

Abstract
In congenital mitochondrial DNA (mtDNA) disorders, a mixture of normal and mutated mtDNA (termed heteroplasmy) exists at varying levels in different tissues, which determines the severity and phenotypic expression of disease. Pearson marrow pancreas syndrome (PS) is a congenital bone marrow failure disorder caused by heteroplasmic deletions in mtDNA. The cause of the hematopoietic failure in PS is unknown, and adequate cellular and animal models are lacking. Induced pluripotent stem (iPS) cells are particularly amenable for studying mtDNA disorders, as cytoplasmic genetic material is retained during direct reprogramming. Here we derive and characterize iPS cells from a patient with PS. Taking advantage of the tendency for heteroplasmy to change with cell passage, we isolated isogenic PS-iPS cells without detectable levels of deleted mtDNA. We found that PS-iPS cells carrying a high burden of deleted mtDNA displayed differences in growth, mitochondrial function, and hematopoietic phenotype when differentiated in vitro, compared to isogenic iPS cells without deleted mtDNA. Our results demonstrate that reprogramming somatic cells from patients with mtDNA disorders can yield pluripotent stem cells with varying burdens of heteroplasmy that might be useful in the study and treatment of mitochondrial diseases.
Keywords: Induced pluripotent stem cells, Pearson's marrow-pancreas syndrome, Mitochondrial DNA, Heteroplasmy, Human genetics, Hematopoiesis
Introduction
Mitochondrial DNA (mtDNA) mutations are implicated in numerous human disorders(1–5), ranging from rare multisystem congenital diseases to common acquired degenerative disorders such as Parkinson's and Alzheimer's(6). There are no curative therapies and consequently mtDNA mutations cause significant morbidity and mortality(7). In congenital mtDNA disorders, a mixture of normal and mutant mtDNA, termed heteroplasmy, is inherited from the oocyte at fertilization and partitioned differentially in tissues during embryogenesis(3, 8). The degree and distribution of heteroplasmy in adult tissues determines the severity and marked phenotypic heterogeneity of the disease. Pearson marrow pancreas syndrome (PS) is a congenital multisystem disorder caused by large deletions in the mitochondrial genome and is characterized by life-threatening bone marrow failure and metabolic derangements(9, 10). The cause of the hematopoietic failure in PS is unknown, and experimental models to reproduce tissue-specific defects in PS and other mtDNA disorders are needed.
Somatic cells can be directly reprogrammed using defined genetic factors to yield iPS cells, which have the capacity to differentiate into any tissue(11–14). Direct reprogramming allows the creation of patient-specific pluripotent cells that retain the cytoplasmic contents of donor cells, including disease-associated mtDNA. We sought to generate iPS cells carrying mutant mtDNA in order to investigate tissue-specific effects of mitochondrial dysfunction. Here we describe the derivation of iPS cells bearing a pathogenic mtDNA deletion from a patient with PS. We observed changes in heteroplasmy during culture of PS-iPS cells, which allowed us to isolate isogenic iPS cells with undetectable levels of mutant mtDNA. Comparison of PS-iPS cells with varying degrees of heteroplasmy in vitro revealed defects in growth and mitochondrial function, and directed differentiation into the hematopoietic lineage revealed a tissue-specific phenotype characteristic of PS. Our results demonstrate that reprogramming of somatic cells from patients with Pearson syndrome can yield patient-identical pluripotent stem cells varying in mtDNA heteroplasmy, providing unique tools to study tissue-specific effects of mtDNA mutations.
Materials and Methods
Patient material
Biological samples were procured under protocols approved by the Institutional Review Board at Boston Children's Hospital. Standard histological evaluations were performed by the Department of Pathology, Boston Children's Hospital.
DNA isolation
Genomic DNA was isolated from peripheral blood or bone marrow using the QIAamp DNA Blood Maxi Kit or the DNeasy Blood and Tissue Kit (Qiagen). DNA was isolated from fibroblast and iPS cell lines by SDS/Proteinase K lysis followed by phenol/chloroform extraction and ethanol precipitation.
Cell lines and culture
Pearson syndrome patient bone marrow-derived fibroblasts (PS-Fib) were isolated by plating 150 μl of liquid bone marrow in DMEM/15% FCS. Media was changed every three days until outgrowths appeared (approximately two weeks), and thereafter cells were expanded by routine trypsinization and subculture. Cells were characterized for mutant mtDNA at passage 2. Pearson syndrome fibroblasts (GM04516) and lymphocytes (GM04515) and Kearns-Sayre syndrome fibroblasts (GM06225) and lymphocytes (GM06224) were obtained from the Coriell Institute for Medical Research.
