

Abstract
A major challenge in drug discovery is to develop and improve methods for targeting protein-protein interactions. Further exemplification of the REPLACE strategy for generating inhibitors of protein-protein interactions demonstrated that it can be used to optimize fragment alternatives of key determinants, to combine these in an effective way and was achieved for compounds targeting the CDK2 substrate recruitment site on the cyclin regulatory subunit. Phenylheterocyclic isosteres replacing a critical charge-charge interaction provided new structural insights for binding to the cyclin groove. In particular, these results shed light onto the key contributions of a H-bond observed in crystal structures of N-terminally capped peptides. Furthermore the structure-activity relationship of a bisarylether C-terminal capping group mimicking dipeptide interactions, was probed through ring substitutions, allowing increased complementarity with the primary hydrophobic pocket. This study further validates REPLACE as an effective strategy for converting peptidic compounds to more pharmaceutically relevant compounds.
INTRODUCTION
CDKs, the cyclin regulatory subunits and their natural inhibitors, the CDK tumor suppressor proteins (CDKIs), are central to cell cycle regulation and their functions are commonly altered in tumor cells. Deregulation of CDK2 and CDK4 through inactivation of CDKIs such as p16INK4a, p21WAF1, p27KIP1, and p57KIP2 can override the G1 checkpoint 1; 2 and lead to transformation. CDKs interact with certain cell cycle substrates through the cyclin binding motif (CBM) and form a complex with the cyclin groove of the G1 and S phase cyclins, a surface binding site involving a protein-protein interaction. It has been shown that CDK isoform and substrate selective inhibition can be achieved through the use of peptides that block recruitment of both pRb and E2F and potently inhibit CDK2/cyclin A2 kinase activity3. Inhibition of CDKs through the cyclin provides an approach to obtain selectivity against other protein kinases and to specifically block the activity of the G1 and S phase CDKs (only these contain a functional cyclin binding groove). In particular, CDKs including CDK7 and 9, that regulate the RNA polymerase-II (RNAPII) transcription cycle should be unaffected by Cyclin Groove Inhibitory (CGI) compounds. Although it has been shown that cancer cells depend on the RNAPII cycle to express anti-apoptotic genes and that inhibition of transcriptional CDKs leads to potent anti-tumor agents 4, it is at the same time likely that this will lead to effects in normal cells and may be responsible for toxicities observed with CDK inhibitors that have been clinically evaluated.
The cyclin binding motif represents a consensus of the cyclin groove binding sequences found in many cell cycle and tumor suppressor proteins 3; 5. CGI peptides in transducible form have been shown to induce cell cycle arrest and selective apoptosis in tumor cells in vitro6–8. These permeabilized peptides also act as anti-tumor agents in that when administered directly to a SVT2 mouse tumor model, significant tumor growth inhibition was obtained and histological analysis showed that tumors underwent apoptosis9. The significant anti-tumor effects obtained through these peptide studies, provides strong impetus for further drug development with the aim of obtaining more drug-like small molecule CGI inhibitors. To this end, we developed a unique strategy for iteratively modifying and replacing peptide determinants in a step-wise fashion 10.
In the search for more drug-like compounds and in order to iteratively convert effective peptidic cyclin groove inhibitors into more pharmacologically appropriate compounds, the REPLACE (REplacement with Partial Ligand Alternatives through Computational Enrichment) strategy was applied to discover mimetics 10. The peptide sequence, HAKRRLIF was previously identified as an optimized variant of the native p21WAF1 cyclin binding motif sequence 5; 11. It contains three major binding determinants including Ala2 interacting with a minor hydrophobic pocket, Arg4 which forms a charge-charge interaction with acidic cyclin residues, and the Leu-Ile-Phe motifette contacting the primary hydrophobic pocket (Figure 1A). As shown by peptide SAR and truncation of the N-terminal residues, loss of interaction with secondary hydrophobic pocket can result in significant decreases in potency (RRLIF is 10 fold less potent that HAKRRLIF)11; 12.
Figure 1.

REPLACE mediated conversion of the octapeptide cyclin groove inhibitor HAKRRLIF into an N and C-terminally capped dipeptide. (Structures were modeled from PDB ID 2UUE).
Using the REPLACE methodology, low molecular weight fragments are docked into the vacated space of a ligand-receptor complex structure which has been created by truncation of peptide determinant. After fragments with requisite chemical functionality are selected through prioritization through scoring of the non-bonded interactions of the docked complexes, they are then chemically ligated onto the truncated peptide through solid phase parallel synthesis. This approach has been validated through in vitro screening of FLIP fragment ligated inhibitory peptides (FLIPs) where a hit rate of approximately 20% was obtained (the actual number of active FLIPS compared to the total number synthesized and tested) 10. A residue critical for binding and highly sensitive to substitution (Arg4) was successfully replaced with a 5-methyl-1-phenyl-1H-1,2,4-triazole-3-carboxylic acid N-terminal capping group (Ncap). Recently REPLACE has been applied and validated against another anti-cancer kinase drug target and more drug-like non ATP competitive PLK1 inhibitors obtained through fragment replacement 13.
In addition to the N-terminal REPLACE efforts, a 3-phenoxybenzylamine mimic for the C-terminal phenylalanine was identified, thereby providing a fragment based starting point for further lead optimization. In this present study, the optimization and structure-activity relationship determination for the aforementioned N and C-terminal capping groups was undertaken. Further validation for the REPLACE strategy was obtained not only through identification of partial ligand alternatives but also in combination of these optimized fragments through appropriate ligation methods. Efforts to maximize binding of the identified fragments in the context of the peptide were established before incorporating N and C-terminal groups into a single molecule that is considerably more drug-like and significantly less peptidic in nature. Exemplification of the REPLACE approach provides a significant advance in targeting protein-protein interactions, a major challenge in drug discovery.
RESULTS
N-terminal Partial Ligand Alternatives: Synthesis of derivatives and isosteres of 1-phenyl-1H-1,2,4-triazole-3 carboxamide
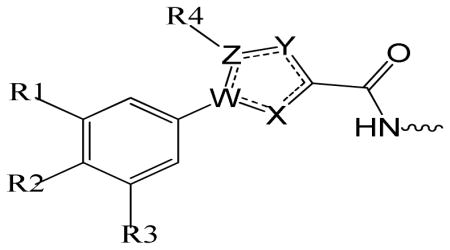
Various heterocyclic isosteres were designed based on the success applying REPLACE to the cyclin groove and in the identification of the 5-methyl-1-phenyl-1H-1,2,4-triazole-3-carboxylic acid capping group to replace Arg4 of the peptide 10. These were pursued further to introduce structural diversity and to improve drug-like properties. In order to more extensively validate the REPLACE strategy and to investigate the optimization of N-terminal capping groups in the Fragment Ligated Inhibitory Peptide (FLIP) context, structural modification of the phenyltriazole scaffold was undertaken. Furthermore in order to determine the sensitivity and importance of the heterocyclic ring for activity and to establish the optimal requirements of the phenyl ring substitution for binding to the cyclin groove, replacement core structures were synthesized and included 5-methyl-1-phenyl-1H-1,2,4-triazole-3-carboxylic acid (phenyltriazole), 5-methyl-1-phenyl-1H-pyrazole-3-carboxylic acid (phenylpyrazole), 1-phenyl-1H-pyrrole-3-carboxylic acid (phenylpyrrole), 2-phenyl-2H-imidazole-4-carboxylic acid (phenylimidazole), 5-phenylfuran-2-carboxylic acid (phenylfuran), and 2-phenylthiazole-4-carboxylic acid (phenylthiazole) as shown in table 1. The phenylpyrazole and phenylfuran isosteres in particular were selected as versatile core structure that could be exploited in order to generate diverse substitutions of the phenyl ring contacting the secondary hydrophobic pocket.
