2, PGJ2/Δ12- PGJ2, 15d-PGJ2, and 15d- PGD2 was performed by monitoring their m/z transitions, 351 → 271, 333 → 271, 315 → 271, and 333 → 271, respectively. Parameters were optimized to obtain the highest [M-H]− ion abundance for each analyte. Data was acquired using Xcalibur Software version 2.0.6.
Measurement of 15d-PGJ2 concentrations in primary neuron culture medium
Primary neurons were seeded in 10cm dishes and 15d-PGJ2 was added to the culture medium to a final concentration of 5 μM at DIV12. Cell culture medium from each plate was collected at 0 h, 1 h, 4 h, 8 h and 24 h. The concentration of free 15d-PGJ2 was measured using UPLC- MS/MS as described above.
Cortical primary neuron-enriched culture and treatment
Cortical primary neuron-enriched cultures were prepared from E17 fetal rats (Sprague-Dawley, Charles River, Wilmington, MA) as previously described (Li et al., 2008) and used for experiments after 9 days in vitro (DIV). Cells were grown in serum-free Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with B27 and GlutaMAX (Invitrogen). During treatment, cells were divided into 4 groups and treated with vehicle (methyl acetate, 0.79 μl/ml cell culture medium), CAY10410 (2.5 μM), or 15d-PGJ2 (0.5 μM or 2.5 μM) for 24 h to 96 h. Cell culture medium freshly prepared with 15d-PGJ2, CAY10410 or vehicle was changed twice daily, and cells were harvested for subsequent experiments at indicated time points. In some experiments, a potent proteasome inhibitor, MG132 (at 10 μM for 16h), was incubated with primary neurons to serve as a positive control to induce ubiquitinated protein accumulation and cell apoptosis.
Cell viability measurement
Cell death was quantitatively assessed indirectly through the MTT cell viability assay according to the manufacturer’s protocol. Culture medium was changed twice daily with freshly prepared medium containing 15d-PGJ2 (0.5 μM or 2.5 μM), CAY10410 (2.5 μM) or vehicle (methyl acetate, 0.79 μl/ml cell culture medium), to retain stable extracellular concentrations. Cells were treated for 96 h and cell viability was normalized to vehicle control.
Western blot
Western blots were performed as previously described (Liu et al., 2011). For Ub-protein detection, cell lysates were resolved on a 4–20% linear gradient polyacrylamide gel (BioRad, Hercules, CA) before detection with anti-poly-ubiquitinated conjugates antibody (1:1000). For PARP, caspase-3, UCH-L1, and LC3B protein detection, cell lysates were resolved on 10% or 12% SDS-PAGE and transferred to PVDF membrane. After blocking with 5% non-fat milk in TBS/Tween-20, membranes were incubated with anti-PARP (1:1000), anti-caspase-3 (1:1000), or anti-UCH-L1 (1:5000) antibodies at 4 °C overnight. Blots were washed and the appropriate secondary antibodies applied. Protein signal was visualized with ECL reagents (Pierce). Blots were then stripped and re-probed using anti-β-actin antibody as a loading control.
Immunofluorescence and confocal microscopy
Fluorescent immunocytochemical staining was performed as previously described (Liu et al., 2011). Primary neurons were seeded on L-poly D-lysine-coated glass coverslips and incubated with 15d-PGJ2 (2.5 μM), CAY10410 (2.5 μM) or methyl acetate (veh) before fixation. Cells were fixed with either 4% paraformaldehyde (PFA) in PBS followed by permeabilization with blocking buffer (2% BSA, 1% fish skin gelatin and 0.02% saponin in PBS) for cleaved Caspase-3 staining or with methanol at −20 °C for 10 min followed by blocking in 20%FBS/PBS for ubiquitinated protein detection. Cells were then incubated with anti-cleaved caspase-3/Asp175 (1:6000), anti-NeuN (1:100), anti-MAP-2 (1:100), anti-ubiquitinated proteins antibody/FK2 (1:200), anti-LC3B/D11 (1:400), or anti-UCH-L1 antibodies (1:100) and Alexafluor 488 or Dye549 conjugated secondary antibodies. Aggresome and aggresome-like inclusion bodies were detected using a ProteoStat aggresome detection kit (Enzo) following manufacturer’s instructions. Confocal images were acquired with a FluoView FV1000 confocal microscope (Olympus, Tokyo, Japan).
Activation of caspase-3 and caspase-7 in primary neurons was also evaluated using CellEvent Caspase-3/7 green detection reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. In brief, after 15d-PGJ2 treatment, cells were incubated with 2 μM caspase-3/7 detection reagent in culture medium for 30 min before fixation with 4% PFA for 15 min at room temperature. Coverslips were then mounted to slides with Prolong gold anti-fade reagent (Invitrogen) and photographed as described above. Double-stranded DNA breaks in apoptotic cells were detected using terminal deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) assay (Invitrogen) following manufacturer’s directions. Staurosporin (10 μM) -treated cells were included as a positive control.
Preparation of detergent-insoluble fraction from cell lysates
The cell lysate detergent insoluble protein aggregate fraction was prepared as previously described (Zhou et al., 2009). In brief, primary neurons were treated with vehicle (methyl acetate), CAY10410 (0.5 μM or 2.5 μM) or 15d-PGJ2 (0.5 μM or 2.5 μM) for 24 h to 96 h. Cells were lysed in radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (Pierce, Rockford, IL) containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Pierce). Cells were incubated with RIPA buffer for 15 min on ice and centrifuged at 16,800g at 4 °C for 15 min. The supernatants were collected and the insoluble pellets were washed twice with cold phosphate-buffered saline (PBS), and then re-suspended in RIPA buffer for 1h on ice and sonicated for protein solubilization. Protein concentrations were determined by the bicinchoninic acid (BCA) method (Pierce).
20S proteasome activity assay
20S proteasome activity was measured using a Chemicon Proteasome Activity Assay kit following manufacturer’s instructions (Millipore, Billerica, MA). Briefly, primary neurons were treated with vehicle (methyl acetate, 0.79 μl/ml cell culture medium), 15d-PGJ2 (0.5 μM or 2.5 μM), or CAY10410 (2.5 μM) for 72 h. Cells were then harvested in cell lysis buffer containing: 50mM Hepes (pH 7.4), 5mM EDTA, 150mM NaCl, 1% Triton X-100 and 2 mM ATP. Protein concentration was measured using the BCA method (Pierce). Cell lysate (50 μg) was incubated with labeled 20S proteasome substrate LLVY-AMC for 2h at 37 °C. Free AMC fluorophore was read using a BioTek Flx800 multi-detection micro-plate reader (Winooski, Vermont) with λex 360nm and λem 460 nm. Relative Fluorescence Units (RFU) for each sample were recorded as the index of proteasome activity.