Long range PCR
Long range PCR to detect mitochondrial DNA deletions was performed by amplifying 100–500 ng of template DNA using the Expand Long Template PCR system (Roche Diagnostics) according to manufacturer's instructions and using the primers huMito5328F and huMito3608R (Supplementary Table 1). The deletion location was mapped using PCR, restriction digests, and Sanger sequencing. Nucleotide positions were assigned per the revised Cambridge Reference Sequence of human mitochondrial DNA. Sequence analysis was performed using data from www.mitomap.org.
Mitochondrial DNA FISH
Templates for probes were amplified by PCR using primers COMMON5' and COMMON3' (for the probe hybridizing to both deleted and undeleted mtDNA species), and CHBMDF1 FISH5' and CHBMDF1 FISH3' (for the probe complementary to a deleted portion common to all deleted mtDNA species examined in this study) (Supplementary Table 1). COMMON probe was labeled with digoxigenin using the DIG-Nick Translation Mix (Roche) and CHBMDF1 FISH probe was labeled using the Biotin-Nick Translation Mix (Roche). Fibroblasts were prepared on coverslips as previously described(15). COMMON and CHBMDF1 probes were simultaneously hybridized on the coverslips in 50% formamide, 2× SSC by heating to 85° C for 2.5 minutes followed by incubation at room temperature overnight. Coverslips were washed in TBS 0.05% Tween (TBST) and incubated in TBST with 0.05% W/V Blocking Reagent (Roche) with FITC conjugated anti-Dig and Alexa Fluor 594 conjugated streptavidin at room temperature for one hour. Coverslips with washed in TBST, dehydrated and mounted in Prolong Gold (Invitrogen), and analyzed by epifluorescence microscopy.
Heteroplasmy determination by quantitative real-time PCR
Quantitative real-time PCR measurements were performed using 30 ng of template DNA and Brilliant SYBR Green QPCR Master Mix (Stratagene) with primers CHBMDF1F and CHBMDF1R (500 nM) for the mutant mtDNA species and WTmitoF and WTmitoR for all mtDNA molecules (Supplementary Table 1). Primer pairs were verified for linear amplification over a 100-fold range of input DNA.
Single-cell multiplex real-time PCR
Primer pairs and probe sets were optimized and validated for sensitivity and specificity. For PS-Fib single-cell multiplex real-time PCR, the following primer/probe sets were used simultaneously: CHBMDF1Set9F (300 nM)/CHBMDF1Set9R (300 nM)/CHBMDF1 probe (200 nM) and WTmito(CHset)F (150 nM) /WTmito(CHset)R (150 nM) /WTmito(CHset)probe (200 nM). For GM04516 single-cell multiplex real-time PCR, the following primer/probe sets were used simultaneously: 04516Set1F (150 nM)/04516Set1R (150 nM)/04516 probe (200 nM) and WTmitoSet3F(150 nM)/WTmitoSet3R(150 nM)/ WTmitoSet3 probe (200 nM). (See Supplementary Table 1 for sequences). Fibroblasts were collected by trypsinization and single cells were FACS sorted into individual wells of a 96-well QPCR plate containing 10 μl of 10% SideStep Lysis buffer. Amplification was performed with appropriate negative and positive controls using primers/probes at the concentrations described above with Brilliant II QPCR Master Mix in a Stratagene MX3000P QPCR system, and results were graphed and scored as described in the figure legends.
Mitochondrial complex quantity and activity assays
Quantification of complex I, complex IV, frataxin and PDH was performed using the MetaPath MitoDisease 4-Plex Dipstick Array (MitoSciences, Abcam) according to manufacturer's instructions. Complex I, III and IV activity were respectively determined using the Complex I Enzyme Activity Dipstick Assay Kit, Cyt C Reductase Human Profiling ELISA Kit, and Complex IV Enzyme Activity Dipstick Assay Kit (MitoSciences, Abcam)(16). A standard curve was established for each assay using a range of normal fibroblast protein concentrations. 0.5 and 5 ug protein extract was used for each sample in duplicate for MetaPath quantity assays. 1 and 10 μg protein extract was used for each sample in duplicate for complex I and IV activity assays. Quantification was performed by scanning the dipsticks followed by image analysis using Adobe Photoshop as described in the manufacturer's instructions. 34 μg protein extract was used in triplicate for the complex III activity assay, and quantitation was performed using a microplate reader. Results are shown as normalized to quantity or activity of complexes in WT cells.