Table 1.
Structure activity of phenylheterocyclic N-terminal Partial Ligand Alternatives
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SCCP ID | R1 | R2 | R3 | R4 | W | X | Y | Z | CDK2/cyclin A IC50 (μM) |
CDK4/cyclin D1 IC50 (μM) |
|
| Triazole | 5843 | H | H | H | H | N | N | N | C | 16.2±3 | 49.2 ±7.60 |
| 5773 | Cl | H | Cl | CH3 | N | N | N | C | 4.0±0.6 | 27.3 ± 3.40 | |
| 5774 | H | Cl | H | CH3 | N | N | N | C | 11.5±3.3 | 12.0 ± 2.06 | |
| 5906 | Cl | H | H | CH3 | N | N | N | C | 5.7±0.99 | 26.5±7.49 | |
| 5907 | CH3 | H | H | CH3 | N | N | N | C | 13.8±1.13 | 51.05±16.05 | |
| Pyrazole | 5762 | H | H | H | CH3 | N | N | C | C | 40.3±6.5 | 54.2 ± 3.05 |
| 5763 | Cl | H | Cl | CH3 | N | N | C | C | 21.8±13.7 | >100 | |
| 5764 | Cl | H | H | CH3 | N | N | C | C | 11.9±2.0 | 70.3 ±12.79 | |
| 5771 | F | H | H | CH3 | N | N | C | C | 29.6±12.2 | 65.9±6.60 | |
| 5765 | H | Cl | H | CH3 | N | N | C | C | 33.7±8.1 | 54.7 ±20.43 | |
| 5766 | OCH3 | H | H | CH3 | N | N | C | C | 64.1±4.2 | >100 | |
| 5767 | H | OCH3 | H | CH3 | N | N | C | C | >180 | >180 | |
| Pyrrole | 5776 | H | Cl | H | H | N | C | C | C | >180 | >180 |
| 5775 | Cl | H | Cl | H | N | C | C | C | >180 | >180 | |
| Furan | 5768 | Cl | H | Cl | H | C | O | C | C | >180 | >180 |
| 5769 | Cl | H | H | H | C | O | C | C | >180 | >180 | |
| Imidazole | 5760 | H | H | H | CH3 | C | N | C | N | >180 | >180 |
| 5852 | F | H | H | H | C | N | C | N | 34.3±0.6 | 68.5 ±14.84 | |
| Thiazole | 5583 | H | Cl | H | H | C | N | C | S | >180 | >180 |
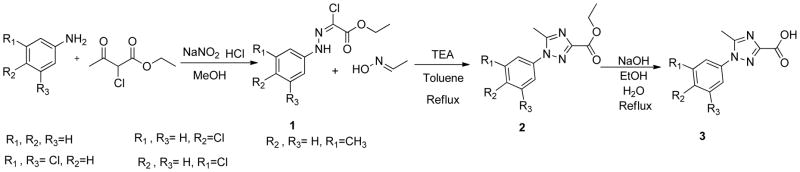
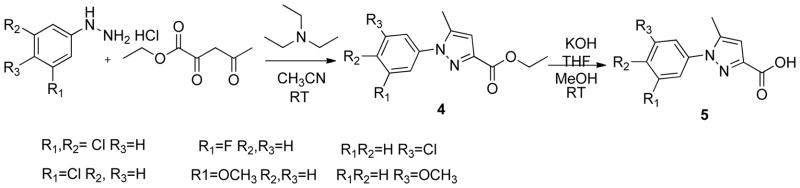
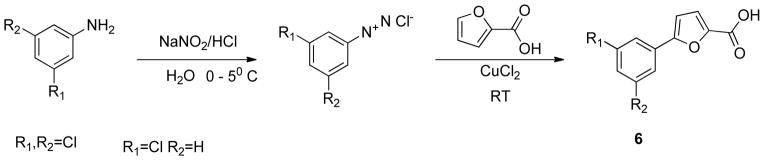
The phenyltriazole derived capping groups were generated in a diazonium reaction starting with the appropriate aniline and ethyl 2-chloroacetoacetate, followed by cyclization in the presense of acetaldehyde oxime (Scheme 1)14; 15. Synthesis of the phenylpyrazole scaffold was achieved through (Scheme 2) reaction of ethylacetopyruvate with the corresponding substituted phenyl hydrazine 16. Initial attempts involved base catalysis of the reaction upon which two isomers were obtained. The desired isomer was identified and confirmed through 1-D NOE analysis where irradiation of the R4 methyl group led to an enhancement of the two ortho aromatic hydrogens. This reaction was further optimized by performing the cyclization in acidic conditions thereby protonating the hydrazine and suppressing formation of the non-desired regioisomer. The versatile pyrazole synthesis allowed generation of a variety of analogs including the unsubstituted phenyl, the 3-methoxy and 4-methoxy phenyl as well as the 3,5-dichloro, 3 chloro and the 4 chlorophenyl compounds. The phenylfuran Ncaps were also synthesized through a diazotization of the requisite aniline followed by reaction with furoic acid (scheme 3) 17, the phenylpyrrole generated through cyclization of appropriate aniline with 2,5-dimethoxytetrahydrofuran-3-carbaldehyde in acetic acid (Scheme 4) 18, and the phenylimidazole through cyclization of 3-fluoro-N′-hydroxybenzimidamide and ethylpropiolate (scheme 5) 19.
Scheme 1.
Synthesis of phenyltriazole N-capping groups
Scheme 2.
Synthesis of phenylpyrazole N-capping groups
Scheme 3.
Synthesis of phenylfuran N-capping groups
Scheme 4.
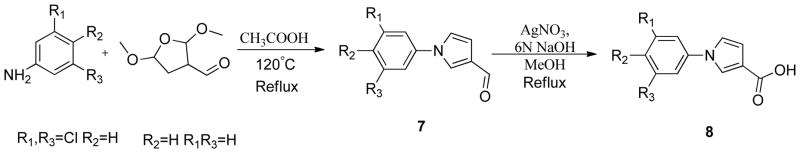
Synthesis of phenylpyrrole N-capping groups
Scheme 5.