Statistical analysis
Data were analyzed using one-way ANOVA with Fisher LSD post hoc analysis to calculate differences between groups. Results were considered to be significant when p < 0.05.
Results
PGD2 and its metabolite CyPG concentrations are increased in rat brain after temporary focal ischemia via a cyclooxygenase (COX) -activation dependent pathway
A mass spectrometric (MS) method was developed to detect and quantify PGD2 and its metabolite J2 series of CyPGs including 15d-PGJ2, PGJ2, and Δ12-PGJ212 in post-ischemic rat brain. Adult male Sprague Dawley rats underwent 90 minutes of MCAO with or without pretreatment with the nonselective COX inhibitor, piroxicam. Rats were sacrificed 24 h after reperfusion and core infarct and penumbral brain regions were rapidly dissected and eicosanoids were extracted and further purified with UPLC. PGD2 and its metabolites were measured using UPLC- MS/MS with SRM of selected product ion fragments of the precursor mass as determined by authentic standards. Concentrations of PGD2 and its metabolite CyPGs increased significantly after MCAO in ischemic penumbral tissue and were attenuated by pre-treatment with the COX inhibitor piroxicam, suggesting an important role for COX activation in the post-ischemic generation of PGD2 and CyPGs. The concentration of PGD2 increased over 4-fold 24 h after MCAO in penumbra as compared to naive (465.5 ± 340.4 nM and 102.3 ± 15.1 nM, respectively), diminishing to 146.7 ± 119.7 nM in the piroxicam-treated MCAO rats (Fig. 1). Similar changes in concentration were observed for free 15d-PGD2 in penumbra (2.7 ± 0.8 nM in naïve, 43.0 ± 35.8 nM in MCAO vehicle-treated, and 7.8 ± 5.8 nM in MCAO piroxicam-treated rats). More dramatic increases in free CyPGs PGJ2, Δ12-PGJ2 and 15d-PGJ2 concentrations were observed in post-ischemic penumbral tissue. Because Δ12-PGJ2 and PGJ2 are not independently resolved using this method of detection, the data presented here are shown as the sum of both PGJ2 and Δ12-PGJ2 (PGJ2/Δ12 -PGJ2). In a previously performed study we found that PGJ2 accounted for 83.6 ± 2.5% vs 16.4 ± 4.3% of Δ12-PGJ2 in rat brain penumbral tissue 24h after MCAO (n=4, data not shown). [PGJ2/Δ12 -PGJ2] increased from 4.4 ± 0.5 nM (3.7 nM PGJ2 and 0.7 nM Δ12-PGJ2) in naïve to 113.3 ± 92.0 nM (94.7 nM PGJ2 and 18.6 nM Δ12-PGJ2) in vehicle-treated MCAO rats; similarly, free endogenous [15d-PGJ2] was below detection limits in samples from naïve brain tissue, but was elevated to 155.9 ± 39.2 nM in vehicle-treated rats having undergone MCAO. PGD2 and its metabolite CyPGs were also induced in samples of post-ischemic core tissue, but to a lesser extent than that of penumbra. These results are the first report that we are aware of demonstrating significant increases in in vivo concentrations of PGD2 and its metabolite CyPGs in post-ischemic rat brain using an MS/MS technique.
Fig. 1.

Brain concentrations of PGD2 and its metabolite cyclopentenone prostaglandins are increased after middle cerebral artery occlusion (MCAO) in rat in a COX activity-related manner. Rats were treated via oral gavage with vehicle (2% methyl cellulose) or COX inhibitor piroxicam (30 mg/kg) 1 hour prior to MCAO (n=6 per group; naïve, n=4). Rats were sacrificed 24 h after reperfusion and ipsilateral brains dissected into penumbra and core samples. Free PGD2 and its metabolites in each infarct area were detected and quantified by UPLC-MS/MS. Pir: piroxicam; Veh: vehicle; NS: not significant. * P < .05; &: not detected. Data are means +/− SD
Because most of the research regarding CyPGs’ biological effects has been performed using in vitro models of cell culture, we initially investigated the stability of 15d-PGJ2 concentrations in primary neuron-enriched cell culture medium over 24 h. Primary neuronal cells were incubated with culture medium spiked with 5 μM 15d-PGJ2. Culture medium samples were assayed at various time points to determine free 15d-PGJ2 concentrations using UPLC-MS/MS. Free 15d-PGJ2 concentrations in culture medium decreased rapidly from 5 μM to 3.53 ± 0.13 μM at 1h, 2.97 ± 0.10 μM at 4h, 2.66 ± 0.09 μM at 8 h, and 1.25 ± 0.05 μM at 24h (n=3). These quantitative data indicate that the concentration of 15d-PGJ2 decreases significantly in the in vitro system over 24 h, prompting us to modify our CyPG treatment paradigm, thus providing steady long-term extracellular exposure to 15d-PGJ2. Endogenous free 15d-PGJ2 was not detectable in neuronal cultures; however, 15d-PGJ2 produced within cells in culture may readily diffuse across membranes and be dramatically diluted in medium.
15d-PGJ2 induces neuronal cell death via a caspase-3 independent mechanism
To test the biological effects of long-term exposure of low-dose CyPGs on neuronal cell viability, primary neuron-enriched cultures were incubated with 15d-PGJ2 for 96 h. Cell culture medium containing freshly added 15d-PGJ2 was changed twice daily to maintain a steady 15d-PGJ2 concentration in the culture medium. As shown in Fig. 2, primary neuronal cell viability decreased slightly, but insignificantly, when treated with 0.5 μM 15d-PGJ2, and significantly with 2.5 μM 15d-PGJ2 after 96 h incubation. To determine whether these effects are mediated through a cyclopentenone ring moiety-dependent mechanism, an analog of 15d-PGJ2, CAY10410 was used to treat primary neurons. CAY10410 has a structure similar to that of 15d-PGJ2 but lacks the reactive cyclopentenone ring, while retaining PPARγ ligand activity. Treatment with 2.5 μM CAY10410 for 96 h did not reduce neuronal cell viability significantly as compared to vehicle control, suggesting the cyclopentenone ring structure is important for 15d-PGJ2 toxicity. However, toxic effects of 15d-PGJ2 using this in vitro dosing paradigm are not manifested until 96 h after treatment; no significant decrease in viability was observed in cells when treated with either 0.5 μM or 2.5 μM 15d-PGJ2 for 24 h, 48 h or 72 h (data not shown).
