

Abstract
We examined the impact of early-life exposure to methylmercury (MeHg) on Caenorhabditis elegans (C. elegans) pdr-1 mutants, addressing gene-environment interactions. We tested the hypothesis that early-life exposure to MeHg and knockout (KO) of pdr-1 (mammalian: parkin/PARK2) exacerbates MeHg toxicity and damage to the dopaminergic (DAergic) system. pdr-1KO worms showed increased lethality and decreased lifespan following MeHg exposure. Mercury (Hg) content, measured with Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) was increased in pdr-1KO worms compared to wildtype (N2) controls. 2′7′ dichlorodihydrofluorescein diacetate (H2DCF-DA) assay revealed a significant increase in reactive oxygen species (ROS) in both strains following MeHg exposure; however, while N2 worms showed an increase in skn-1 transcript levels following MeHg exposure, there was no difference in skn-1 induction in pdr-1KO worms. Dopamine-dependent behavioral analysis revealed an effect of MeHg on N2 wildtype worms, but no effect on pdr-1KO worms. Taken together, these results suggest that pdr-1KO worms are more sensitive to MeHg than wildtype worms, but MeHg does not exacerbate behavioral changes related to the absence of pdr-1.
Keywords: Methylmercury, parkin, early-life exposure, dopamine, large amino acid transporter
INTRODUCTION
Aging, early-life toxicant exposure and genetic predisposition, all likely contribute to the etiology of neurodegenerative disease. Several studies support an association between fish consumption or occupational exposure to mercury (Hg) and increased prevalence of Parkinson’s disease (PD) [1–4]. Challenges in determining the contributions of early-life exposures to etiologies of neurodegenerative diseases are the long aging period and presence of several confounds that can occur during the aging process. The nematode, C. elegans, is a unique model system, with a short lifecycle allowing temporal resolution in determining long-term effects of early-life exposures. Hg is a persistent environmental, bioaccumulative metal. Studies with methylmercury (MeHg) show that developmental exposure has long-term consequences. Pathophysiological effects may manifest years or even decades after exposure making it imperative to study the effects of early-life exposure in later life stages. We tested whether early-life exposure to MeHg, and genetics, are important factors in toxicity and neurodegeneration later in life.
Dopamine neurons are known to be sensitive to environmental neurotoxicants [5, 6] and high Hg levels have been detected in PD patients [1, 2]. Autosomal recessive juvenile Parkinson’s disease (ARJP) has been linked to mutations in the human parkin (PARK2) gene [7, 8]. C. elegans contain an ortholog for parkin called pdr-1. pdr-1 shares homology and conserved function to the human gene and is expressed in most neurons [9]. C. elegans hermaphrodites possess eight DAergic neurons, with a fully characterized genome that contains all genes responsible for DA biosynthesis, packaging and reuptake [10] and behavioral assays, including the basal slowing response, have been shown to be good indicators of DAergic function [11].
Several studies have highlighted the effects of MeHg on the dopaminergic (DAergic) system. While DAergic neurons are not the only subpopulation susceptible to MeHg, MeHg inhibits DA uptake, and systemic or intrastriatal MeHg administration increases rat striatal DA release [12, 13]. In vitro rat synaptosomes show age-dependent sensitivity to MeHg, characterized by higher DA release and lower DAT activity [4, 14]. Delayed effects on a number of DAergic parameters, including DA levels, turnover and uptake, occur in rat offspring at weaning following in utero MeHg exposure [15]. Transient effects on DA receptor number associated with behavioral dysfunctions are noted in rat pups exposed at late gestation to a single high-dose of MeHg [16].
MeHg also modifies kinase signaling pathways, like activation of c-jun N-terminal kinases [17]; shown to play an important role in the degeneration of DAergic neurons [16]. Oxidative stress and lipid peroxidation are shared mechanisms in mediating neuronal death in both MeHg and neurodegenerative diseases [1]. SKN-1 (mammalian: Nrf2) is also posited to be an important mediator of DAergic loss due to its involvement in the stress response and its expression DAergic neurons [18].
MeHg entry into cells occurs through the large amino acid transporter (LAT1), and levels of LAT1 are highly expressed during the prenatal period due to its role in amino acid transport during neurodevelopment [19], making the developing brain particularly vulnerable to MeHg. The MeHg-cysteine complex closely mimics the structure of methionine, making it a substrate for LAT1 [20, 21]. C. elegans have nine genes [aat-1 to aat-9 (amino acid transporter catalytic chain)] that encode homologues of LAT1. Three of these genes (aat-1, aat-2, and aat-3) have a much higher degree of similarity to the mammalian homologs [22].
The studies described herein were designed to establish novel and translational information on mechanisms associated with DAergic neurodegeneration, especially after early life exposure to MeHg and loss of the activity of parkin. Owing to parkin’s expression in neurons and MeHg’s involvement in DA neurodegeneration we tested the hypothesis that early-life exposure to MeHg and knockdown of pdr-1 exacerbates MeHg toxicity, and loss of DAergic function later in life.
MATERIALS AND METHODS
Note: Methylmercury is toxic and all mercurials were handled as potentially highly toxic compounds and disposed of properly.