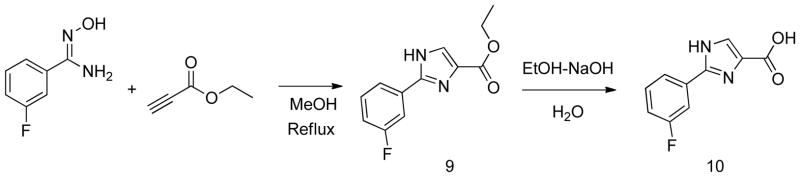
Synthesis of phenylimidazole N-capping groups
Structure-activity relationship of substituted phenylheterocyclic isosteres as N-terminal capping groups
Prior to testing of the library of phenylheterocyclic Ncaps, studies on the binding affinity of the native p21WAF1 pentapeptide RRLIF were undertaken. This peptide is an important reference point since modified peptides and FLIPs are compared directly to the native ligand. The results showed IC50 values of 1.4 and 16.1 μM for CDK2/cyclin A2 and CDK4/cyclin D1 in the fluorescence polarization assay respectively demonstrating that RRLIF binds about 10 fold tighter to cyclin A. With this established, direct comparison of the efficacy of the various heterocycles was determined through the competitive binding of FLIP variants with identical phenyl substitutions. To this end, variants of each heterocyle were generated with 3,5-dichloro and 4-chloro phenyl modifications and incorporated to make the FLIPs. The resulting SAR around the heterocyclic scaffolds determined that while isosteric ring systems are presented, significant differences are observed in the in vitro potencies as measured in the fluorescence polarization (FP) binding assay (Table 1). After testing of each of these, the phenyltriazole series was revealed to be the most successful among all six isosteric capping groups (5773, 5774) where these were found to have IC50 values for CDK2/cyclin A2 of 4 μM and 11.5 μM, indicating slight loss of potency compared to RRLIF. In contrast, a potency increase for 5774 was observed for the binding to CDK4/cyclin D relative to RRLIF (12 μM vs. 16.1 μM) whereas 5773 has a decreased inhibitory activity with an IC50 of 27.3 μM. Direct comparison of the corresponding phenylpyrazole FLIPs 5763 (3,5-dichloro, 21.8 μM) and 5765 (4 chloro, 33.7 μM) against CDK2/cyclin A2 indicates that a 3–5 fold potency loss is obtained for the FLIP bioisostere incorporating one less nitrogen. With the exception of the phenylimidazole scaffold, all other heterocyclic derivatives were found to be marginally or completely inactive at the concentrations tested. The 3-fluorophenylimidazole containing FLIP analog (5852) was found to be of comparable potency to the corresponding phenylpyrazole (5771) for both CDK2/cyclin A2 and CDK4/cyclin D1. These results as a whole suggest that appropriate positioning of a viable H-bond donor atom in the heterocyclic ring is a key determinant of activity.
Further to the direct comparison of identically substituted heterocyclic variants, the SAR of the phenyl ring and its interaction with the secondary hydrophobic pocket were examined. Firstly in the phenyltriazole series, substitutions included the 3-chloro, 3-methyl and the unsubstituted phenyl ring. Of these analogs, the 3-chloro was determined to be the most active (5906, 5.7 μM) and of comparable potency to the 35DCPT. Despite the similar size of the 3-methyl derivative (5907), it was found to be of considerably lower activity and was less active than the unsubstituted version. This was found to be true both in the CDK2/cyclin A2 and the CDK4/cyclin D1 contexts. Further testing of the derivatives synthesized in the phenylpyrazole series provided additional insights into the structure-activity relationship. As indicated from the data and consistent with the SAR for the triazole, the 3,5-dichloro analog (5763) is a stronger binder to CDK2/cyclin A2 than the 4-chloro (5765) however the opposite is true for activity against CDK4/cyclin D1. In the latter case, the relative potencies are reversed compared to cyclin A in that 5765 is approximately two-fold more potent than 5763. In the pyrazole context, the unsubstituted analog (5762, IC50 = 40.3 μM for CDK2/cyclin A2 and 54.2 μM for CDK4/cyclin D1) was of similar potency to the 4-chloro analogs. The 3-chloro derivative (5764) was found to be the most potent in this series being 2-fold more active for CDK2/cyclin A2 relative to the 3,5-dichloro (5763). In addition the 3-fluoro substituted phenylpyrazole FLIP (5771) was also made and found to be a weaker binder for CDK2/cyclin A2 (IC50 = 29.6 μM) and comparable inhibitor for CDK4/cyclin D1 (IC50 = 65.9 μM) relative to 5764. The 3-methoxy analog (5766) is about 6 fold less potent than the 3-chloro (5764) and is also a marginal inhibitor of CDK4/cyclin D1. The 4-methoxy substitution (5766) was observed to be detrimental since no appreciable inhibition of either CDK complex was evident.
C-terminal Partial Ligand Alternatives (Ccaps): derivatives of 1-phenyl-1H-1,2,4-triazole-3 carboxamide
Previously the computational enrichment strategy was applied to identify potential non-peptidic replacements for the C-terminal residues (Ile7 and Phe8) previously verified as critical determinants for binding to the cyclin groove. A SAR study of various bis aryl compounds revealed that 3-phenoxybenzylamine was the most effective C-capping group (Table 2). Further to this 3-phenoxybenzylamine and its isostere (4-phenoxypyridin-2-yl)methanamine were introduced as C-cap core structures to examine the binding requirements with the primary hydrophobic pocket. Structural modifications were undertaken to determine if potency enhancements observed after addition of halogens to the aromatic sidechain of phenylalanine in the peptide context were relevant to this C-cap 11; 12. In this effort, phenyltriazole Ncaps were utilized since these were shown to be the most effective as described (Table 1). Combination of the N-cap and C-caps was undertaken to corroborate that the activities identified in the peptide context were retained in the less peptidic and more drug-like compound. The C-cap core structures synthesized and tested were made in two very similar contexts presenting no significant structural differences: phenoxybenzylamine (PBA) (Table 2, 5807 and 5849) and 3-phenoxypyridylmethylamine (PPMA) (Table 2, 5824, 5823, 5822, 5825, and 5848). Substitutions incorporated primarily halogens at the 3 and 4 positions of the phenoxy group interacting with the lipophilic pocket but also included a 2-methyl and 3,5-dichloro substituted versions.
Table 2.
Structure activity of C-terminal Partial Ligand Alternatives
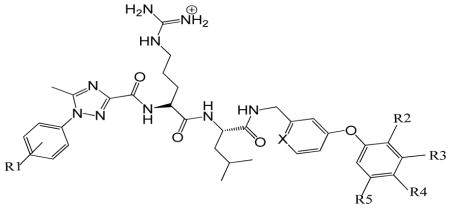
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| SCCP | R1 | X | R2 | R3 | R4 | R5 | CDK2/cyclin A IC50 (μM) |
CDK4/cyclin D1 IC50 (μM) |
| 5807 | 4-chloro | C | H | H | H | H | 106.1±26.2 | >200 |
| 5824 | 4-chloro | N | H | F | H | H | 53.2±11.6 | >200 |
| 5823 | 4-chloro | N | H | H | F | H | 18.1±4.0 | >200 |
| 5822 | 4-chloro | N | H | H | Cl | H | 54.4±0.9 | >500 |
| 5825 | 4-chloro | N | CH3 | H | H | H | 60±3.5 | >500 |
| 5848 | 3,5-dichloro | N | H | Cl | H | H | 61±10.8 | 120 |
| 5849 | 3,5-dichloro | C | H | Cl | H | Cl | >180 | 120 |
| 5926 | 3,5-dichloro | N | H | F | F | H | >180 | >180 |
Overall, the binding affinities of the seven FLIPs for CDK2/cyclin A2 (table 2) were in general shown to be greater than for CDK4/cyclin D1. IC50 values for the active compounds range from 18.1 μM to 106.1 μM. A compound with 1-(4-chlorophenyl)-5-methyl-1H-1,2,4-triazole-3-carboxylic acid (4CPT) as the N-cap structure and 3PBA as the C-cap, (Table 2, 5807) was found to be the weakest inhibitor of CDK2/cyclin A2. Testing of the FLIP compound bearing a fluoro substitution at 3-position (PPMA, 5824) resulted in an IC50 of 53.2 μM and the 3-chloro substituted analog (5848) had comparable potency. Movement of the fluoro substitution to the 4-position (5823) provided a 4-fold increase in binding activity (18.1 μM) and resulted in the most potent compound in this series however the 4-chloro did not result in a similar enhancement (5822, IC50 = 54.4 μM). Incorporation of a methyl group at the 2-position (5825) led to an IC50 value of 60 μM and therefore was not as successful as other analogs. Testing of the 3,5-dichloro PBA derivative (5849) in the 3,5-DCPT context suggested a different activity profile from the other compounds and indicating that this FLIP is not a potent inhibitor either for CDK4/cyclin D1 or CDK2/cyclin A2. In order to assess drug-likeness of the most active N and C cap combination, ClogP and polar surface area were calculated (ChemBioOffice 12, CambridgeSoft) for RRLIF and SCCP5823 and found to be −2.75, 5.3 and 303, 199 Å2 respectively.