Fig. 2.

The cyclopentenone prostaglandin 15d-PGJ2 induces cell death through a cyclopentenone ring moiety-dependent mechanism. Primary neuronal cells were treated with vehicle (Veh), CAY10410 (CAY, 2.5 μM) or 15d-PGJ2 for 96 h. Cell viability was measured using the MTT assay. n = 54–72 wells per group (4 plates run on 2 experimental days). * P < 0.001. Data are means +/− SE
The above data indicate that 15d-PGJ2 concentrations decrease rapidly in cell culture medium and that multiple treatments with low molarity 15d-PGJ2 induce primary neuronal cell death through a cyclopentenone ring structure-dependent mechanism. Previous studies suggest the minimum concentration for a single dose of 15d-PGJ2 to induce in vitro neuronal cell death is 10 μM to 20 μM (Kondo et al., 2002, Liu et al., 2011); however, here we report that the toxic effects of 15d-PGJ2 on primary neurons can be observed at a concentration of 2.5 μM when using a dosing paradigm involving replacing the medium twice daily. Because persistent over-generation of 15d-PGJ2 and other CyPGs can occur under many pathological conditions in the brain (Pierre et al., 2009), subsequent studies were performed changing medium twice daily in an effort to maintain a more steady-state extracellular concentration of 15d-PGJ2.
To examine the role of apoptosis in 15d-PGJ2-induced cell death, Western blots and immunocytochemistry were used to detect activation of caspase-3 and poly ADP-ribose polymerase (PARP) cleavage. TUNEL staining was used to detect apoptotic DNA fragmentation. Culture medium containing methyl acetate (vehicle), 15d-PGJ2 (0.5 μM or 2.5 μM), or CAY10410 (0.5 μM or 2.5 μM) was changed twice daily for 24 h to 96 h. As shown in Fig. 3a, neither 15d-PGJ2 nor CAY10410 significantly induced caspase-3 or PARP cleavage. Additionally, there were no differences in full length caspase-3 or PARP expression levels between 15d-PGJ2, vehicle, or CAY10410 -treated groups. Consistent with Western blot results, immunocytochemical detection of cleaved caspase-3 indicated no increase in cleaved caspase-3 positive cells in neurons treated with 2.5 μM 15d-PGJ2 for 48 h as compared to vehicle control. MG132, a proteasome inhibitor reported to induce cell apoptosis via caspase-3 activation, was used as a positive control (Janen et al., 2010, Zhou et al., 2009). As shown in Fig. 3b (upper panel), cleaved caspase-3 positive cells were visible only in primary neurons treated with MG132 (10 μM) for 16 h.
Fig. 3.

15d-PGJ2 induces primary neuronal cell death via a caspase-3 independent mechanism. Cells were treated with vehicle (Veh, methyl acetate), CAY10410 (CAY), 15d-PGJ2 for 24 h – 96 h. Pro-apoptosis reagents, MG132 and staurosporin, were incubated with primary neurons at 10 μM for 16 h as positive controls. (a). Caspase-3 and PARP cleavage: Cells were harvested and immunoblotted using anti-cleaved caspase-3 and anti-PARP antibodies. β-actin was used as a loading control. Full: full length protein; Clvd: cleaved. (b). Representative immunocytochemical photos of rat primary neurons after treatment with Veh or 2.5 μM 15d-PGJ2. Upper panel: Cleaved caspase-3 was detected using anti-cleaved caspase-3 antibody (green) and anti-NeuN (red) antibodies (48 h after treatment). Middle panel: Activated caspase 3/7 was detected using a fluorogenic substrate for activated caspases-3 and 7 (green, 72 h after treatment). Lower panel: Double-stranded DNA breaks were detected using TUNEL (green, 72 h after treatment). All photos: 60X using a confocal microscope; scale bar = 40 μm. Blue is DAPI nuclear stain
Caspase-3 activation and apoptosis were further investigated in primary neurons using a fluorogenic substrate for activated Caspase-3/7 and TUNEL assay. Primary neurons were treated with 2.5 μM 15d-PGJ2 or vehicle for 72 h, while 16 h incubation with 10 μM MG132 or staurosporin was used as a positive control. 15d-PGJ2 did not induce activation of caspase-3/7 (Fig. 3b, middle panel) or result in apoptosis as indicated by TUNEL when compared to vehicle control (Fig. 3b, lower panel). Taken together, neuronal necrotic cell death, but not caspase-3 activation-mediated apoptosis is likely to be the major mechanism for 15d-PGJ2-induced cell death in primary neurons.
15d-PGJ2 increases ubiquitinated protein accumulation and aggregation
Previous work has shown that CyPG-induced cell toxicity is usually associated with increased Ub-protein levels in the cell (Myeku and Figueiredo-Pereira, 2011, Li et al., 2004b, Wang and Figueiredo-Pereira, 2005, Arnaud et al., 2009, Liu et al., 2011). To further detect the effects of continuous low-dose 15d-PGJ2 treatment on Ub-protein accumulation and aggregation, rat primary neurons were treated with vehicle (methyl acetate), CAY10410 (0.5 μM or 2.5 μM) or 15d-PGJ2 (0.5 μM or 2.5 μM) for 96 h. Cells were harvested and cell homogenates were separated into RIPA buffer-soluble and RIPA buffer–insoluble fractions. As shown in Fig. 4a, treatment of primary neurons with 15d-PGJ2 resulted in a dose-dependent increase in the accumulation of RIPA-soluble Ub-proteins as compared to vehicle control. Interestingly, a robust increase in high molecular weight Ub-proteins was detected in RIPA-insoluble fractions from 15d-PGJ2-treated primary neurons compared to those from vehicle-treated neurons and CAY 10410 (Fig. 4b). Collectively, these data suggest that 15d-PGJ2 induces Ub-protein accumulation and aggregation in neuronal cells after 96 h continuous exposure to 15d-PGJ2. Because CAY10410 lacks the reactive cyclopentenone ring in the 15d-PGJ2 structure, the above data indicate that the effect of 15d-PGJ2 on Ub-protein accumulation and aggregation relies on protein modifications via its cyclopentenone ring structure.