1.1 C. elegans maintenance
C. elegans strains were handled and maintained at 20°C as previously described [23]. Worms were grown on plates containing nematode growth medium (NGM) or 8P seeded with either Escherichia coli strain OP50 or NA22, respectively, as previously described [23]. The hermaphroditic wildtype N2 Britol strain was used as a control for all experiments. The VC1024 (pdr-1 (gk448) III) strain was used for pdr-1 knockout (KO) experiments. VC1024 was backcrossed four times. All strains were obtained from the Caenorhabditis Genetics Center, Minneapolis, MN.
1.2 MeHgCl treatments
To obtain a synchronous population prior to treatment, worms were treated with an alkaline bleach solution. Methylmercuric chloride (CH3HgCl; Sigma-Aldrich) treatments (0–50μM) were performed for 30 minutes using synchronized L1 worms to determine appropriate dosing. Five thousand (lifespan, lethality, behavior and broodsize), 10,000 (DCF assay), 20,000 (RNA), 50,000 (MeHgCl analyses) or 150,000 (dopamine) nematodes were treated with 0, 10 or 20μM MeHgCl. After treatment, worms were washed three times with M9 buffer (KH2PO4; Na2HPO4; and NaCl) and either plated on seeded NGM plates or collected for immediate analysis. A sample size of six (n = 6) represents the total number of independent worm preparations; each independent experiment was carried out with 5,000–150,000 worms (see above).
1.3 Lethality
Following treatment and washing, worms were plated on seeded 60 mm NGM plates and allowed to grow for 24 hours. Worms were then counted and scored using a grid system. Nematodes on 4 of the 64 grids were counted and the number of worms per grid was averaged and multiplied by 64, and results expressed as percent control.
1.4 Lifespan and brood size
For determination of lifespan, 40 nematodes from each dose group were picked to a fresh NGM plate 24 hours following treatment. The worms were counted every day and scored as live or dead. Live C. elegans were picked to new plates every day during the egg-laying period of their lifecycle and once egg-laying ceased they were picked every other day until no live C. elegans remained.
For broodsize, four worms were placed individually on four NGM plates per treatment concentration 24 hours after MeHgCl treatment. Every 24 hours, the worm was transferred to a new NGM plate until no new progeny were generated in a 24 hour period. The progeny on each plate were counted and all plates from one hermaphrodite were added together and expressed as total progeny.
1.5 Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) for Mercury content
The protocol used for ICP-MS was carried out as previously described [23]. Briefly, worms were treated with MeHgCl, followed by three washes. Samples were then allowed to air dry until a dehydrated pellet was obtained. Next, the dried sample was transferred to preweighed Teflon jars and reweighed. Concentrated acids HNO3, HCl, H2SO4 were added, and the samples were placed in heat block at 100°C under the hood overnight. The following day digested samples were reweighed and placed in preweighed metal-free 15mL centrifuge tubes. They were then analyzed by ICP-MS, which has a Hg limit-of-detection in the parts per trillion range.
1.6 RNA isolation
Following treatments, synchronized L1 stage worms were washed 3 times with M9 and frozen at −80°C until RNA purification. Worm pellets were resuspended in Trizol (200μL/100μL pellet); the protein and other impurities were separated from nucleic acids using chloroform. The pellet was then washed with 70% EtOH; DNA was digested using RNAse free DNAse kit (Ambion). RNA concentrations were determined (OD 260nm) using a ND-1000 spectrophotometer (NanoDrop®). Purified mRNA was stored at −80°C.
1.7 cDNA synthesis
Reverse transcription reactions were performed in 20 μL following the manufacturer’s protocol (Applied Biosystems). A 10μL reaction mixture containing, 10X RT buffer, 25X dNTP mix (100mM), 10X random primers, Multiscribe reverse transcriptase and sterile nuclease-free water was added to 10μL of RNA (75ng). The contents were mixed gently and incubated at 25°C for 10 minutes, 37°C for 120 minutes followed by inactivation by heating at 85°C for 5 minutes. Synthesized cDNA was stored at −20°C until used. The suitability of the cDNA for PCR was determined using 2 μL of cDNA added to a 40 μL PCR. A control reaction containing 2μl of purified mRNA was run without reverse transcription to test for presence of DNA contamination in the purified mRNA samples. The PCR was analyzed on a 1.5% (w/v) agarose gel and cDNA was stored at −20°C.
1.8 Primers
Oligonucleotide sequences for pdr-1:5′GGACGGAGAAGCATGATTTG3′ 5′GCACATGACTGCGAGGACTA3′; aat-1: 5′TATTCCGGTGCTCGTGAAGGACAA3′ and 5′GCTGTTCCGATTGCAAGCCAGTAA3′; aat-2: 5′ ACTTCCTTCTGCTACATCGGGCAT3′ and 5′ CGCCGGATGCATAATTGCTGTTCT3′; aat-3: 5′ ACTGGATTGCTATTGGAGGAGCCA3′ and 5′ ACAAGAAGCACGCACCCAATGAAG3′; skn-1: 5′AGTGTCGGCGTTCCAGATTTC3′ and 5′GTCGACGAATCTTGCGAATCA3′; gapdh (housekeeping):5′CAATGCTTCCTGCACCACTA3′ and 5′CTCCAGAGCTTTCCTGATGG3′. The specificity of each primer pair was confirmed by the identification of a single PCR product of predicted size on 1.5% (w/v) agarose gels. Oligonucleotide sequences were designed based on published literature.