DISCUSSION
The cyclin binding groove is composed of a primary hydrophobic pocket (interacting with Leu6 and Phe8 of HAKRRLIF), a secondary hydrophobic pocket (binding to Ala2) as well as an acidic region (contacting Lys3 and Arg4). The REPLACE strategy has been applied in order to identify N-and C-caps replacements for peptide determinants and generate non-peptidic inhibitors with greater drug-likeness. The SAR of phenylheterocyclic N-caps and bisarylether C-caps was therefore studied to optimize binding affinity for cyclin A and D and further explore the structural basis for binding and protein and ligand determinants in the regions described above.
Previously it has been shown in SAR studies that peptide affinity can be significantly impacted through substitution of the N-terminal residues contacting the secondary hydrophobic pocket and acidic region in both cyclin A2 and D1 contexts and also that this can lead to preferential binding to one cyclin over the other 20. In order to explore this further and to optimize inhibitor binding, an extensive SAR was generated for the phenylheterocyclic capping groups described. In prior studies crystallography revealed that the phenyltriazole Ncap makes significant complementarity with the CBG through interactions with the secondary hydrophobic pocket and through observed H-bonds to the side chains of Trp217 and Gln254 10. The 1-phenyl-1H-1,2,4-triazole-3 carboxylic acid Ncap represents an excellent template to generate isosteres with very similar steric complementarity while investigating the requirements for substitution of the aryl ring and the H-bond interactions observed in the crystal structure. The isosteric series provides a rare opportunity to directly compare similarly substituted heterocyclic isosteres and investigate direct contributions of key H-bond interactions. In the first instance, this analysis provides considerable insights into need for the heteroatoms in the 1,2,4-triazole ring and in particular the H-bond observed between N2 and the indole NH of Trp217. The inactivity of the phenylfuran series and the general decreased potency of the phenylpyrazole (2–5 fold relative to the phenyltriazoles) suggest not only a role for N2 as a binding determinant but also for the N4 heteroatom. As the sp2 hybridized oxygen atom of a furan in general is not a good H-bond acceptor due to participation of the lone pairs in the aromatic character of the ring and increased electronegativity compared to nitrogen, the decreased activity of this series is in line with expectations. While the triazole ring also is aromatic, the total of 3 nitrogen lone pairs are available and therefore any individual one is more available to participate in H-bonding. The lower relative potency of the phenylpyrazole series, which possess the N2 acceptor atom, but lack the N4 heteroatom was however an unexpected result. Crystal structures of the phenyltriazoles bound to cyclin A did not suggest a role for the N4 atom in binding and the potency loss is not easily explained since the pyrazole and triazole rings would be expected to have similar physicochemical properties. A probable contribution of N4 to binding is suggested by modeling studies in which a water molecule was placed between this atom and the carboxylate sidechain of Asp283 or Asp129 in two cyclins (Figure 2). This putative water forms appropriate interactions in forming a bridging H-bond between the ligand and the cyclin in the correct geometry and distance, suggests a role for N4 and therefore explains the decreased potency observed in the phenylpyrazole series. The phenylpyrrole containing FLIPs place an H-bond donor adjacent to the indole NH and unsurprisingly do not retain detectable levels of activity. While the phenylimidazole capped peptide generated did not have an identically substituted ring (3-fluoro) to the other analogs in this evaluation, it can nonetheless be compared to the counterpart synthesized in the phenylpyrazole series. Both of these FLIP isosteres possess similar activity suggesting that their H-bonding properties to the indole NH are equivalent. Overall, comparison of the phenylheterocyclic N-capping groups provides key insights into the contributions of H-bonds observed in crystal structures with cyclin A while suggesting a structural rationale for molecular differences between similar bioisosteres.
Figure 2.

Modeled structure of SCCP5773 in complex with CDK4/cyclin D1 (brown carbon atoms) overlayed with the crystal structure of the same Ncap bound to CDK2/cyclin A2 (yellow carbon atoms). A Connolly surface representation of cyclin A is shown along with the putative water molecule contributing bridging hydrogen bonds.
Further insights into the SAR of the aryl ring of the phenylheterocycles and also the molecular determinants of binding to the minor hydrophobic pocket of the cyclin groove is provided through the data obtained for the triazole and pyrazole series. Overall the activity of phenyltriazole containing FLIPs and phenylpyrazole compounds were well aligned in terms of their relative potency in binding to both CDK2/cyclin A2 and CDK2/cyclin D1 with the phenyltriazoles generally being more potent. Selectivity for cyclin A or cyclin D was observed depending on the aryl substitution. The 3,5-dichloro analogs (phenyltriazole, 5773; phenylpyrazole, 5763) are more potent than their 4-chloro counterparts in the binding to CDK2/cyclin A2 whereas 4-chloro substituted FLIPS (phenyltriazole, 5774; phenylpyrazoles, 5765) showed greater affinity to CDK4/cyclin D1 thus reversing the order of selectivity. Preferential binding to cyclin A by 3-substituted N-caps and for cyclin D by 4-chloro derivatives was therefore observed in both series of phenylheterocyclic isosteres. In order to determine the structural basis for cyclin selectivity, modeled complexes of the 3,5 and 4-chloro subsituted phenyltriazole FLIPs bound to cyclin D1 were generated based on crystal structures of these analogs in complex with cyclin A and apo structures of cyclin D1. Previously the cyclin grooves of cyclins A2 and D1 have been systematically compared and shown to have significant differences in the volume of the secondary hydrophobic pocket and leading to selectivity in the peptide context depending on the size of the contacting sidechain20. The smaller pocket results in displacement of the 3-substituted analogs from the subsite at the expense of hydrophobic complementarity and thus interact with weaker affinity for cyclin D1 in contrast to that for cyclin A (Figure 2). The improved binding of the 4-substitutions (relative to the 3 and 3,5-subsituted derivatives) to cyclin D1 can also be explained by a binding mode more conducive to close contact of the phenyl ring with the minor pocket and interactions of the 4-chloro atom with Glutamates 66 and 70. The decreased potency of the 3-fluoro derivatives relative to the 3-chloro in both cyclin contexts is in line with the expectation based on forming the best complementarity with the volume of the minor hydrophobic pocket. The activity decrease observed with the 3-methyl analog in the triazole context is harder to explain although the slightly different volume of the methyl may be less optimal for interaction. Furthermore the increase in activity of the 3-chloro relative to the dichloro only in the pyrazole context can likely be explained by a slightly different binding mode as a result of the additional H-bond in the phenyltriazole FLIPs. In this alternate conformation, the 5 substitution leads to a steric clash and less favorable interactions result from the necessity of avoiding these non-optimal contacts. Marginal binding of the methoxy substituted pyrazole compounds is consistent with results obtained for the other 3 and 4 modified FLIPs and the expected interactions of the larger substituents with the available space for interaction.