Fig. 4.

Ubiquitinated proteins accumulate and aggregate in primary neurons after chronic treatment with 15d-PGJ2. A and B: Cells were incubated with Vehicle (Veh, methyl acetate), CAY10410 (CAY), or 15d-PGJ2 for 96 h (n = 3 per group) then harvested with RIPA buffer. Both RIPA -soluble and -insoluble fractions were collected. Poly-ubiquitinated proteins were detected by Western blot using an anti- poly-ubiquitinated conjugates antibody. Poly-ubiquitinated proteins were quantified and normalized to vehicle-treated groups. (a) RIPA-soluble fraction. (b): RIPA-insoluble fraction. Data are means +/− SE. * P < 0.05. (c) Detection of ubiquitinated protein by immunocytochemical staining using anti-ubiquitinated proteins (red) and anti-MAP-2 (green) antibodies. Primary neurons were treated with vehicle (Veh), 2.5 μM CAY10410, or 2.5 μM 15d-PGJ2 for 72 h. MG132 (10μM, 16 h incubation), a protease inhibitor, was used as a positive control. Blue is DAPI nuclear stain. Photos are at 240X. (d) Primary neurons were treated with vehicle (Veh) or 2.5 μM 15d-PGJ2 for 24 h or 72 h prior to fixation and immunocytochemical staining with anti-ubiquitinated proteins antibody (green). Photos are at 60X and 240X. All photos: scale bar = 25μm
To observe the distribution of Ub-proteins in neuronal cells after exposure to 15d-PGJ2, immunocytochemistry was performed using primary neurons treated with vehicle, CAY10410 (2.5 μM), or 15d-PGJ2 (2.5 μM) for 24 h and 72 h. As shown in Fig. 4c, the distribution of Ub-proteins in the neuron was dramatically altered after 72 h of 15d-PGJ2 treatment; Ub-proteins were distributed evenly throughout the cytoplasm and were seen as small discrete punctate structures in vehicle and CAY10410-treated neurons, while large perinuclear Ub-protein-positive aggregates were frequently observed in 15d-PGJ2-treated neurons and MG132-treated cells (positive control). Time-course formation of punctuate Ub-protein aggregates was seen after 24 h of 15d-PGJ2 treatment, while the accumulation of larger clumped Ub-protein structures in the cytoplasm were not observed until 72 h incubation with 15d-PGJ2 (Fig. 4d).
15d-PGJ2 impairs proteasome activity without activation of autophagy
The removal of Ub-proteins in cells relies mainly on two major systems: the ubiquitin-proteasome system and the autophagy-lysosome system (Rubinsztein, 2006). The 26S proteasome is composed of a “gatekeeper” 19S cap subunit and a 20S proteasome core, where substrate degradation occurs (Lehman, 2009). To evaluate the effects of sustained 15d-PGJ2 treatment on neuronal UPS function, a 20S proteasome activity assay was performed using primary neurons treated with vehicle, CAY10410 (2.5 μM), or 15d-PGJ2 (0.5 μM or 2.5 μM) for 72 h. Treatment with 15d-PGJ2 for 72 h significantly inhibited 20S proteasome activity in a dose-dependent manner compared to vehicle control (Fig. 5a). Furthermore, because the inhibitory effect of 15d-PGJ2 on proteasome activity is absent in cells treated with CAY10410, this suggests that the inhibition of proteasome substrate degradation is reliant on the cyclopentenone ring structure.
Fig. 5.
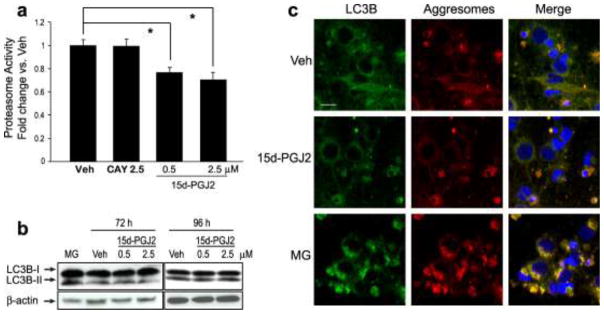
Long-term treatment of primary neuronal culture with 15d-PGJ2 impairs proteasome activity without autophagy activation. (a) Proteasome activity 72 h after treatment with vehicle (Veh), CAY10410 (CAY) or 15d-PGJ2 normalized to the vehicle-treated group. Data are means +/− SE. n = 8 per group. * P < 0.01 vs Veh. (b) Cells were treated with vehicle or 15d-PGJ2 for 72 h or 96 h, or 10 μM MG132 (MG, positive control, 16 h treatment). Lysates were then immunoblotted with anti-LC3B antibody. β-actin was used as a loading control. (c) Primary neurons were treated with Veh, 2.5 μM 15d-PGJ2 for 96 h or MG for 16 h then fixed with formalin and immunostained using anti-LC3B antibody (green). Aggresomes were detected using a ProteoStat aggresome detection kit (red). Blue is DAPI nuclear stain. Photos are 180X taken with an Olympus confocal microscope; scale bar = 20 μm
In response to Ub-protein accumulation, cells can also protectively activate autophagy to enhance lysosomal degradation (Banerjee et al., 2010). To evaluate the direct effect of 15d-PGJ2 treatment on primary neuronal autophagy, primary neurons were treated with vehicle or 15d-PGJ2 (0.5 μM or 2.5 μM) for 72 h and 96 h and Western blot and immunocytochemistry were performed to detect the autophagy marker LC3B. During autophagy, cytosolic light chain 3-I (LC3-I) is converted to LC3-II by phosphatidylethanolamine conjugation, which can be used as an index of autophagy (Lee et al., 2010). As shown in Fig. 5b, conversion of autophagy marker LC3B-I to LC3B-II in 15d-PGJ2-treated neurons is similar to that of the vehicle-treated group, suggesting that 15d-PGJ2 does not significantly induce autophagy activation.