1.9 Semi-quantitative determination of transcript levels by Real-time PCR
Real-time PCR was conducted in a Gene Amp 7300 sequence detection system (Applied Biosystems) using a 96-well plate (MicroAmp™ Fast). The relative quantification was determined with the 2−ΔΔCt method [24]. GAPDH was used as an internal control.
1.10 DCF assay for oxidative stress
The formation of reactive oxygen species (ROS) was evaluated with 2′7′ dichlorodihydrofluorescein diacetate (H2DCF-DA). Synchronized L1s were treated with MeHg as described earlier, and washed 4 additional times in M9 buffer. H2DCF-DA (1mM in DMSO) was then added to the tube with worms (in M9) for one hour in the dark. After 1h, the worms were washed three times with M9 to remove all H2DCF-DA content outside the worms. Worms were frozen and thawed twice and homogenized by sonication and then centrifuged. The supernatants were transferred to a 96-well plate and their fluorescence levels (excitation: 485 nm; emission: 535 nm) detected with a FLEXstation III (Molecular Devices) pre-heated at 37°C. The fluorescence from each well was measured initially and at 1 hour. Values from final readings (one hour time point) minus initial readings were adjusted for background signal and reported as percent fluorescence relative to their respective control strain.
1.11 Basal slowing behavioral analysis
The behavioral analysis was adapted from a previously published protocol [11]. Well-fed L4 worms from the same treatment group were placed on two sets of 60×15mm plates: one with ~20μL bacteria spread in a ring with an inner diameter of ~1cm and an outer diameter of ~3.5cm and one without bacteria (both incubated at 37°C overnight and cooled to room temperature prior to assay). Bacterial mechanosensation induces the dopamine-mediated slowing of locomotion in the presence of food (bacteria), and can be measured by counting the number of body bends per 20-second interval. Locomotor rates were then compared between the well-fed worms placed on plates of food versus those placed on plates without food and is referred to as the change (Δ) in body bends/20 seconds. A lower value represents less slowing on food, indicating deficits in DAergic function. The cat-2 strain is tyrosine hydroxylase deficient and therefore defective in bacterial mechanosensation, making it a positive control [11].
1.12 Statistical analysis
Statistics were carried out and graphs were generated with GraphPad Prism. Briefly, we used a sigmoidal dose-response model with a top constraint at 100% to draw the curves and determine the LD50. The Kaplan-Meier method was used to estimate survival curves, and curves were compared using the log rank test or Wald test of coefficients from a Cox proportional hazards regression. Statistical analysis of significance was carried out by one-way ANOVA for brood size, and change in DA-mediated locomotor activity, and two-way ANOVA for ROS content, transporter levels, skn-1 and MeHg content, followed by post-hoc Bonferroni or student’s t-test when p<0.05. In all figures, error bars are ±SEM.
RESULTS
2.1 MeHg-induced lethality in N2 and pdr-1KO worms (Figure 1)
Figure 1.
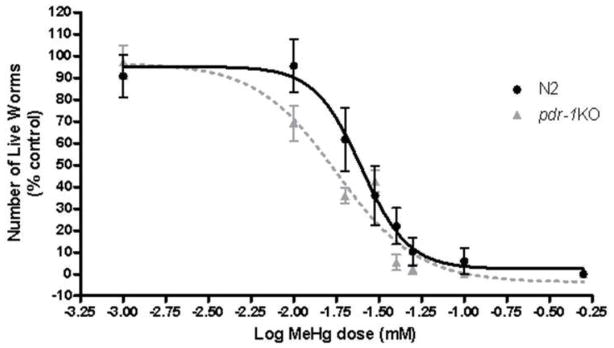
Dose-response curve of MeHg-induced lethality in N2 and pdr-1KO worms. pdr-1KO worms (LD50= 17 μM, n=10) were more sensitive to MeHg than worms wildtype worms (LD50 for N2 = 25 μM; n=8).
Assessment of lethality following a 30 minute exposure revealed a leftward-shift in the curve for pdr-1 mutants compared to N2 wildtype worms. LD50 for N2 = 24 ± 0.059 μM; LD50 for pdr-1KOs = 17 μM ± 0.051. These data suggest that pdr-1 KO worms are more sensitive to MeHg than N2 wildtypes. The doses used in this study, 10 and 20 μM, were chosen based on the LD50 values for each strain.
2.2 Lifespan and brood size (Figures 2 and 3)
Figure 2.

Lifespan is affected in pdr-1KO worms but not wildtype after MeHg exposure. (A) N2 wildtype worms do not show a decrease in lifespan following MeHg exposure (B) pdr-1KOs exhibit a reduced lifespan overall (compared to wildtype worms) and a further reduction in lifespan (**p<0.05) following 10 or 20 μM MeHg exposure. Plotted values represent averages of three independent experiments, and the curves represent the best sigmoidal fit using log rank statistics.
Figure 3.