Direct reprogramming and iPS cell characterization
Derivation, culture, characterization and differentiation of induced pluripotent stem (iPS) cells was as described(17) with modifications to protocol as noted in the text. iPS lines were cultured on irradiated MEF feeder cells or hESC-qualified Matrigel (BD Biosciences). PS-iPS cells were generally passaged every 6–7 days, with manual removal of differentiated cells under a dissecting microscope, release of colonies with Collagenase IV (Invitrogen) or Dispase (Stem Cell Technologies), and fragmentation and collection using a cell scraper. Teratomas were formed as described(12) by intramuscular injection of iPS cells in immunodeficient mice under approved animal use protocols. Histology was performed at the Dana-Farber Harvard Cancer Center Rodent Histopathology core facility.
Immunostaining of iPS cells
iPS lines were grown on glass coverslips and stained with: (1) Oct4 rabbit polyclonal antibody (1:300; Abcam) with Alexa-Fluor 488 chicken-anti-rabbit secondary antibody (1:1000; Life Technologies); (2) SSEA4 mouse monoclonal antibody pre-conjugated to Alexa Fluor 647 (1:100; BD Biosciences); (3) Nanog rabbit polyclonal antibody (1:200; Abcam) with Alexa-Fluor 488 chicken-anti-rabbit secondary antibody; (4) Tra-1-60 mouse monoclonal antibody (1:100; Millipore) with Alexa-Fluor 594 goat-anti-mouse secondary antibody (1:1000; Life Technologies); (5) Tra-1-81 mouse monoclonal antibody (1:100; Millipore) with Alexa-Fluor 594 goat-anti-mouse secondary antibody.
Reverse-transcription PCR
RNA was made using Trizol, and cDNA was made with Superscript III (Invitrogen), according to manufacturer's protocols. PCR was performed using the gene-specific primers in Supplemental Table 1 using SYBR green SsoAdvanced polymerase (Bio-rad).
Southern blots
10 μg of genomic DNA was digested with NcoI, separated on a 0.6% agarose gel and transferred to a positively charged nylon membrane. For evaluation of heteroplasmy, hybridization was performed using a probe corresponding to nt 14840–15261 (cytochrome b) of the mitochondrial genome, which detects a 7.5 kb band from the intact mitochondrial genome and a 5 kb band from the deleted mitochondrial genome in the PS patient. Hybridization was performed with Rapid-Hyb buffer (GE Healthcare) according to manufacturer's instructions. For proviral integration pattern analysis, the OCT4, SOX2, KLF4, and MYC-encoding retroviruses used for reprogramming each contains an IRES-GFP cassette a single NcoI restriction site. The proviral integration patterns were determined by probing Southern blots with a GFP probe.
Live-cell imaging
iPS cells were plated on Matrigel (BD Biosciences) in mTESR medium (Stem Cell Technologies) in a glass bottom dish (MatTek) on a microscope outfitted with a thermo/CO2-regulated chamber and a computer-controlled motorized stage. Images of iPS colonies were obtained at 6-hour intervals in a tiled array and stitched to form a single composite image. The surface area of the colony was measured using Elements AR (Nikon) software. Growth analysis ended at 6 days or when the colony grew into an adjacent colony or when the colony outgrew nine low-power fields.
Mitochondrial membrane potential
iPS cells were incubated in hES cell medium containing 7 nM tetramethylrhodamine, ethyl ester (TMRE) (Invitrogen, T669) and 100 nM MitoTracker Green (MTG) (Invitrogen, M7514) for 90 minutes. Immediately prior to imaging this medium was replaced with phenol red-free hES cell growth medium containing 100 nM TMRE, and live cells were imaged by epifluorescence microscopy.
Extracellular flux analysis
Extracellular flux analysis was performed using the Seahorse platform as previously described(18). PS-iPS cells cultured on Matrigel were trypsinized to single cells and plated onto Seahorse 24-well analysis plates at a density of 50,000 single cells per well in mTESR media containing 10 μM compound Y-27632 (Sigma). Each cell line was assayed in quadruplicate. One day later, the cells were analyzed for OXPHOS function (Injection 1: 20 μM oligomycin; Injection 2: 3 μM CCCP; Injection 3: 10 μM antimycin and 10 μM rotenone) and glycolysis function (Injection 1: 250 mM glucose; Injection 2: 20 μM oligomycin; Injection 3: 1.5 M 2-DG). The listed injection concentrations are 10× the final concentration. Results were normalized for protein concentration in each well and analyzed by Seahorse XF24 software.
To quantify extracellular flux analyses, background signals were removed by subtracting final OCR or ECAR values from signals. Standard deviations were calculated by analytically combining averaged data points and their errors from each well into a composite distribution, and then summing the variances of the estimated signal and background distributions. Figure 4E and 4G were normalized to PS-iPS1 to account for expected inter-experiment variability in raw data. Significant differences in basal respiration rates were identified by t-test of the normalized data (n=12) averaged over three replicates. Percentage of oxygen used for ATP production (Figure 4F) and glycolysis activity (Figure 4G) and the appropriate standard deviations were calculated using Taylor series approximations of the mean and variance of the ratio of random variables.