In addition to the formation of autophagosomes for the degradation of misfolded proteins in lysosomes, cells might also protectively form aggresomes to sequester toxic Ub-protein aggregates (Olzmann et al., 2008). To confirm the effect of 15d-PGJ2 on neuronal autophagy and to explore its effect on aggresome formation, primary neurons were incubated 96 h with 15d-PGJ2 vehicle or the positive control MG132 (16 h incubation). Cells were immunostained with anti-LC3B antibody to identify autophagosomes, and ProteoStat aggresome dye for aggresome detection (Fig. 5c). Consistent with previous reports (Zhou et al., 2009), MG132 treatment induced significant aggresome formation and co-localization with the autophagosome marker LC-3B. In contrast, 96 h treatment with 2.5 μM 15d-PGJ2 induced very little autophagosome or aggresome formation, even though significant Ub-protein accumulation and aggregation were detected by Western blot under these conditions.
15d-PGJ2 modifies endogenous neuronal proteins including UCH-L1 and induces UCH-L1 aggregation
The above data demonstrate that 15d-PGJ2 impairs the neuronal UPS and induces Ub-protein accumulation through a cyclopentenone ring moiety-dependent mechanism, which suggests that protein modification by 15d-PGJ2 could be the underlying mechanism for these cytotoxic effects. To test this hypothesis, primary neurons were treated with 2.5 μM biotinylated 15d-PGJ2 (b-15d-PGJ2) for 24 h to 72 h. Cell lysate protein bands and a characteristic protein smear representing b-15d-PGJ2-modified endogenous proteins were evident in b-15d-PGJ2-treated neuronal samples but not in vehicle control (Fig. 6a). Furthermore, biotin signals representing 15d-PGJ2-adducted proteins increased in a time-dependent manner from 24 h to 72 h, suggesting the accumulation of 15d-PGJ2 -modified proteins in neurons after long-term exposure to low-dose 15dPGJ2. To evaluate the relationship between 15d-PGJ2-modified proteins and accumulated Ub-proteins, immunocytochemistry was performed on primary neurons treated with 2.5 μM b-15d-PGJ2 for 24 h. As shown in Fig. 6b, b-15d-PGJ2 treatment also induced Ub-protein accumulation as compared to the vehicle control group. Additionally, b-15d-PGJ2 -modified proteins extensively co-localized with Ub-proteins in the cytoplasm of primary neurons, suggesting that 15d-PGJ2-modified proteins might be a major source of excessive Ub-protein generation in 15d-PGJ2-treated primary neurons.
Fig. 6.

Continuous incubation with 15d-PGJ2 modifies endogenous neuronal proteins and increases protein aggregates. (a–b) Rat primary neurons were treated with biotinylated (b-) 15d-PGJ2 or vehicle (Veh). (a) b-15d-PGJ2 adducts to endogenous proteins in primary neurons after treatment for 24 h – 72 h. Cell lysates underwent SDS-PAGE and biotin-incorporated proteins (b-proteins) were detected using HRP-conjugated Streptavidin (upper). β-actin was used as a loading control (lower). (b) Immunocytochemical detection (60X, inset 240X) of ubiquitin and b-15d-PGJ2 using anti-ubiquitin conjugates (Ub-conjugates, green) and anti-biotin (red) antibodies. Blue is DAPI nuclear stain. Scale bar = 60 μm; inset = 20 μm. (c) UCH-L1 aggregates in primary neurons after 15d-PGJ2 treatment. Primary neurons were harvested with RIPA buffer 96 h after treatment with 15d-PGJ2 or vehicle. RIPA -soluble (upper) and -insoluble (lower) fractions were collected, and UCH-L1 was detected by Western blot with anti-UCHL-1 antibody and quantified. Graphs are means +/− SE normalized to vehicle. n=3 per group. * P<0.05 vs vehicle. (d) Immunocytochemical staining (180X) of rat primary neurons 96 h after incubation with vehicle, 2.5 μM CAY 10410 or 2.5 μM 15d-PGJ2. UCHL-1 and ubiquitinated proteins were visualized with anti-UCHL-1 (red) and anti-ubiquitinated conjugates (Ub-conjugates, green) antibodies, respectively. Blue is DAPI nuclear stain. Scale bar = 20 μm. Photos (b,d) were taken with an Olympus confocal microscope
UCH-L1, an abundant protein constituting about 2% of total soluble proteins in the brain, was previously reported to be a modification target of 15d-PGJ2 in the primary neuron (Liu et al., 2011). Our previous work has shown that modification of 15d-PGJ2 results in unfolding of the protein structure and the loss of hydrolase activity of UCH-L1 (Koharudin et al., 2010, Liu et al., 2011). Since UCH-L1 is a component in abnormal protein aggregates found in Lewy bodies in Parkinson’s disease and neurofibrillary tangles in Alzheimer’s disease, we hypothesized that UCH-L1 also participates in 15d-PGJ2-induced Ub-protein aggregations (Choi et al., 2004). To test this hypothesis, primary neurons were treated with 15d-PGJ2 (0.5 μM and 2.5 μM) for 96 h. Both RIPA -soluble and -insoluble fractions were collected and UCH-L1 levels in each fraction were determined by Western blot. As shown in Fig. 6c, 15d-PGJ2 treatment resulted in reduced UCH-L1 levels in the soluble fraction and increased UCH-L1 levels in the insoluble fraction, indicating that UCH-L1 is present in protein aggregates in 15d-PGJ2-treated primary neurons.
Furthermore, immunocytochemistry with 15d-PGJ2-treated primary neurons revealed that UCH-L1 is a component in 15d-PGJ2- induced Ub-protein aggregates. In this experiment, primary neurons were treated with vehicle, CAY10410 (2.5 μM) or 15d-PGJ2 for 96 h. UCH-L1 (red) and Ub-proteins (green) were distributed evenly throughout the cytoplasm in control neurons, with very limited co-localization between the two proteins. A similar distribution pattern of UCH-L1 and Ub-proteins was also detected in neurons treated with CAY10410. Conversely, clustered perinuclear aggregates containing both UCH-L1 protein and Ub-proteins were detected in 15d-PGJ2-treated neurons, exhibiting extensive co-localization (Fig. 6d). Taken together, 15d-PGJ2 decreases UCH-L1 solubility and enhances its aggregation in primary neurons via its cyclopentenone ring structure; UCH-L1 is a component of Ub-protein aggregates induced by treatment with 15d-PGJ2.