Brood size of wildtype and pdr-1KO nematodes following exposure to 10 or 20μM MeHg. (A) Wildtype worms treated with 0, 10, or 20μM MeHg (B) pdr-1KOs treated with 0, 10, or 20μM MeHg. Total progeny was significantly decreased in both strains following 20μM MeHg compared to 0μM control. Two-way ANOVA revealed a significant Dose effect (<0.0001) and Strain effect (<0.0001) but not a Dose x Strain interaction (p>0.05). Post-hoc analysis revealed a significant difference between N2 0μM and N2 20μM (**p=0.0055), pdr-1KO 0μM control and pdr-1KO 20μM (##p=0.0099) and between 20μM N2 and 20μM pdr-1KO (^^^p=0.0009).
Log-rank test revealed that MeHg had a significant effect on lifespan in pdr-1KO (**p<0.05), worms which was not seen in wildtype worms (Χ2 (2, N = 120) = 0.9708, p = 0.6154, n.s.) (fig. 2A/B). Knockout of pdr-1 alone had an effect on lifespan, which was exacerbated by MeHg (fig. 2) (Χ2 (2, N = 120) = 11.99, **p = .0025). Overall, there was a Dose effect (p<0.001) and a Strain effect (p=0.003). The broodsize analysis revealed a significant effect of Dose (F(2,30)=13.99, ***p<0.0001) and Strain (F(1,30)=20.37, ***p<0.0001) but not a Dose x Strain interaction (F(2, 30) = 1.741, p>0.05, n.s.) (fig. 3). Post-hoc analysis revealed a significant difference between N2 0μM and N2 20μM (**p=0.0055), pdr-1KO 0μM and pdr-1KO 20μM (##p=0.0099) and between 20μM N2 and 20μM pdr-1KO (^^^p=0.0009).
2.3 Mercury content (Figure 4)
Figure 4.

Hg content in wildtype and pdr-1KO worms. The was a significant effect of Dose and a trend toward a Strain effect. Hg content measured by ICP-MS. Two-way ANOVA revealed a statistically significant effect of Dose (p<0.0001) but Strain (p=0.06) and Dose x Strain interaction (p>0.05) failed to reach significance. Post-hoc analysis revealed a significant difference between pdr-1KO 0μM control and pdr-1KO 20μM (*p=0.016) and the difference between the 20μM wildtype and 20μM pdr-1KO failed to reach significance (p=0.06). Results are presented as mean ± SEM of 3–5 independent experiments.
Hg accumulation was higher in pdr-1KO worms compared to wildtype worms at the 20 μM dose, but failed to reach significance at the 10 μM dose. Overall, there was a significant effect of Dose (F(2,20)=15.83, ***p<0.0001). Although there was a difference in pdr-1KO worms compared to wildtypes, the Strain effect failed to reach statistical significance (F(1,20)=3.912, p=0.06, n.s.), as did the Dose x Strain interaction (F(2, 20) = 2.51, p>0.05, n.s.). There was a significant difference between pdr-1KO 0μM and pdr-1KO 20μM (*p=0.016), and the difference between the 20μM N2 wildtype and 20μM pdr-1KO failed to reach statistical significance (p=0.06).
2.4 aat-1/aat-2/aat-3 expression (Figure 5)
Figure 5.


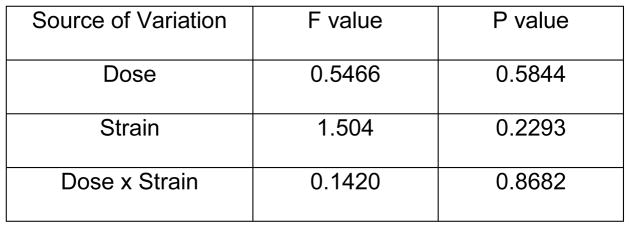
aat-1 and aat-2 but not aat-3 expression is higher in pdr-1KO worms than in wildtype worms. (A) aat-1 expression in wildtype and pdr-1KO worms. Two-way ANOVA revealed a statistical significant Dose effect (p=0.0196) but not a Strain, or Dose x Strain interaction (p>0.05). Post-hoc analysis showed significance. **denotes significance from 0μM wildtype control (*p<0.05, **p<0.005). (B) aat-2 expression in wildtype and pdr-1KO worms. Two-way ANOVA analysis showed no significant effect of any of the parameters measured (p>0.05). Post-hoc analysis showed significance. **denotes significance from 0μM wildtype control (*p<0.05, **p<0.005). (C) aat-3 expression in wildtype and pdr-1KO worms. Two-way ANOVA revealed no significant Dose or Strain effects, or Dose x Strain interaction (p>0.05). Post-hoc analysis showed significance. **denotes significance from 0μM wildtype control (*p<0.05). **denotes significance from 0μM wildtype control (*p<0.05, **p<0.005). Results are presented as mean ± SEM of 9–10 independent experiments.