After the optimization of the phenylheterocyclic Ncaps, the study of bisarylether as C-caps was carried out to gain insights into the binding of fragments to the primary hydrophobic pocket. N-caps and C-caps were incorporated into a single molecule in order to further investigate and validate the key aspect of REPLACE that individual PLAs found in the peptide context can then be combined to generate less peptidic and more drug-like compounds. In the first instance, combination of the 4-CPT and 3-PBA as N and Ccaps for an Arg-Leu dipeptide (Table 2, 5807) suggested that potency loss is occurring through a variant binding mode. Addition of halogen atoms to the phenoxy group of the Ccap however results in recovery of lost binding and in the particular case of the 4-fluoro derivative, the in vitro activity is less than 2-fold of the expected potency for combination of the two capping groups while substantially improving the drug-like character of the cyclin groove inhibitor. Drug-likeness is improved by incorporation of the bisarylether system which replaces two residues with a small molecule capping group. Similar to the spacing function of Ile in the peptide context (RRLIF increased activity relative to RRLF), the C-caps have an aromatic ring as a spacer group.
Further modeling studies of the PBA and PBMA Ccap core structures indicates the structural basis for increased binding affinity of the halogen substituted FLIPs (Figure 1D). The bisaryl ether Ccap replaces the interaction of the Phe sidechain and its contacts with the primary hydrophobic pocket adjacent to those of the Leu residue. Previous peptide SAR studies have demonstrated that 3 and 4 halogen substituted Phe residues have increased potencies through more optimal contacts with this sub-site 11; 12. As can be seen (Table 2), all 3 and 4 modified Ccaps resulted in better binding to the cyclin groove compared to 3PBA (5807) and are therefore consistent with this result. The 5 fold increase observed for the 4-fluoro (5823) indicates that the phenoxy aryl ring adopts a very similar conformation to that in the peptide context (i.e. RRLIX where X is 4-fluorophenylalanine) and therefore leads to comparable increase in complementarity with this pocket. It is perhaps an even more optimal geometry since the only a 2-fold increase is observed in the peptide context. The larger 4-chloro substitution (5822) decreases the FLIP potency therefore suggesting the fluorine is the optimal size. The lack of activity of the C-capped cyclin groove inhibitors for CDK4/cyclin D1 is largely consistent with the previous observation that the phenylalanine subsite (in addition to the differences in the secondary hydrophobic pocket) has a decreased volume relative to cyclin A. As none of the bis-aryl ethers have appreciable activity, it is apparent that interactions of the Ccap with the smaller lipophilic site are suboptimal and therefore lead to significantly reduced activity (Figure 1D).
In this study, further validation for the REPLACE strategy has been obtained from the standpoint that partial ligand alternatives can be optimized when linked to and in the context of the peptide. In addition, both N and C PLAs have been combined to create a more drug like and less peptidic compound. With respect to the pentapeptide, three residues of the cyclin groove inhibitor have been removed in introducing small molecule functionality into the molecule. Specifically, the most potent analog (5823) has physicochemical properties that are more consistent with Lipinski’s 21 and other rules for drug-likeness. It has a ClogP of 5.3 (between −0.4 and +5.6 for Ghose modified rules 22) in contrast to the RRLIF value of −2.75, a polar surface area of 100 Å2 less than the parent peptide and 6 and 10 H-bond donors and acceptors respectively. Further REPLACE efforts centered on switching out the Arg and Leu residues for a non-peptidic core will lead to additional optimization in terms of potency, stability and cellular permeability through bringing the above parameters further in line. In particular, the inclusion of citrullene, the non charged isostere of arginine and betahomoleucine will be undertaken. These have been shown in the peptide context to retain potency 23 and will further improve stability and drug-likeness. These results also demonstrate that in the iterative conversion of peptides into small molecules, potential changes in binding mode, while detrimental, can be circumvented by reoptimization of protein-ligand affinity in the molecule combining individual partial ligand alternatives. As a whole, this validation of REPLACE further serves to exempify this strategy in drug discovery efforts to generate potent and selective protein-protein interaction inhibitors of kinase-substrate interactions.
EXPERIMENTAL SECTION
All solvents and reagents were used as obtained. 1H NMR and 13C NMR spectra were recorded with a Varian Mercury 300 and 400 Spectrometer, respectively. Mass spectra were measured with Micromass QTOF: Tandem quadruple-time of flight mass spectrometer electrospray ionization (ESI) and VG 70S: Double-focusing magnetic sector mass spectrometer (EI). Analytical purities of evaluated compounds were >95% unless stated otherwise. The following analytic method (unless stated otherwise) was used on a Waters Alliance 2695 HPLC with a 2996 diode-array detector and equipped a C18 (2) 100 A, 250 x 4.6mm, 5μ column (Phenomenox Luna). A gradient from 100% water (0.1% trifluoroacetic acid) to 60% acetonitrile (0.1% trifluoroacetic acid) was run over 30 mins and held for 4 mins. The chromatograms were extracted at 226 and 254 nm.
Preparation of phenyltriazole N-capping groups (Scheme 1)14; 15 (5843, 5773, 5774, 5906, 5907)
Ethyl 2-chloro-2-(2-(3,5-dichlorophenyl)hydrazono)acetate (1)
To a solution of 3,5-Dichloroaniline (1.62g, 10mmol) in 10 ml methanol, 10 ml of 6 N hydrochloric acid was added while maintaining the temperature at 0°C. Sodium nitrate (1.38g, 20mmol) was then slowly added as a solid. The reaction mixture was stirred for 15 mins at 0°C after which sodium acetate was added as a solid to adjust the reaction to pH 5. Subsequent to this, a solution of ethyl 2-chloroacetoacetate (1.64, 10mmol) in methanol was slowly added while maintaining the temperature at 0°C, after which the reaction mixture was allowed to warm to ambient temperature and stirred for 12 h. The reaction was monitored by TLC using 25% ethyl acetate in hexanes, for completion. Methanol was then removed under reduced pressure, diethyl ether was added and the organic layer was separated and washed with saturated sodium bicarbonate and water prior to drying over sodium sulfate. The organic layer was concentrated and the product crystalized from ethanol to give an orange solid 2.29g (77.7%).
1H NMR (methanol-d4, 300 MHz) δ (ppm) 7.2 (s, 3H), 7.0 (s, 1H), 4.32–4.39 (q, J=3.52, 3H) 1.35–1.4 (t, J=3.41Hz, 3H).
MS (EI+) 294
Ethyl 1-(3,5-dichlorophenyl)-5-methyl-1H-1,2,4-triazole-3-carboxylate (2)
The step 1 product, Ethyl 2-chloro-2-(2-(3,5-dichlorophenyl)hydrazono)acetate (1) (2.29g, 7.7mmol), and acetaldehyde oxime (0.473g, 7.7mmol) were dissolved in toluene and heated to reflux for 2h after which triethylamine (0.81g, 7.7mmol) was added. The reaction was monitored by TLC using 25% ethyl acetate in hexanes. After completion, the reaction mixture was then concentrated and partitioned between ethyl acetate and water. The layers were separated and the aqueous layer was washed with ethyl acetate. The combined organic layers were washed with water and brine, dried with sodium sulfate, filtered, and then concentrated. The residue was crystallized from diethylether/hexanes to give the product as fine, tan needles weighing 1.97g (84.5%).