Discussion
The major novel findings of this study are: (1) This study is the first to use the highly sensitive and specific UPLC-MS/MS technique to characterize the concentrations of all of the free J2-series CyPGs in different regions of rat brain after temporary focal ischemia. In addition to 15d-PGJ2, the concentrations of the CyPGs PGJ2 and Δ12-PGJ2 are elevated in rat brain after temporary focal ischemia and 15d-PGJ2 is the most abundant J2-series CyPG present in post-ischemic rat brain; (2) CyPG formation primarily depends upon COX activity: CyPG production is abrogated when rats are pretreated with the COX inhibitor piroxicam; (3) [CyPGs] are greatest in the penumbral cortex where ischemia is incomplete. The concentration of free 15d-PGJ2 is about 160nM in the penumbra 24h post-ischemia, which is approximately 3 times higher than that in the core region, and 7 times higher than whole hemisphere as reported previously (Liu et al., 2011); (4) The cytotoxic effects of 15d-PGJ2 on neuronal cells are cumulative as these effects (cell death and excess Ub-protein aggregation) were observed at lower doses but required a longer incubation time, indicating an important role for CyPGs in chronic inflammation-related neurodegeneration; (5) By using detergent-rich RIPA buffer to collect both detergent -soluble and -insoluble fractions, 15d-PGJ2 was found to induce both Ub-protein accumulation and aggregation in primary neurons; this effect was related to over-generation of Ub-proteins due to protein misfolding and impaired Ub-protein degradation caused by decreased proteasome activity and failed induction of compensative autophagy. The above findings provide important new evidence to support the physiological and pathological relevance of CyPGs in the pathogenesis and progression of many neurological disorders, as chronic inflammation, excess Ub-protein aggregation, and cell death have long been considered as important factors contributing to secondary post-ischemic injury and many neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases.
Brain concentrations of prostaglandins are increased after cerebral ischemia due to the release of free arachidonic acid via activation of phospholipase A2 and induction and activation of COX-2 (Nakayama et al., 1998, Lin et al., 2006, Li et al., 2008, Perez-Sala, 2011). PGD2 is the most prevalent prostaglandin found in brain and its concentration is also dramatically increased after cerebral ischemia (Uchida and Shibata, 2008). Shibata et al showed that PGD2 is spontaneously converted to CyPGs by an enzyme independent mechanism (Shibata et al., 2002). Thus, it is not surprising that there are increased concentrations of CyPGs found in the rat brain after ischemia. In this study, we found the concentration of free 15d-PGJ2 to be significantly greater in brain infarct penumbral tissue, which has the highest level of COX-2 induction, than in our previous data from whole rat hemisphere after MCAO (Liu et al., 2011). It is noteworthy that the tissue concentrations of 15d-PGJ2 detected in the current experiments are much less than those required to elicit significant in vitro effects. However, direct comparison of in vitro to in vivo concentrations is problematic. Concentrations of CyPGs at specific sites of action within the cell may be even higher than the average tissue concentration. Furthermore, in the current study we also measured concentrations of PGJ2/Δ12-PGJ2 and 15d-PGD2 in penumbral tissue, and found that these prostaglandins are also increased after MCAO. The total tissue concentration for each species of CyPG was on the order of 0.1 μM. Other CyPGs such as the PGE2 metabolite PGA2 and the isoprostane prostaglandins may also be produced in post-ischemic brain (Musiek et al., 2006, Gharbi et al., 2007, Brooks et al., 2008), but were not measured in this study. CyPGs are highly reactive compounds that may interact with cysteine sulfhydryl groups in glutathione (GSH) and other peptides and proteins (Perez-Sala, 2011). Brunoldi et al showed that CyPGs are formed in liver in response to oxidative stress, but rapidly react with GSH to form stable adducts (Brunoldi et al., 2007). Additional experiments are necessary to quantify these important adducts in vivo. Thus the measured concentration of free CyPGs in brain may not reflect the overall production of these compounds since they can rapidly adduct to GSH and other nucleophiles.
Whether CyPGs are toxic or protective in cerebral ischemia in vivo remains controversial. CyPGs such as 15d-PGJ2 are known to have potent anti-inflammatory effects and thus may be protective in many injury paradigms, but they also may produce toxicity and cell death (Rohn et al., 2001, Kondo et al., 2002, Xiang et al., 2007). The protective effects of CyPGs are believed to be mediated via activation of anti-inflammatory pathways including those modulated by PPARγ. PPARγ-mediated effects may be important in the endothelial and microglial response to ischemia; however, our in vitro primary neuronal cell culture system does not include these cellular components. A number of previous studies have found that CyPGs are toxic to neurons and other cell types including neuron-like cell lines (Li et al., 2004a, Li et al., 2004b, Ogburn et al., 2006, Xiang et al., 2007). These studies have generally employed significantly higher concentrations of CyPGs, typically in the 20 to 50 M range. Since free CyPG concentrations rapidly decline in culture media, such high single doses may be necessary to maintain toxic levels for extended periods of time. Accordingly, we maintained lower doses of 15d-PGJ2 in the medium through frequent changes of medium and found that this paradigm produced toxicity. Previous studies have also found that CyPGs may induce cell death via apoptotic pathways (Kondo et al., 2002, Nencioni et al., 2003, Saito et al., 2003, Xiang et al., 2007), although many of these studies employed immortal cell lines that have significantly different cell death pathways than primary neurons. In the current study, chronic exposure to 15d-PGJ2 induced cell death in neurons but did not activate caspase-3 or induce TUNEL staining. These results suggest that chronic low level exposure to 15d-PGJ2 induces cell death in neurons by non-apoptotic mechanisms.
CyPGs have a number of other mechanisms by which they may produce toxicity in cells. Sub-chronic injections of 15d-PGJ2 into the striatum of rats produced accumulation of ubiquitin and α–synuclein protein aggregates similar to those found in sporadic Parkinson’s disease (Pierre et al., 2009). CyPGs disrupt the trafficking and processing of Ub-proteins resulting in the accumulation of Ub-proteins into cytosolic aggregates (Wang and Figueiredo-Pereira, 2005, Wang et al., 2006). This may occur as a result of modification of cytoskeletal elements, including actin, by CyPGs resulting in the collapse of the cytoskeleton and the endoplasmic reticulum (Ogburn and Figueiredo-Pereira, 2006). Other data suggest that CyPGs may bind to the 19S regulatory subunit of the proteasome and directly inhibit proteasome function, resulting in accumulation of Ub-proteins (Ishii et al., 2005). Autophagy may also play a role in removing aggregated Ub-proteins from the cell. Autophagy may protect the cell from excessive accumulation of Ub-proteins by either degrading the proteins in lysosomes or sequestering the proteins in aggresomes (Banerjee et al., 2010, Zhou et al., 2009). Thus, failure of autophagy could also contribute to toxicity produced by excessive Ub-proteins. The current data demonstrate that 15d-PGJ2 treatment results in accumulation of Ub-proteins in primary neurons. Confocal microscopy shows that the Ub-proteins accumulate in a similar perinuclear clumped pattern as described by Ogburn et al in SK-N-SH cells (Ogburn and Figueiredo-Pereira, 2006). 15d-PGJ2 treatment partially inhibited proteasome activity, but had no effect upon LC3-I/LC3 II conversion or any detectable effect upon aggresome formation. These data suggest that 15d-PGJ2 inhibits proteasome activity without activation of autophagy.