Expression of these putative MeHg transporters was measured to determine if higher intraworm Hg concentration in pdr-1KOs was due to increased transporter expression. aat-1 expression There was a significant effect of Dose (F(2,45) = 4.30, *p < 0.05) but no significant effect of Strain (F(1,45) = 1.51, n.s), nor was there a Dose x Strain interaction (F(2,45) = 0.07, n.s.). Post-hoc analysis revealed a significant difference between 0μM wildtype worms and 10μM (*p<0.05) and 20μM (**p<0.005) wildtype worms, and there was a trend toward an increase in expression in pdr-1KOs. aat-2 expression There was no significant effect on any of the parameters measured; Dose (F(2,32) = 0.50, n.s.), Strain (F(1,32) = 2.42, n.s), or Dose x Strain interaction (F(2,32) = 0.36, n.s.). Post-hoc analysis revealed a significant difference between 0μM wildtype worms and 10μM and 20μM wildtype worms (*p<0.05), and a significant difference between wildtype 0μM and pdr-1KO 0μM (**p<0.005). aat-3 expression There was no significant effect on any of the parameters measured; Dose (F(2,31) = 0.5466, n.s.), Strain (F(1,31) = 1.504, n.s), or Dose x Strain interaction (F(2,31) = 0.1420, n.s.). Post-hoc analysis revealed significant differences between 0μM wildtype and pdr-1KO 0μM (*p<0.05).
2.5 Reactive oxygen species (ROS) (Figure 6)
Figure 6.

Reactive oxygen species (ROS) are elevated following MeHg treatment. Wildtype and pdr-1KO worms exhibit significantly higher ROS upon MeHg treatment. Two-way ANOVA showed a significant effect of Dose (p<0.05), but the effect of Strain and the Dose x Strain interaction were not significant (p>0.05). *denotes significance from 0μM wildtype (*p<0.05). ##denotes significance from pdr-1KO 0μM (# p<0.05; ##p<0.005). NaNP=sodium nitroprusside (500μM) used as a positive control.
To study MeHg-induced stress response, we measured the production of ROS. Using the DCF assay we show a significant increase in ROS in both strains 1 hour after 10 μM MeHg treatment (fig. 6). The effect of Dose was significant (F(2,14) = 5.029,*p<0.05), but the effect of Strain (F(1,14) = 0.1179, n.s.) and the Dose x Strain interaction (F(2,14) = 0.03061, n.s.), were not significant. Sodium nitroprusside (NaNP) was used as a positive control and MeHg in the absence of worms was used as a negative control. NaNP was significantly different from 0μM wildtype and 0μM pdr-1KO (*p<0.05; #p<0.05).
2.6 MeHg increases skn-1 expression in wildtype but not pdr-1KO worms (Figure 7)
Figure 7.

skn-1 is induced upon exposure to 10 and 20 μM MeHg in wildtype worms but not pdr-1KOs. There was a significant Strain and Dose effect but not a Dose x Strain interaction. Post-hoc analysis revealed a significant difference between the 0μM wildtype worms and the 10μM wildtype (**p=0.008) and 20μM wildtype strain (***p=0.0008). The difference between the10μM wildtype strain vs. the 10μM pdr-1KO strain was also significant (#p=0.02). The difference between the 0μM wildtype strain vs. the 0μM pdr-1KO strain (p=0.056) and the 20μM wildtype strain vs. the 20μM pdr-1KO strain failed to reach significance (p=0.06).
Whole extracts were used to probe for skn-1 transcriptional activity following 30 minute exposure to MeHg. Using qRT-PCR, we show that wildtype worms significantly upregulate skn-1 in response to MeHg (fig. 7), but pdr-1KOs do not. Differences in the average threshold cycle (ΔCt) were determined and normalized to the expression of GAPDH. The Strain effect was significant (F(1,50)=12.71, ***p<0.001), as was the effect of Dose (F(2,50)=5.73, **p<0.01). There was no Dose x Strain interaction (F(2,50)=1.46, n.s.). These data suggest that wildtype worms mounted an antioxidant response, whereas pdr-1KO worms failed to do so in response to MeHg.
2.7 Basal slowing behavioral analysis (Figure 8)
Figure 8.
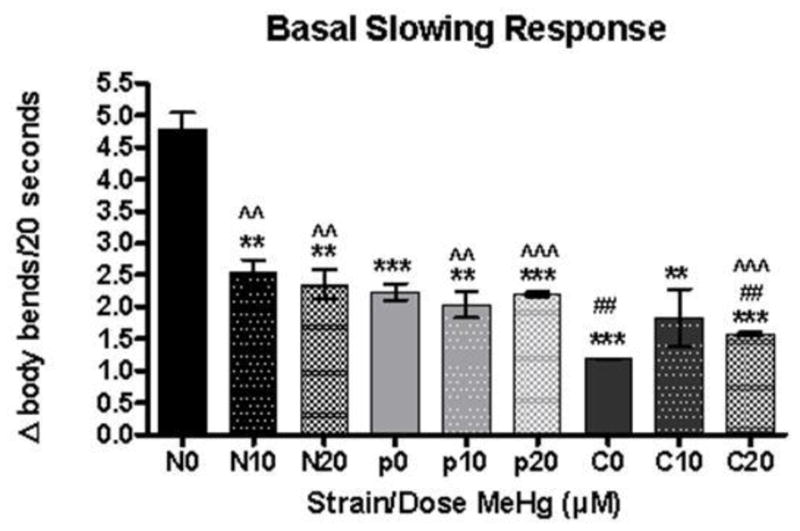
MeHg has an effect of the Basal Slowing Response in wildtype but not pdr-1KO worms. Change in DA-mediated locomotor activity represented by ratio off food/on food. N=N2; p=pdr-1KO; C=cat2 **denotes significance from 0μM wildtype control (**p<0.05, ***p<0.0005). ## denotes significance from 0μM pdr-1KO (##p<0.05). ^^denotes significance from 0μM cat2. Error bars represent the mean ± SEM of 25–40 individual worms.