1H NMR (chloroform-D, 300 MHz) δ (ppm) 7.45 (s, 2H), 7.49 (s, 2H) 4.46–4.53 (q, J=3.36Hz, 2H) 2.62 (s, 3H), 1.41–1.46 (t, J=3.35Hz, 3H)
MS (EI+) 300
1-(3,5-dichlorophenyl)-5-methyl-1H-1,2,4-triazole-3-carboxylic acid (3)
The step 2 product, Ethyl 1-(3,5-dichlorophenyl)-5-methyl-1H-1,2,4-triazole-3-carboxylate (2) (1.9g, 6.33mmol) was refluxed in sodium hydroxide (2.02g, 50.64mmol), 19ml of ethanol and 19ml of water for 2h and the reaction was monitored by TLC (35% ethyl acetate in hexanes). After the completion of reaction, the reaction mixture was cooled, the alcohol was evaporated and diluted with water. The reaction mixture was acidified with 1N hydrochloric acid and stirred to precipitate the product.
1H NMR (chloroform-D, 400 MHz) δ (ppm) 7.86 (t, J=1.04Hz) 7.8 (d, J= 1.12Hz3H) 2.54 (s, 3H)
13C NMR (Dimethyl sulfoxide-D6, 400 MHz) δ (ppm) 160.6, 154.5, 154.11, 138.5, 135.1, 134.7, 129.0, 123.8
HRMS EI+ observed 270.9914, calculated 270.9915, 0.4ppm
Preparation of phenylpyrazole N-capping groups (Scheme 2)16 (5762-5767, 5771)
Ethyl 1-(3,5-dichlorophenyl)-5-methyl-1H-pyrazole-3-carboxylate (4)
1 g (4.68mmol) of 3,5-dichlorophenyl hydrazine was dissolved in 25 ml of acetonitrile in a 100 ml round bottom flask, triethylamine (0.47g, 6.9mmol) and ethyl aceto pyruvate (0.74g, 4.68mmol) were added to the reaction mixture under stirring and left to continue overnight. The reaction was monitored by TLC (25% ethyl acetate in hexanes) for completion. After the reaction was complete, the reaction mixture was diluted with dichloromethane and washed with water. The organic layer was separated, dried over sodium sulfate and concentrated. The crude mixture was then purified by flash chromatography (Biotage SP4) using a SNAP 100g column with a gradient run starting from 6% ethylacetate: 94% hexanes to 50% ethyl acetate and 50% hexanes over 15 column volumes to yield a white solid 0.39g (21% yield).
1H NMR (chloroform-D3, 300MHz) δ (ppm) 7.431 (s, 3H), 6.744 (s, 1H), 4.49-4.44 (t, J=7.0Hz, 2H), 2.39 (s, 3H), 1.39–1.42 (t, J=2.54Hz, 3H),
MS (EI+) 298
1-(3,5-dichlorophenyl)-5-methyl-1H-pyrazole-3-carboxylic acid (5)
The ester obtained from step1, ethyl 1-(3,5-dichlorophenyl)-5-methyl-1H-pyrazole-3-carboxylate (4) (0.39g, 1.3mmol) was dissolved in 10 ml methanol. The ester solution was treated with a 10 ml solvent mixture of tetrahydrofuran, 20% potassium hydroxide and methanol (1:1:1). The solution was stirred at room temperature overnight and after completion, as evidenced by TLC (25% ethylacetate in hexanes), methanol was evaporated and the reaction solution was partitioned between ethylacetate and water. After washing with ethyl acetate, the aqueous layer was collected and acidified to pH 2 using 1N hydrochloric acid. The acidified aqueous layer was then partitioned between dichloromethane and additional water. The organic layer was dried over sodium sulfate and concentrated to get solid product 0.23g (100%).
1H NMR (dimethyl sulfoxide-D6, 400MHz) δ (ppm) 7.78–7.79 (t, J=0.937Hz,) 7.8 (d, J=1.14, 1H), 6.74 (s, 1H), 2.50 (s, 3H)
13C NMR (dimethyl sulfoxide-D6, 400MHz) δ (ppm) 162.9, 144.5, 141.4, 140.8, 134.5, 128.0, 123.5, 11.9
HRMS EI+ Observed 269.9959, calculated 269.9963, 1.5ppm
Preparation of phenylfuran N-capping groups (Scheme 3)17 (5768, 5769)
Synthesis of 5-(3,5-dichlorophenyl)furan-2-carboxylic acid (6)
A mixture of 3,5-dichloro aniline (0.84g, 5mmol), hydrochloric acid (15%, 3ml ) and water (4.5ml), was heated until a clear solution was obtained. This solution was then cooled to 0oC, diazotized with aqueous sodium nitrite (30%, 1.2ml) and filtered. To the filtered solution, water (2.5ml) and furoic acid (0.56g, 5mmol) were added. An aqueous solution of cupric chloride (0.125g in 10 ml water) was added dropwise and stirred for 4h at room temperature and kept aside for 16h. The resulting solid was filtered off, suspended in water and purified by flash chromatography (Biotage SP4) using a SNAP 100g column with a gradient 0% to 40% ethylacetate for 5 column volumes, the concentration of ethyl acetate at 40% was kept constant for 2 column volumes and then increased to 100% in 5 column volume. The purified product obtained weighed 0.36g (28%)
1H NMR (dimethyl sulfoxide-D6, 400MHz) δ (ppm) 7.85 (d, 2H), 7.65 (t, 2H), 7.36 (d, J=3.88Hz, 1H), 7.40 (d, 3.96, 1H)
13C NMR (dimethyl sulfoxide-D6, 400MHz) δ (ppm) 159.0, 152.9, 145.0, 134.9, 132.2, 128.0, 122.7, 119.7, 110.5
HRMS EI+ Observed 255.9698, calculated 255.9694, 1.6ppm
Preparation of phenylpyrrole N-capping groups (Scheme 4) 18 (5775, 5776)
1-(4-chlorophenyl)-1H-pyrrole-3-carbaldehyde (7)
4-chloroaniline (0.51g, 4 mmol), 2,5-dimethoxytetrahydrofuran-3-carbaldehyde (0.76g, 4.8 mmol), acetic acid (16ml) were stirred in a round bottom flask until a red colored solution was observed. The reaction mixture was then refluxed at 120°C for 15 mins and a dark reddish brown solution was obtained. The reaction was monitored by TLC (25% ethyl acetate: 75% hexanes) and worked up when complete. The reaction mixture was cooled, extracted with ethyl acetate and the organic layer was washed with saturated sodium bicarbonate, dried over sodium sulfate and concentrated to get 0.77g (94%)
1H NMR (chloroform-D3, 300MHz) δ (ppm) 9.85 (s, 1H), 7.62–7.63 (t, J=1.8Hz, 1H), 7.44–7.47 (d, J=9Hz, 2H), 7.34–7.37 (d,J=9.3Hz, 2H) 7.05 (t, J=0.9Hz, 1H), 6.57–6.58 (q, J=1.5Hz, 1H)
MS (EI+) 204
1-(4-chlorophenyl)-1H-pyrrole-3-carboxylic acid (8)
1-(4-chlorophenyl)-1H-pyrrole-3-carbaldehyde(7) (0.76g, 3.58mmol) obtained from step 1, silver nitrate (1216.26mg, 7.16mmol), 6 N sodiumhydroxide (15ml) and methanol (5.8ml) were refluxed for 5h and the reaction was monitored by TLC for completion. The reaction mixture was filtered through celite and the aqueous layer was acidified to get a white precipitate. The precipitate was filtered off, dissolved in ethyl acetate, and dried over sodium sulfate to get the product 0.6g (65%)
1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ (ppm) 7.95 (t, J=0.93, 1H), 7.71–7.73 (d, J=4.6Hz, 2H), 7.53–7.56 (d, J= 4.7Hz, 2H), 7.43–7.44 (t, J= 1.33Hz1H), 6.61–6.62 (q, J=0.88Hz, 1H)
13C NMR (dimethyl sulfoxide-d6, 400 MHz) δ (ppm) 165.0, 137.9, 130.5, 129.5, 123.9, 121.7, 120.7, 118.6, 111.5 HRMS
HRMS (EI+) observed 221.0242, calculated 221.0244, 0.9ppm
Preparation of phenylimidazole N-capping group (Scheme 5)19 (5852)
Ethyl 2-(3-fluorophenyl)-1H-imidazole-4-carboxylate (9)
The reactants 3-fluoro-N′-hydroxybenzimidamide (1g, 6.49mmol) and ethylpropiolate (0.636g, 6.49mmol) were dissolved in methanol and heated at reflux for 3 days. Toluene was then added and the methanol was distilled off. The reaction was heated at reflux for 6h after which the toluene was removed by rotary evaporation and the residue was chromatographed on silica gel twice (2% methanol/dichloromethane and 20% ethyl acetate/ Hexanes). The residue was dissolved in phenyl ether and heated to 200°C for 25 mins and then poured into cyclohexane and extracted with 1N hydrochloric acid. The acid layer was basified with sodium bicarbonate and extracted with ethyl acetate. After drying with sodium sulfate, it was filtered, concentrated and purified by flash chromatography on silica gel (Ethylacetate/Hexanes gradient) to give the desired product as an off-white solid.