Figure 4. Phenotypes in PS-iPS cells.
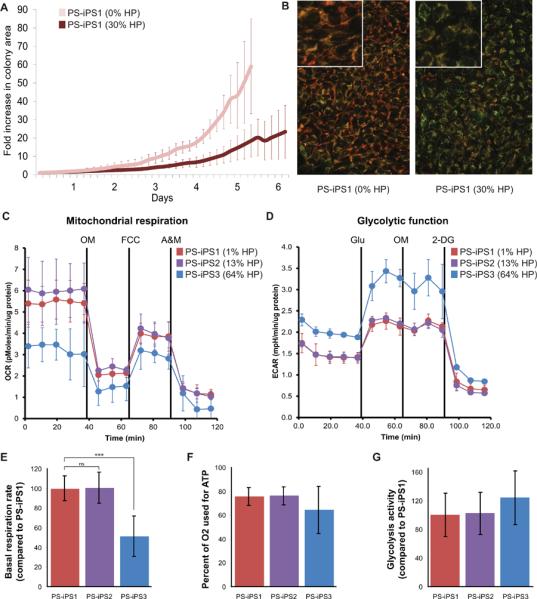
HP = heteroplasmy, the percentage mutant mtDNA. (A): PS-iPS1 growth in culture with and without mutant mtDNA, measured simultaneously by live cell imaging. Fold size increase is relative to size at day 0. (B): Mitochondrial membrane staining with MitoTracker Green (stains all mitochondrial membranes) and TMRE (stains high membrane potential). Left panel shows PS-iPS1 at 0%, right panel shows PS-iPS1 at 30% HP. Insets in both panels are at higher magnification. (C): Representative data from extracellular flux analysis of oxygen consumption. Solid black lines indicate time of chemical injections: the ATP synthase poison oligomycin (OM), mitochondria uncoupler FCCP, and mitochondrial poisons antimycin and rotenone (A&M). (D): Representative data from extracellular flux analysis of glycolytic function. Cells begin in relative starvation before the first injection of glucose (Glu). The second injection is oligomycin (OM), followed by the glycolysis inhibitor 2-Deoxy-D-glucose (2-DG). (E): Quantification of basal oxygen consumption (respiration) compared to PS-iPS1, *** indicates p < 0.0001, n=3. (F): Quantification of the percentage of oxygen which is used to drive ATP synthase, n=3. (G): Quantification of glycolysis activity, compared to PS-iPS1, n=2. All error bars indicate standard deviation.
Hematopoietic colony forming assay and sideroblast quantification
iPS cells were collected as large aggregates and resuspended in EB differentiation medium (80% DMEM, 20% FCS (Stem Cell Technologies 06900), 50 μg/ml ascorbic acid, 0.2 μg/ml holotransferrin) on low attachment dishes. After one day, cytokines were added: hSCF (300 ng/ml), hFlt3L (300 ng/ml), IL-3 (10 ng/ml), IL-6 (10 ng/ml), G-CSF (50 ng/ml), BMP4 (50 ng/ml). Media containing cytokines was replaced every three days for 14–16 days, as described(19). EBs were dissociated and equal number of cells was plated in MethoCult GF H4434 complete methylcellulose medium (Stem Cell Technologies). After 14–16 days of hematopoietic differentiation, CFU colonies were counted by an experienced observer who was blind to the identity of the samples. For sideroblast quantification, CFU-GEMM and BFU-E picked from methylcellulose were washed in PBS, plated on glass slides by Cytospin, and stained using Prussian blue. Erythroid cells without and with iron deposits were scored by a hematopathologist who was blind to the sample identity.