We measured DA-mediated behavior at 72 hours post exposure (adult stage) to determine if there was a decrease in functional DA. For each strain, locomotion rates in the absence and presence of bacteria (supplemental fig. 1) were calculated, and results are presented as Δ body bends/20 seconds (fig. 8). Asterisks indicate values significantly different from the slowing seen in the wildtype untreated N2 worms. Higher values indicate functioning DAergic neurons, while lower values indicate dysfunction. When the change in locomotor activity (Δ body bends/20 seconds) was compared by one way ANOVA, it was statistically significant (F(8,26)=22.60, ***p<0.0001). Post-hoc analysis showed all doses/strains were significantly different from 0 μM wildtype (**p<0.05, ***p<0.0005). There was an effect of MeHg on wildtype worms but not on pdr-1KO worms. cat-2 unexposed worms were significantly different from all MeHg-treated wildtype and pdr-1KO worms irrespective of dose (^^p<0.05).
DISCUSSION
Due to the known effects of MeHg on the DAergic system [4, 12, 13, 16] and the increased risk for damage to the DAergic system with parkin mutations [7, 8], we tested the hypothesis that loss of pdr-1 exacerbates MeHg toxicity, with significant decreases in DAergic function following early-life exposure to MeHg. Because parkin is implicated in ARJP, the juvenile form of PD, our exposures were carried out in early-life stages. Mammals are susceptible to MeHg during the perinatal stage, including gestation and early postnatal periods [25].
Lethality was used as a short-term determinant of toxicity, revealing a leftward-shift in the curve for pdr-1KO worms compared to wildtype worms. The shift is indicative of increased sensitivity of pdr-1KOs to MeHg (fig. 1). This increased sensitivity to MeHg in pdr-1KOs has not been previously described, but is not surprising considering that environmental contaminants, including metals, have been implicated in the pathogenesis of PD [5, 6, 23].
Survival assays revealed decreased lifespan in pdr-1KO worms compared to wildtype worms, which was exacerbated by exposure to MeHg (figs. 2A/B). The lack of a MeHg effect in wildtype worms is consistent with an earlier report [23], and the decrease in lifespan in pdr-1KO worms compared to wildtype worms is corroborated by studies in Drosophila that show significant decreases in lifespan in flies lacking parkin compared to control flies [26]. To our knowledge this is the first report on the effect of MeHg on pdr-1KO lifespan. Broodsize was decreased in both strains at the 20 μM dose of MeHg (fig. 3), consistent with the literature from MeHg-exposed wildtypes [18, 23]. While this is the first report of the effect of early-life MeHg on parkin, studies in parkin mutant mice, show no differences in viability or reproduction compared to controls [27], analogous to our data.
Intraworm Hg burden was higher in pdr-1KO worms compared to wildtypes (fig. 4), suggesting that they either take up more Hg or fail to extrude Hg. To date, there are no studies implicating parkin in regulating LAT1 expression; however, a role for parkin in ubiquitination and degradation of the divalent metal transporter 1 (DMT1) in vitro [28] and in vivo (unpublished observation) has been demonstrated. Accordingly, parkin may have the ability to modulate the aat’s, thereby increasing susceptibility to MeHg toxicity.
The increase in ROS (fig. 6) in both strains is consistent with the literature on MeHg [1]. We, and others, have shown that MeHg triggers oxidative stress and lipid peroxidation in multiple experimental models of neurodegeneration [18, 29]. Additionally, mutations in parkin in the human teratocarcinoma (NT-2) and neuroblastoma (SK-N-MC) cell lines have been shown to increase levels of 8-hydroxyguanine, protein carbonyls and 3-nitrotyrosine, and to decrease levels of glutathione (GSH), in response to H2O2, MPP+ or 4-hydroxy-2-trans-nonenal (HNE) [30], consistent with increased oxidative stress. Post-mortem PD patients show increased lipid peroxidation, protein oxidation, 3-nitrotyrosine formation, DNA oxidation and breaks, and a decrease in the activities of ROS-scavenging enzymes and glutathione peroxidase [31]. An additional study in a human DA neuroblastoma cell line (SH-SY5Y) demonstrated that overexpression of parkin decreased the level of ROS and protein carboynyls and attenuated DA-induced activation of c-Jun N-terminal kinase (JNK) and caspase-3 [32]. We didn’t observe a significant increase in ROS levels in pdr-1KOs compared to wildtypes in response to MeHg exposure; this doesn’t exclude the possibility that the DAergic neurons are more affected as we are measuring whole worm levels. The lack of a significant increase in ROS at the 20μM dose could be due to the increased death of worms at this dose and therefore a decrease in active mitochondria at the time point measured. An alternative explanation could be that the species that are measured by the dye are not the primary ROS that are generated at the 20μM dose.