2-(3-fluorophenyl)-1H-imidazole-4-carboxylic acid (10)
The step 2 product, ethyl 2-(3-fluorophenyl)-1H-imidazole-4-carboxylate (9) (0.3g, 1.281mmol was refluxed in sodium hydroxide (0.45g, 11.25 mmol), ethanol (5ml) and water (5ml) for 2h and the reaction was monitored by TLC. After the completion of reaction, the reaction mixture was cooled, the alcohol was evaporated and diluted with water. The reaction mixture was acidified with 1N hydrochloric acid and stirred to precipitate the product.
1H NMR dimethyl sulfoxide-D6, 400MHz) δ (ppm) 13.28 (d, 1H), 7.52 (q, 2H) 7.24(t, 2H)
13C NMR (DMSO, 400MHz) δ (ppm) 163.57, 161.16, 133.87, 132.02, 131.00, 130.92. 124.90, 121.42, 115.68, 111.98
HRMS ES+ Observed 207.0576, calculated 207.0570, 2.9ppm
(4-(3-chlorophenoxy)pyridine-2-yl)methanamine (5848)
This material was sourced commerically from UORSYS Ltd and fully characterized in house as follows:
1H NMR (chloroform-D3, 400 MHz) δ (ppm) 3.95, s 2H, 6.75, m, 1H, 6.87, d, 1H, 7.01, m, 1H, 7.13, m, 1H, 7.25, m, 1H, 7.37, m, 1H, 8.45, d, 1H
13C NMR (chloroform-D3, 400 MHz) δ (ppm) 164.8, 164.4, 154.9, 151.0, 135.5, 130.9, 125.6, 121.2, 118.9, 110.7, 109.7, 47.7
HRMS EI+ observed 234.0561, calculated 234.0560, 0.4 ppm
Peptide & FLIP Synthesis
All peptides and N-terminally capped FLIPs were synthesized and purified using standard Fmoc chemistry by GenScript (Piscataway, NJ). HPLC and MS were used to confirm the purity and structure of each peptide (see Supplementary Information Table 1). Compounds in Table 2 were synthesized by the following procedure used for compound 5926: The N-terminally capped and protected Arg-Leu peptide (16.1 mg, 0.02 mmol) was dissolved in DCM and (4-(3-chlorophenoxy)pyridine-2-yl)methanamine was added along with HBTU (16.7 mg, 0.02 mmol) and DIPEA (6.5 mg, 0.02 mmol). The reaction was stirred and monitored until TLC/HPLC indicated the complete consumption of starting material. Once complete, the reaction mixture was concentrated and then partitioned between EtOAc and H2O. The organic layer was washed with 1N NaOH, 1N HCl, and brine and dried with Na2SO4. The resulting product was deprotected with 95:2.5:2.5 TFA:H2O:TIPS over night, then concentrated and triturated with cold diethyl ether. The final N and C capped peptide was purified using semi-preparative HPLC (0–80 CH3CN over 20 mins).
Fluorescent Polarization Binding Assay
CDK4/cyclin D1 FP assay
This assay was performed using black 384-well plates using a previously described procedure24 with the following modifications. To each well were added: 5 μl CDK4/cyclin D1 (0.3 Dg/well purified recombinant human kinase complex from Invitrogen), 5 μl compound solution, 5 μl 30 nM fluoresceinyl-Ahx-Pro-Val-Lys-Arg-Arg-Leu-(3ClPhe)-Gly tracer peptide. Compounds and kinase complexes were diluted using assay buffer (25 nM HEPES pH 7, 10 nM NaCl, 0.01% Nonidet P-40, 1mM dithiothretiol (DTT). Plate was centrifuged for 1 min at 500 rpm and then incubated with shaking for 45 mins at room temperature. Fluorescence polarization was read on DTX880 multimode detector (Beckman Coulter, Brea, CA) fitted with 485 nm/535 nm excitation/emission filters and a dichroic mirror suitable for fluorescein. Relative mp was calculated for each concentration tested using the equation showing below. IC50 values were determined by logarithmic regression by correlating relative mps and testing concentrations.
CDK2/cyclin A2 FP assay
This assay was performed using black 384-well plates. To each well were added: 5 μl CDK4/cyclin D1 (0.3 Dg/well purified recombinant human kinase complex), 5 μl compound solution, 5 μl 30 nM fluoresceinyl-Ahx-Pro-Val-Lys-Arg-Arg-Leu-Phe-Gly tracer peptide. Compounds and kinase complexes were diluted using assay buffer (25 nM HEPES pH 7, 10 nM NaCl, 0.01% Nonidet P-40, 1mM dithiothretiol (DTT). Plate was centrifuged for 1 min at 500 rpm and then incubated with shaking for 45 mins at room temperature. Fluorescence polarization was read on DTX880 multimode detector (Beckman Coulter, Brea, CA) fitted with 485 nm/535 nm excitation/emission filters and a dichroic mirror suitable for fluorescein. Relative mp was calculated for each concentration tested using the equation showing below. IC50 values were determined by logarithmic regression by correlating relative mps and testing concentrations.