Results
Induced pluripotent stem cells from a patient with Pearson syndrome
A 3 year-old patient presented with transfusion-dependent anemia from birth, metabolic acidosis, pancreatic exocrine insufficiency, insulin dependent diabetes mellitus, hypothyroidism, and transfusion-related iron overload. Examination of the patient's bone marrow showed vacuolated hematopoietic precursors and ringed sideroblasts, which are erythroid progenitors containing inappropriate iron granules in mitochondria (Figure 1A). Together with her clinical presentation, these findings suggested the diagnosis of Pearson syndrome. We confirmed this diagnosis by identifying a 2501 base pair deletion in a subset of the patient's mtDNA from peripheral blood and bone marrow (Figure 1B, C, and Supplemental Figure 1). The deletion from base pairs 10949–13449 interrupts the mitochondrial oxidative phosphorylation (OXPHOS) complex I NADH dehydrogenase genes ND4 and ND5, and also deletes three tRNAs (LCUN, SAGY, and H) that are required for translation of all products of the mitochondrial genome (encoding components of OXPHOS complexes I, III, IV, and V). From the patient's bone marrow we derived a primary fibroblast culture (PS-Fib), which carried mutant mtDNA in a proportion comparable to that found in the patient's blood and bone marrow (60–80% of all mitochondrial genomes) (Figure 1D). By mtDNA FISH and single-cell multiplex PCR analyses, we determined that over 95% of cells in the fibroblast population harbored deleted genomes (Figure 1C, E). As expected, we found quantitative and functional defects in OXPHOS complexes I, III, and IV in PS-Fib fibroblasts compared to wild-type (WT) fibroblasts (Figure 1F). These results describe patient somatic cells carrying the characteristic genetic and functional defects of a mtDNA deletion disorder.
Figure 1. Diagnosis of a patient with Pearson syndrome.
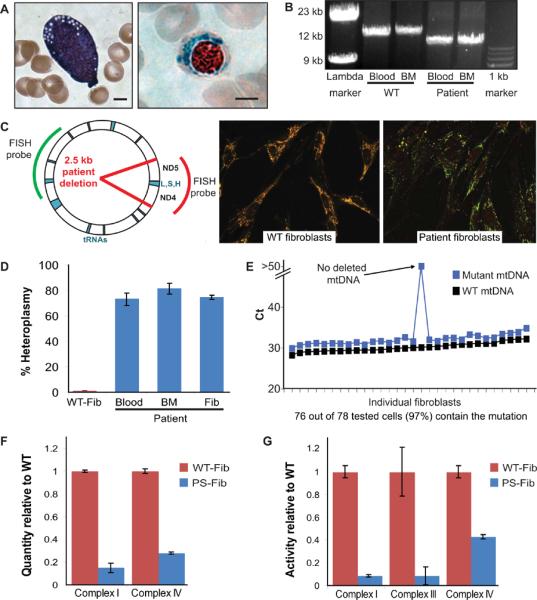
(A): A vacuolated hematopoietic precursor (left, hematoxylin and eosin stain) and ringed sideroblast (right, Prussian blue stain) from the patient's bone marrow aspirate (scale bar = 4 um). (B): Mitochondrial genome long range PCR revealing the patient's large mitochondrial deletion. (C): Location of the patient's 2.5 kb mutation in the mitochondrial genome and fluorescence in situ hybridization comparing WT and patient fibroblasts. The red probe covers a stretch of the mtDNA lost in the patient while the green probe covers an unaffected region. (D): Heteroplasmy of patient peripheral blood, bone marrow, and bone marrow-derived fibroblasts as measured by qPCR. (E): Multiplex qPCR performed at the single cell level on the patient's bone marrow derived fibroblasts. (F): Quantification of electron transport chain protein complexes by sandwich ELISA. Quantity is normalized to frataxin. (G): Activity of electron transport chain complexes. Complex I activity is measured by a nitrotetrazolium blue-based assay, complex III and complex IV activity are measured by cytochrome c assays coupled to horseradish peroxidase (HRP) and di-amino benzidinetetrachloride (DAB) staining. All error bars indicate standard deviation.
105 PS-Fib cells were infected with retroviruses encoding OCT4, SOX2, KLF4, and MYC. No pluripotent colonies had emerged after the typical 3–4 weeks of culture under human embryonic stem (ES) cell conditions. However, continued culture for 8–12 weeks yielded 3 colonies, which displayed the morphological and functional characteristics of pluripotent stem cells (Figure 2). The same low efficiency and delayed kinetics of iPS cell derivation from patient fibroblasts was observed in three independent reprogramming experiments (Supplementary Table 2). We focused our analyses on three Pearson syndrome iPS clones (PS-iPS, clones 1–3), each derived from an independent reprogramming experiment. iPS clones demonstrated ES-like cell morphology and self-renewal under ES cell culture conditions (Figure 2A). They expressed high levels of genes associated with pluripotency, as measured by immunofluorescence and PCR (Figures 2B–2C). Each line was capable of in vitro differentiation into embryoid bodies and, when injected into immunodeficient mice, showed the capacity to yield teratomas that included tissues from all three embryonic germ layers (Supplementary Figure 2). All iPS clones carried the pathological mtDNA deletion found in the parent fibroblasts as demonstrated by PCR using primers flanking the deletion junction (Figure 2D). These data demonstrate the derivation of human iPS cell lines from a patient with Pearson syndrome.