Given the increase in ROS in both strains, we measured skn-1 expression to determine if the presence of ROS was activating a stress response. skn-1 induction was dose-dependent in wildtype worms, but there was a lack of a significant response in pdr-1KO worms (fig. 7). This lack of response by pdr-1KOs is important and likely contributes to the reduction in lifespan after MeHg exposure (fig. 2). Extended longevity correlates with enhanced resistance against oxidative damage [33, 34]; therefore, failure to initiate an oxidative stress response would decrease longevity which is evident in our lifespan data. skn-1 mutants are sensitive to oxidative stress and have shortened lifespans [35]. Evidence from induced pluripotent stem cells (iPSCs) derived from two PARK2 patients showed increases in NRF2 with decreases in GSH compared to their control lines [36]. We did not observe increased skn-1 levels in pdr-1KOs compared to wildtypes as was shown in the Imaizumi study, which may be due to the fact that we are measuring whole worm levels. While we did not directly measure GSH levels, we would predict that levels of GSH would be decreased in our pdr-1KOs in response to MeHg as many of the glutathione s-transferases (gsts) are under the control of the SKN-1-activated antioxidant response element (ARE) promoter. GSH and several gsts were reported to increase following MeHg exposure in wildtype C. elegans [18, 37]. skn-1 has been shown to affect lifespan [35] and may participate in pdr-1KO-induced shortened lifespan. The lack of antioxidant response in pdr-1KOs can be exacerbated by the propensity of MeHg to bind to GSH thereby attenuating the cellular redox status and leaving neurons more susceptible to damage by ROS [38].
Functioning DAergic neurons are required for food sensing (mechanosensation) [11]. The basal slowing response behavioral assay revealed a decrease in the Δ body bends/20 seconds in wildtype but not in pdr-1KO worms (fig. 8), suggesting a decrease in DA-dependent function by 72 hours in wildtype worms but not pdr-1KOs. The absence of a MeHg effect in pdr-1KO worms may be due to the decreased functioning of the DAergic neurons even at the 0μM control dose and the presence of a ‘floor’ effect, as the control dose in pdr-1KO worms was similar to the 20μM MeHg dose in wildtype worms. Studies in parkin mutant mice indicate deficits in DA function, including abnormal baseline motor activity lack of amphetamine-induced increased locomotor activity, reduced amphetamine-induced dopamine release, decreased levels of VMAT2 and DAT protein and reduced [3H]-DA uptake [27]. An additional rodent study showed decreased DA release and uptake in young parkin knockout animals (Oyama et al., 2010). Full characterization of the DAergic system in pdr-1KOs following early-life MeHg will be necessary to understand if the lack of MeHg effect is limited to DA-mediated behavior or extends to other measurements in the DAergic system. Future studies will assess structural integrity of the DAergic neurons as well as attempt to quantify DA directly, which has proven to be difficult in worms (unpublished observations).
In summary, the combination of early-life exposure to MeHg and pdr-1KO had significant effects on oxidative stress and aging. Our data suggest that early-life MeHg exposure is a risk factor for loss of DAergic function later in life in wildtype worms but the combination of pdr-1KO and early-life MeHg does not further exacerbate the already reduced DAergic function produced by pdr-1KO alone.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) [R01ES07331, T32 ES007028] and the National Institutes of Health Loan Repayment Program. We would like to acknowledge the C. elegans Reverse Genetics Core Facility at UBC, which is part of the International C. elegans Gene Knockout Consortium and the Caenorhabditis Genetic Center (CGC) for providing the strains used in this manuscript. The authors would also like to acknowledge contributions from Chris Muller.