Supplementary Material
Acknowledgments
We thank Dr’s. Michael Walla and William Cotham in the Department of Chemistry and Biochemistry at the University of South Carolina for assistance with Mass Spectrometry, Helga Cohen and Dr. Perry Pellechia for NMR spectrometry. Dr Michael Wyatt and Ms. Kara Estes provided assistance with the FP assay. This work was partly funded by the National Institutes of Health through the research project grant, 5R01CA131368.
Footnotes
Notes: The authors declare no competing financial interest.
Supplementary Table 1 provides analytical characterization data for FLIPs shown in Table 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature Reviews Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 3.McInnes C, Andrews MJI, Zheleva DI, Lane DP, Fischer PM. Peptidomimetic design of CDK inhibitors targeting the recruitment site of the cyclin subunit. Current Medicinal Chemistry-Anti-Cancer Agents. 2003;3:57–69. doi: 10.2174/1568011033353506. [DOI] [PubMed] [Google Scholar]
- 4.Oelgeschlager T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. Journal Of Cellular Physiology. 2002;190:160–169. doi: 10.1002/jcp.10058. [DOI] [PubMed] [Google Scholar]
- 5.Zheleva DI, McInnes C, Gavine AL, Zhelev NZ, Fischer PM, Lane DP. Highly potent p21WAF1 derived peptide inhibitors of CDK-mediated pRb phosphorylation: delineation and structural insight into their interactions with cyclin A. Journal of Peptide Research. 2002;60:257–270. doi: 10.1034/j.1399-3011.2002.21014.x. [DOI] [PubMed] [Google Scholar]
- 6.Chen YN, Sharma SK, Ramsey TM, Jiang L, Martin MS, Baker K, Adams PD, Bair KW, Kaelin WG., Jr Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci U S A. 1999;96:4325–4329. doi: 10.1073/pnas.96.8.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulman BA, Lindstrom DL, Harlow E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1998;95:10453–10458. doi: 10.1073/pnas.95.18.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ball KL, Lain S, Fahraeus R, Smythe C, Lane DP. Cell-cycle arrest and inhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Current Biology. 1996;7:71–80. doi: 10.1016/s0960-9822(06)00029-7. [DOI] [PubMed] [Google Scholar]
- 9.Mendoza N, Fong S, Marsters J, Koeppen H, Schwall R, Wickramasinghe D. Selective Cyclin-dependent Kinase 2/Cyclin A Antagonists that Differ from ATP Site Inhibitors Block Tumor Growth. Cancer Research. 2003;63:1020–1024. [PubMed] [Google Scholar]
- 10.Andrews MJ, Kontopidis G, McInnes C, Plater A, Innes L, Cowan A, Jewsbury P, Fischer PM. REPLACE: a strategy for iterative design of cyclin-binding groove inhibitors. Chembiochem. 2006;7:1909–1915. doi: 10.1002/cbic.200600189. [DOI] [PubMed] [Google Scholar]
- 11.Kontopidis G, Andrews MJ, McInnes C, Cowan A, Powers H, Innes L, Plater A, Griffiths G, Paterson D, Zheleva DI, Lane DP, Green S, Walkinshaw MD, Fischer PM. Insights into cyclin groove recognition: complex crystal structures and inhibitor design through ligand exchange. Structure. 2003;11:1537–1546. doi: 10.1016/j.str.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Castanedo G, Clark K, Wang S, Tsui V, Wong M, Nicholas J, Wickramasinghe D, Marsters JC, Jr, Sutherlin D. CDK2/cyclinA inhibitors: targeting the cyclinA recruitment site with small molecules derived from peptide leads. Bioorg Med Chem Lett. 2006;16:1716–1720. doi: 10.1016/j.bmcl.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 13.McInnes C, Estes Kara, Yang Zhengguan, Farag Doaa Boshra, Johnson Paul, Lazo John, Wyatt Michael D. Targeting sub-cellular localization through the Polo-Box Domain: non-ATP competitive Inhibitors recapitulate a PLK1phenotype. Molecular Cancer Therapeutics. 2012;8:1683–1692. doi: 10.1158/1535-7163.MCT-12-0006-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfefferkorn JA, Choi C, Larsen SD, Auerbach B, Hutchings R, Park W, Askew V, Dillon L, Hanselman JC, Lin Z, Lu GH, Robertson A, Sekerke C, Harris MS, Pavlovsky A, Bainbridge G, Caspers N, Kowala M, Tait BD. Substituted pyrazoles as hepatoselective HMG-CoA reductase inhibitors: discovery of (3R,5R)-7-[2-(4-fluoro-phenyl)-4-isopropyl-5-(4-methyl-benzylcarbamoyl)-2H-pyrazo l-3-yl]-3,5-dihydroxyheptanoic acid (PF-3052334) as a candidate for the treatment of hypercholesterolemia. J Med Chem. 2008;51:31–45. doi: 10.1021/jm070849r. [DOI] [PubMed] [Google Scholar]
- 15.Li-Ya Wang WCT, Lin Hui-Yi, Wong Fung Fuh. An Effective Nitrilimine Cycloaddition for the Synthesis of 1, 3, 5-Trisubstituted 1, 2, 4-Triazoles from Oximes with Hydrazonoyl Hydrochlorides. Synlett. 2011;10:1467–1471. [Google Scholar]
- 16.Atkinson RNDI, Gregg RJ, Gross MF, Kort ME, Shi L. Preparation of pyrazole-carboxamides and -sulfonamides as sodium channel modulators. US 2005020564 US Pat Appl Publ. 2005
- 17.Prasad D Jagadeesh, PBK, Poojary Boja, Kumari N Suchetha. Synthesis of some thiadiazolotriazinone derivatives as possible antimicrobial agents. Phosphorus, Sulfur, Silicon. 2007;182:1083–1091. [Google Scholar]
- 18.Gianluca Sbardella AM, Artico Marino, Loddo Roberta, Setzu Maria Grazia, Colla Paolo La. Synthesis and in vitro antimycobacterial activity of novel 3-(1H-pyrrol-1-yl)-2-oxazolidinone analogues of PNU-100480. Bioorganic and Medicinal chemistry letters. 2004;14:1537–1541. doi: 10.1016/j.bmcl.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Rolf Paul JAB, Hallett WA, Hanifin John W, Ernestine Tarrant M, Torley Lawrence W, Callahan Francis M, PFF, Johnson Bernard D, Lenhard Robert H, Schaub Robert E, Wissner Allan. Imidazo[1, 5-d][1, 2, 4]triazines as Potential Antiasthma Agents. J Med Chem. 1985;28:1704–1716. doi: 10.1021/jm00149a029. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Bolger JK, Kirkland LO, Premnath PN, McInnes C. Structural and functional analysis of cyclin D1 reveals p27 and substrate inhibitor binding requirements. ACS Chem Biol. 2010;5:1169–1182. doi: 10.1021/cb1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 22.Ghose AK, Viswanadhan VN, Wendoloski JJ. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. Journal of Combinatorial Chemistry. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 23.Kontopidis G, Andrews MJ, McInnes C, Plater A, Innes L, Renachowski S, Cowan A, Fischer PM. Truncation and optimisation of peptide inhibitors of cyclin-dependent kinase 2-cyclin a through structure-guided design. ChemMedChem. 2009;4:1120–1128. doi: 10.1002/cmdc.200900093. [DOI] [PubMed] [Google Scholar]
- 24.Andrews MJ, McInnes C, Kontopidis G, Innes L, Cowan A, Plater A, Fischer PM. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Org Biomol Chem. 2004;2:2735–2741. doi: 10.1039/B409157D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.