Figure 2. Generation of PS-iPS cells.
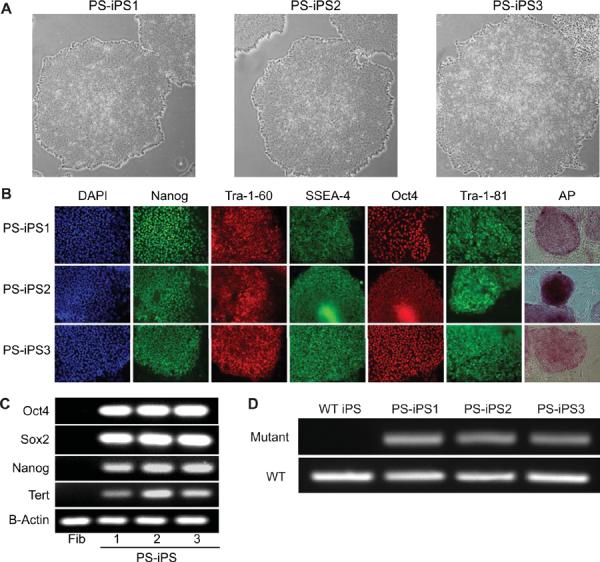
(A): Pearson syndrome iPS (PS-iPS) cell lines 1, 2, and 3 grown on Matrigel (40×). (B): Immunofluorescence staining for human pluripotency markers (100×). (C): RT-PCR of pluripotency associated genes demonstrates expression in iPS lines but not in the patient's bone marrow derived fibroblasts. (D): Mitochondrial PCR of PS-iPS lines using primers specific for the patient's mutation (upper) or primers that amplify an unaffected region of the mitochondrial genome (lower).
Pearson syndrome iPS cells vary in mtDNA heteroplasmy as a function of passage
In early passage PS-iPS cells, initial burdens of deleted mtDNA varied between 55–70% when assessed by Southern blot (Figure 3A). As expected, the PS-iPS cells could be continually propagated well beyond the point of senescence compared to the original fibroblast population. When we analyzed the degree of heteroplasmy during culture, we found varying changes from clone-to-clone: heteroplasmy in PS-iPS3 remained unchanged at approximately 50%, but PS-iPS1 and PS-iPS2 purged mutant mtDNA over time (Figure 3). Importantly, proviral integration analysis performed on each PS-iPS line showed a unique pattern that was maintained across passages, demonstrating that each line was descended from a single fully reprogrammed PS-Fib cell (Supplementary Figure 3). In the case of PS-iPS1, this analysis also demonstrates that the late passage cells arose by loss of the mutant mtDNA species from the earlier highly-heteroplasmic clone (Figure 3C). Upon continued culture of PS-iPS1, we obtained iPS cells with undetectable levels of the mutant mtDNA genome (Figure 3B, C). These data demonstrate clonal variation in changes in mtDNA heteroplasmy during culture, and derivation of mutation-free iPS cells from a patient with a mtDNA deletion disorder.
Figure 3. Characterization of PS-iPS cell heteroplasmy.
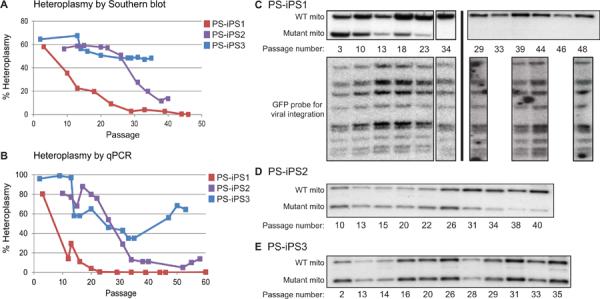
(A): Changes in heteroplasmy over passage number as measured by Southern blot. iPS cells were generally passaged every 6–7 days. (B): Changes in heteroplasmy over passage number as measured by qPCR. (C) Southern blots of PS-iPS1. Upper blots are probed for wild-type and mutant mtDNA (quantified in Figure 3A). Lower blots show a retroviral integration pattern created by viruses encoding reprogramming factors. Southern blots of PS-iPS1. Black lines indicate individual blots. Left panel shows a single blot probed for wild-type and mutant mtDNA (upper, quantified in Figure 3A) and virus integrations (lower). Right panel shows two independent blots probed for wild-type and mutant mtDNA (upper) and virus integrations (lower). (D): Southern blot of PS-iPS2 probed for wild-type and mutant mitochondrial DNA (quantified in Figure 3A, retroviral integration patterns in Supplementary Figure 3). (E): Southern blot of PS-iPS3 probed for wild-type and mutant mitochondrial DNA (quantified in Figure 3A, retroviral integration patterns in Supplementary Figure 3).