References
- 1.Gorell JM, et al. Occupational metal exposures and the risk of Parkinson’s disease. Neuroepidemiology. 1999;18(6):303–8. doi: 10.1159/000026225. [DOI] [PubMed] [Google Scholar]
- 2.Kirkey KL, et al. Occupational categories at risk for Parkinson’s disease. Am J Ind Med. 2001;39(6):564–71. doi: 10.1002/ajim.1055. [DOI] [PubMed] [Google Scholar]
- 3.Ngim CH, Devathasan G. Epidemiologic study on the association between body burden mercury level and idiopathic Parkinson’s disease. Neuroepidemiology. 1989;8(3):128–41. doi: 10.1159/000110175. [DOI] [PubMed] [Google Scholar]
- 4.Wermuth L, et al. Prevalence and incidence of Parkinson’s disease in The Faroe Islands. Acta Neurol Scand. 2008;118(2):126–31. doi: 10.1111/j.1600-0404.2007.00991.x. [DOI] [PubMed] [Google Scholar]
- 5.Brooks AI, et al. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999;823(1–2):1–10. doi: 10.1016/s0006-8993(98)01192-5. [DOI] [PubMed] [Google Scholar]
- 6.McCormack AL, et al. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10(2):119–27. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 7.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno Y, et al. Parkin and Parkinson’s disease. Curr Opin Neurol. 2001;14(4):477–82. doi: 10.1097/00019052-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Springer W, et al. A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum Mol Genet. 2005;14(22):3407–23. doi: 10.1093/hmg/ddi371. [DOI] [PubMed] [Google Scholar]
- 10.Sulston J, Dew M, Brenner S. Dopaminergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol. 1975;163(2):215–26. doi: 10.1002/cne.901630207. [DOI] [PubMed] [Google Scholar]
- 11.Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26(3):619–31. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 12.Bartolome J, et al. Exposure to methylmercury in utero: effects on biochemical development of catecholamine neurotransmitter systems. Life Sci. 1984;35(6):657–70. doi: 10.1016/0024-3205(84)90261-3. [DOI] [PubMed] [Google Scholar]
- 13.Dreiem A, et al. Methylmercury inhibits dopaminergic function in rat pup synaptosomes in an age-dependent manner. Neurotoxicol Teratol. 2009;31(5):312–7. doi: 10.1016/j.ntt.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Petersen MS, et al. Increased prenatal exposure to methylmercury does not affect the risk of Parkinson’s disease. Neurotoxicology. 2008;29(4):591–5. doi: 10.1016/j.neuro.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 15.Castoldi AF, et al. Brain monoaminergic neurotransmission parameters in weanling rats after perinatal exposure to methylmercury and 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB153) Brain Res. 2006;1112(1):91–8. doi: 10.1016/j.brainres.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 16.Dare E, et al. Effects of prenatal exposure to methylmercury on dopamine-mediated locomotor activity and dopamine D2 receptor binding. Naunyn Schmiedebergs Arch Pharmacol. 2003;367(5):500–8. doi: 10.1007/s00210-003-0716-5. [DOI] [PubMed] [Google Scholar]
- 17.Fujimura M, et al. Methylmercury exposure downregulates the expression of Racl and leads to neuritic degeneration and ultimately apoptosis in cerebrocortical neurons. Neurotoxicology. 2009;30(1):16–22. doi: 10.1016/j.neuro.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Vanduyn N, et al. SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci. 2010;118(2):613–24. doi: 10.1093/toxsci/kfq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boado RJ, Li JY, Pardridge WM. Developmental regulation of the rabbit blood-brain barrier LAT1 large neutral amino acid transporter mRNA and protein. Pediatr Res. 2004;55(4):557–60. doi: 10.1203/01.PDR.0000113461.07950.72. [DOI] [PubMed] [Google Scholar]
- 20.Kerper LE, Ballatori N, Clarkson TW. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am J Physiol. 1992;262(5 Pt 2):R761–5. doi: 10.1152/ajpregu.1992.262.5.R761. [DOI] [PubMed] [Google Scholar]
- 21.Yin Z, et al. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem. 2008;107(4):1083–90. doi: 10.1111/j.1471-4159.2008.05683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veljkovic E, et al. Functional characterization of Caenorhabditis elegans heteromeric amino acid transporters. J Biol Chem. 2004;279(9):7655–62. doi: 10.1074/jbc.M309528200. [DOI] [PubMed] [Google Scholar]
- 23.Helmcke KJ, et al. Characterization of the effects of methylmercury on Caenorhabditis elegans. Toxicol Appl Pharmacol. 2009;240(2):265–72. doi: 10.1016/j.taap.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Goulet S, Dore FY, Mirault ME. Neurobehavioral changes in mice chronically exposed to methylmercury during fetal and early postnatal development. Neurotoxicol Teratol. 2003;25(3):335–47. doi: 10.1016/s0892-0362(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 26.Saini N, et al. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol Dis. 2010;40(1):82–92. doi: 10.1016/j.nbd.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Itier JM, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12(18):2277–91. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- 28.Roth JA, et al. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J Neurochem. 2010;113(2):454–64. doi: 10.1111/j.1471-4159.2010.06607.x. [DOI] [PubMed] [Google Scholar]
- 29.Ni M, et al. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol Sci. 2010;116(2):590–603. doi: 10.1093/toxsci/kfq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyun DH, et al. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J Neurosci Res. 2005;82(2):232–44. doi: 10.1002/jnr.20638. [DOI] [PubMed] [Google Scholar]
- 31.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson’s disease. Mov Disord. 1998;13(Suppl 1):24–34. [PubMed] [Google Scholar]
- 32.Jiang H, et al. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13(16):1745–54. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- 33.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 34.Lithgow GJ, Walker GA. Stress resistance as a determinate of C. elegans lifespan. Mech Ageing Dev. 2002;123(7):765–71. doi: 10.1016/s0047-6374(01)00422-5. [DOI] [PubMed] [Google Scholar]
- 35.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003;17(15):1882–93. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imaizumi Y, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. 2012;5(1):35. doi: 10.1186/1756-6606-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmcke KJ, Aschner M. Hormetic effect of methylmercury on Caenorhabditis elegans. Toxicol Appl Pharmacol. 2010;248(2):156–64. doi: 10.1016/j.taap.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bramanti E, et al. Application of mercury cold vapor atomic fluorescence spectrometry to the characterization of mercury-accessible -SH groups in native proteins. Anal Biochem. 1999;274(2):163–73. doi: 10.1006/abio.1999.4257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.