CyPGs may modify a number of other proteins that have important effects on cell function and survival, including UCH-L1 (Perez-Sala, 2011, Liu et al., 2011). UCH-L1, also known as PARK5, is mutated in a form of familial Parkinson’s disease (Andersson et al., 2011). Loss of UCH-L1 activity has also been implicated in the pathogenesis of memory loss in Alzheimer’s disease (Lombardino et al., 2005, Gong et al., 2006). CyPGs covalently modify UCHL1 in vitro, resulting in unfolding of the protein and the formation of high molecular weight aggregates (Koharudin et al., 2010, Liu et al., 2011). Neuronal UCH-L1 hydrolase activity is inhibited by treatment with 15d-PGJ2, and inhibition of UCH-L1 activity exacerbates hypoxic neuronal death (Liu et al., 2011). UCH-L1 is found within aggresomes associated with Parkinson’s and other neurodegenerative diseases (Choi et al., 2004, Liu et al., 2009). Loss of UCH-L1 activity may contribute to hypoxic neuronal cell death (Liu et al., 2011). In the current study, we found that continuous low-dose 15d-PGJ2 treatment resulted in a perinuclear accumulation of Ub-proteins that colocalize with UCH-L1. We also found that treatment with 15d-PGJ2 resulted in an increased concentration of UCH-L1 in the detergent-insoluble fraction of neuronal cell lysates, consistent with the notion that CyPGs bind to UCH-L1 and result in the formation of insoluble protein aggregates (Koharudin et al., 2010, Liu et al., 2011). These studies suggest that the binding of CyPGs and other oxidized lipids to UCH-L1 could be important in the pathogenesis of neurodegenerative diseases and stroke.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke R01NS37459 and the Veteran’s Administration Merit Review program (SHG). The authors thank Pat Strickler for secretarial support.
Footnotes
Disclosure/Conflict of Interest
The authors declare no conflict of interest. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- Ahmad M, Zhang Y, Liu H, Rose ME, Graham SH. Prolonged opportunity for neuroprotection in experimental stroke with selective blockade of cyclooxygenase-2 activity. Brain Res. 2009;1279:168–173. doi: 10.1016/j.brainres.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson FI, Werrell EF, McMorran L, Crone WJ, Das C, Hsu ST, Jackson SE. The effect of Parkinson’s-disease-associated mutations on the deubiquitinating enzyme UCH-L1. Journal of molecular biology. 2011;407:261–272. doi: 10.1016/j.jmb.2010.12.029. [DOI] [PubMed] [Google Scholar]
- Arnaud LT, Myeku N, Figueiredo-Pereira ME. Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem. 2009;110:328–342. doi: 10.1111/j.1471-4159.2009.06142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends in neurosciences. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma. J Clin Invest. 2003;112:945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JD, Milne GL, Yin H, Sanchez SC, Porter NA, Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J Biol Chem. 2008;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunoldi EM, Zanoni G, Vidari G, Sasi S, Freeman ML, Milne GL, Morrow JD. Cyclopentenone prostaglandin, 15-deoxy-Delta12,14-PGJ2, is metabolized by HepG2 cells via conjugation with glutathione. Chem Res Toxicol. 2007;20:1528–1535. doi: 10.1021/tx700231a. [DOI] [PubMed] [Google Scholar]
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- Dohm CP, Kermer P, Bahr M. Aggregopathy in neurodegenerative diseases: mechanisms and therapeutic implication. Neurodegener Dis. 2008;5:321–338. doi: 10.1159/000119459. [DOI] [PubMed] [Google Scholar]
- Gharbi S, Garzon B, Gayarre J, Timms J, Perez-Sala D. Study of protein targets for covalent modification by the antitumoral and anti-inflammatory prostaglandin PGA1: focus on vimentin. J Mass Spectrom. 2007;42:1474–1484. doi: 10.1002/jms.1291. [DOI] [PubMed] [Google Scholar]
- Gispert-Sanchez S, Auburger G. The role of protein aggregates in neuronal pathology: guilty, innocent, or just trying to help? J Neural Transm Suppl. 2006:111–117. doi: 10.1007/978-3-211-45295-0_18. [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Hochrainer K, Jackman K, Anrather J, Iadecola C. Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke. 2012;43:2229–2235. doi: 10.1161/STROKEAHA.112.650416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry. 2005;44:13893–13901. doi: 10.1021/bi051336u. [DOI] [PubMed] [Google Scholar]
- Janen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia. 2010;58:1766–1774. doi: 10.1002/glia.21047. [DOI] [PubMed] [Google Scholar]
- Koharudin LM, Liu H, Di Maio R, Kodali RB, Graham SH, Gronenborn AM. Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proc Natl Acad Sci U S A. 2010;107:6835–6840. doi: 10.1073/pnas.1002295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M, Sasaki S, Iwata M, Noguchi N, Uchida K. 15-Deoxy-Delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc Natl Acad Sci U S A. 2002;99:7367–7372. doi: 10.1073/pnas.112212599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009;118:329–347. doi: 10.1007/s00401-009-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wu S, Hickey RW, Rose ME, Chen J, Graham SH. Neuronal cyclooxygenase-2 activity and prostaglandins PGE2, PGD2, and PGF2 alpha exacerbate hypoxic neuronal injury in neuron-enriched primary culture. Neurochem Res. 2008;33:490–499. doi: 10.1007/s11064-007-9462-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Jansen M, Ogburn K, Salvatierra L, Hunter L, Mathew S, Figueiredo-Pereira ME. Neurotoxic prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal cells through the p38MAPK pathway: a death wish? J Neurosci Res. 2004a;78:824–836. doi: 10.1002/jnr.20346. [DOI] [PubMed] [Google Scholar]
- Li Z, Melandri F, Berdo I, Jansen M, Hunter L, Wright S, Valbrun D, Figueiredo-Pereira ME. Delta12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochem Biophys Res Commun. 2004b;319:1171–1180. doi: 10.1016/j.bbrc.2004.05.098. [DOI] [PubMed] [Google Scholar]