Functional characterization of PS-iPS cells
With isogenic, pluripotent cell lines that varied only in their degree of mtDNA heteroplasmy, we attempted to ascertain the effects of mitochondrial function on pluripotent cell growth and differentiation in vitro. When we used time-lapse imaging to compare colony growth between isogenic iPS lines with and without mutant mtDNA, we found that early passage PS-iPS1 cells containing 30% deleted mtDNA grew significantly more slowly than isogenic late passage PS-iPS1 cells which were purged of deleted genomes (Figure 4A). These differences in growth were not attributable to different rates of spontaneous differentiation, as judged by our observation of cellular and colony morphology during the experiment (Supplementary Figure 4). We also found that iPS cells purged of mutant mtDNA maintained a higher mitochondrial membrane potential than isogenic culture of iPS cells carrying 30% mutant mtDNA (Figure 4B). These results show the restoration of mitochondrial function and growth after elimination of mutant mtDNA in cultured PS-iPS cells.
To investigate the physiological basis of the growth defect in the cells, we analyzed mitochondrial respiration and glycolytic function by measuring oxygen consumption and media acidification rates (Figure 4C–G). iPS cells carrying 64% mutant mtDNA (PS-iPS3) showed a 50% lower baseline oxidative respiration rate compared to iPS cells carrying low or no mutant mtDNA (Figure 4C and E). This significant oxygen consumption defect could be caused by either impaired electron transport chain (ETC) function or by an uncoupling of ETC from ATP synthase activity. To distinguish between these possibilities, we added oligomycin to inhibit ATP synthase (Injection 1, OM). Upon oligomycin addition, all three lines reduced their oxygen consumption by similar degrees (64–76%, no significant difference, Figure 4C and F), indicating that ATP synthesis efficiency is equivalent between lines. These results suggest that the decreased oxygen consumption found in iPS cells with a high burden of mutant mtDNA likely results from impaired ETC function.
We next measured glycolytic rates in PS-iPS cells with varying degrees of mutant mtDNA. Glycolysis activity was assessed by measuring the extracellular acidification rate (ECAR) which varies depending on the cells' production of lactate, a metabolic byproduct of glycolysis. To measure glycolysis levels, cells were starved, then glucose was added to the media (Figure 4D, Injection 1 Glu). PS-iPS3 showed a trend toward higher glycolytic activity compared to iPS cells with negligible or low burdens of mutant mtDNA (PS-iPS1 and PS-iPS2), but the differences did not achieve statistical significance in aggregate analysis of replicates (Figure 4G). Taken together, the extracellular flux analyses suggest that pluripotent stem cells carrying high proportions of deleted mtDNA are defective in oxygen consumption due to reduced ETC function, which may be partly compensated by increasing glycolytic activity.
Hematopoietic differentiation of PS-iPS cells
Given the hematologic defects in Pearson syndrome, we next assessed the capacity of PS-iPS cells to form hematopoietic progenitors in vitro. We generated EBs from PS-iPS cells in the presence of mesoderm- and hematopoiesis-inducing factors, followed by dissociation and suspension in methylcellulose containing hematopoietic factors. We found that all PS-iPS lines were capable of forming progenitor colonies with typical myeloid, erythroid or mixed myeloid-erythroid morphology, verified at the single cell level by Wright-Giemsa staining (Figure 5A and data not shown). We were unable to detect statistically significant differences in colony number among PS-iPS lines carrying varying degrees of mutant mtDNA, possibly due to high intrinsic variability in ES-derived hematopoietic colony forming assays, but we noted a trend towards reduced numbers of colonies in samples carrying deleted mtDNA (Figure 5B, n = 4 differentiation experiments). To investigate whether sideroblasts were present, we performed iron staining on the hematopoietic cells isolated from the methylcellulose cultures. PS-iPS3 cells, which carry a high burden of mutant mtDNA, yielded high numbers of erythroid precursors with pathologic iron granule deposition compared to PS-iPS cells with less mutant mtDNA (Figure 5C, D). Collectively, these results demonstrate the recapitulation of a tissue-specific phenotype by directed differentiation of iPS cells carrying mutant mtDNA, and the amelioration of the phenotype in mutation-free, patient-identical iPS cells derived in vitro.
Figure 5. Hematopoietic differentiation of PS-iPS cells.