- Lin TN, Cheung WM, Wu JS, Chen JJ, Lin H, Liou JY, Shyue SK, Wu KK. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:481–487. doi: 10.1161/01.ATV.0000201933.53964.5b. [DOI] [PubMed] [Google Scholar]
- Liu H, Li W, Ahmad M, Miller TM, Rose ME, Poloyac SM, Uechi G, Balasubramani M, Hickey RW, Graham SH. Modification of ubiquitin-C-terminal hydrolase-L1 by cyclopentenone prostaglandins exacerbates hypoxic injury. Neurobiol Dis. 2011;41:318–328. doi: 10.1016/j.nbd.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Meray RK, Grammatopoulos TN, Fredenburg RA, Cookson MR, Liu Y, Logan T, Lansbury PT., Jr Membrane-associated farnesylated UCH-L1 promotes alpha-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc Natl Acad Sci U S A. 2009;106:4635–4640. doi: 10.1073/pnas.0806474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardino AJ, Li XC, Hertel M, Nottebohm F. Replaceable neurons and neurodegenerative disease share depressed UCHL1 levels. Proc Natl Acad Sci U S A. 2005;102:8036–8041. doi: 10.1073/pnas.0503239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Donnelly MK, Crago EA, Roman DM, Sherwood PR, Horowitz MB, Poloyac SM. Rapid, simultaneous quantitation of mono and dioxygenated metabolites of arachidonic acid in human CSF and rat brain. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3991–4000. doi: 10.1016/j.jchromb.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–22440. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 2009;14:455–468. doi: 10.1007/s10495-008-0301-y. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nencioni A, Lauber K, Grunebach F, Van Parijs L, Denzlinger C, Wesselborg S, Brossart P. Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J Immunol. 2003;171:5148–5156. doi: 10.4049/jimmunol.171.10.5148. [DOI] [PubMed] [Google Scholar]
- Ogburn KD, Bottiglieri T, Wang Z, Figueiredo-Pereira ME. Prostaglandin J2 reduces catechol-O-methyltransferase activity and enhances dopamine toxicity in neuronal cells. Neurobiol Dis. 2006;22:294–301. doi: 10.1016/j.nbd.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ogburn KD, Figueiredo-Pereira ME. Cytoskeleton/endoplasmic reticulum collapse induced by prostaglandin J2 parallels centrosomal deposition of ubiquitinated protein aggregates. J Biol Chem. 2006;281:23274–23284. doi: 10.1074/jbc.M600635200. [DOI] [PubMed] [Google Scholar]
- Olanow CW, McNaught KS. Ubiquitin-proteasome system and Parkinson’s disease. Mov Disord. 2006;21:1806–1823. doi: 10.1002/mds.21013. [DOI] [PubMed] [Google Scholar]
- Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem. 2008;15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira MP, Hurtado O, Cardenas A, Bosca L, Castillo J, Davalos A, Vivancos J, Serena J, Lorenzo P, Lizasoain I, Moro MA. Rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2 cause potent neuroprotection after experimental stroke through noncompletely overlapping mechanisms. J Cereb Blood Flow Metab. 2006;26:218–229. doi: 10.1038/sj.jcbfm.9600182. [DOI] [PubMed] [Google Scholar]
- Perez-Sala D. Electrophilic eicosanoids: Signaling and targets. Chemico-biological interactions. 2011;192:96–100. doi: 10.1016/j.cbi.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Pierre SR, Lemmens MA, Figueiredo-Pereira ME. Subchronic infusion of the product of inflammation prostaglandin J2 models sporadic Parkinson’s disease in mice. J Neuroinflammation. 2009;6:18. doi: 10.1186/1742-2094-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Wong SM, Cotman CW, Cribbs DH. 15-deoxy-delta12,14-prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor-gamma, induces neuronal apoptosis. Neuroreport. 2001;12:839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Saito S, Takahashi S, Takagaki N, Hirose T, Sakai T. 15-Deoxy-Delta(12,14)-prostaglandin J2 induces apoptosis through activation of the CHOP gene in HeLa cells. Biochem Biophys Res Commun. 2003;311:17–23. doi: 10.1016/j.bbrc.2003.09.161. [DOI] [PubMed] [Google Scholar]
- Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem. 2002;277:10459–10466. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- Shibata T, Yamada T, Kondo M, Tanahashi N, Tanaka K, Nakamura H, Masutani H, Yodoi J, Uchida K. An endogenous electrophile that modulates the regulatory mechanism of protein turnover: inhibitory effects of 15-deoxy-Delta 12,14-prostaglandin J2 on proteasome. Biochemistry. 2003;42:13960–13968. doi: 10.1021/bi035215a. [DOI] [PubMed] [Google Scholar]
- Uchida K, Shibata T. 15-Deoxy-Delta(12,14)-prostaglandin J2: an electrophilic trigger of cellular responses. Chem Res Toxicol. 2008;21:138–144. doi: 10.1021/tx700177j. [DOI] [PubMed] [Google Scholar]
- Wang Z, Aris VM, Ogburn KD, Soteropoulos P, Figueiredo-Pereira ME. Prostaglandin J2 alters pro-survival and pro-death gene expression patterns and 26 S proteasome assembly in human neuroblastoma cells. J Biol Chem. 2006;281:21377–21386. doi: 10.1074/jbc.M601201200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Figueiredo-Pereira ME. Inhibition of sequestosome 1/p62 up-regulation prevents aggregation of ubiquitinated proteins induced by prostaglandin J2 without reducing its neurotoxicity. Mol Cell Neurosci. 2005;29:222–231. doi: 10.1016/j.mcn.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Lin T, Reeves SA. 15d-PGJ2 induces apoptosis of mouse oligodendrocyte precursor cells. J Neuroinflammation. 2007;4:18. doi: 10.1186/1742-2094-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 2010;1:779–786. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ikenoue T, Chen X, Li L, Inoki K, Guan KL. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proc Natl Acad Sci U S A. 2009;106:8923–8928. doi: 10.1073/pnas.0903621106. [DOI] [PMC free article] [PubMed] [Google Scholar]