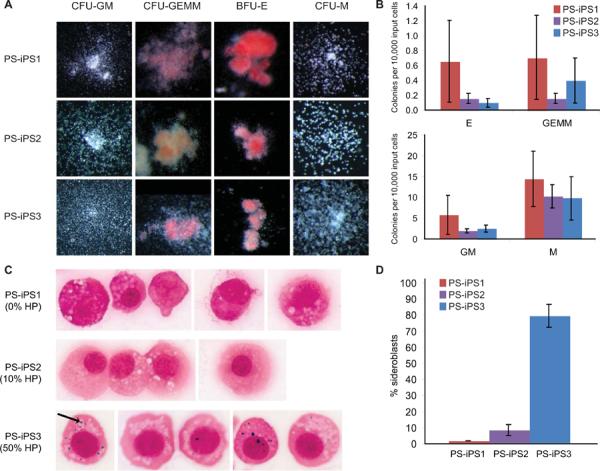
HP = heteroplasmy, the percentage mutant mtDNA. (A): Images of hematopoietic colonies derived from each PS-iPS line (25–63×). (B) Quantification of hematopoietic colony forming efficiencies, n=4. (C): Erythroid progenitors derived from PS-iPS cells, stained for iron with Prussian blue. The arrow indicates an example iron granule (1000×). (D): Quantification of sideroblasts, scored as percent of erythroid progenitors containing visible iron granules on Prussian blue stain.
Reprogramming of fibroblasts from other patients with mtDNA deletion syndromes
We obtained fibroblasts from two other patients suspected to have mitochondrial disease from the Coriell repository: one with symptoms consistent with Pearson syndrome (GM04516), and one with the later-onset variant Kearns-Sayre syndrome (GM06225). We confirmed these clinical diagnoses by detection and mapping of mtDNA deletions in both lines (Supplementary Figure 2A). The burden of mutant mtDNA at a population level in the skin-derived fibroblasts was low: 6% for GM04516 and 3% for GM06225. We reprogrammed these samples as described above and obtained with relatively high efficiency and normal kinetics several iPS cell lines bearing the hallmarks of pluripotency (Supplementary Figure 2B). However, of the resulting lines, nearly none carried the mtDNA mutation: only 1 of 61 lines derived from GM04516, and 0 of 21 lines analyzed from GM06225 (Supplemental Figure 2C). The reprogramming efficiencies and proportion of lines carrying the mtDNA deletion of samples from all three patients are documented in Supplementary Table 2. Collectively, these data demonstrate the efficient derivation of disease-free patient-specific iPS lines from patients with a low burden of a mosaic genetic abnormality.
Discussion
We have generated pluripotent stem cell lines by direct reprogramming of somatic cells from a patient with Pearson syndrome, a rare multisystem mitochondrial DNA deletion disorder. iPS cells carrying mutant mtDNA were derived with low efficiency and delayed kinetics, and showed impaired mitochondrial function and growth. During culture, heteroplasmy varied between PS-iPS cell lines, allowing us to isolate pluripotent cells with the same nuclear genome but with and without mutant mtDNA. PS-iPS cells were differentiated towards the hematopoietic lineage in vitro and were capable of generating erythroid and myeloid colonies. Moreover, differentiation of cells from the PS-iPS line with the highest mutant mtDNA burden yielded erythroid cells with inappropriate iron deposits which are characteristic of Pearson syndrome. Thus, we were able to observe a mitochondrial disease phenotype in a cell lineage affected in the human disease.
Mitochondrial function varies widely in different cells types of the body and therefore mitochondrial disorders in patients manifest with myriad syndromes and conditions. In the case of mtDNA defects, symptoms also depend on both the severity of the heteroplasmy and the mosaic of cell types affected. Such heterogeneity makes mitochondrial disorders a challenge to diagnose, treat and study. mtDNA sequences are difficult to manipulate directly in cells, leading to the widespread use of cytoplasmic hybrid (cybrid) cell lines(20, 21). Cybrids are created by fusion of cytoplasts harboring mutant mtDNA with a cell depleted of mitochondria (ρ0). Cybrids have provided valuable insights into the role of mtDNA in cancer, the effects of specific mtDNA mutations, and inter-species mitochondrial complementarity(21). However, the cybrid approach is heavily dependent upon the cell type chosen to receive the affected mitochondria(22, 23). Pearson syndrome mutations have been studied only using cybrids created from ρ0 cancer cell lines(24, 25). Because cell type-specific mitochondrial function is important to understand disease processes, an experimental system in which the effects of mtDNA mutations could be studied in specific human cell types would be of great value.
The advent of direct reprogramming technology has opened new possibilities for the study of mitochondrial disorders, making available abundant and versatile stem cell lines carrying a patient's genetic lesion. Two recent papers have employed iPS cells to investigate cell-type specific mitochondrial function in disease. Both groups created iPS cells from patients with early-onset versions of Parkinson's disease and subsequently differentiated the cells into neurons(22, 23). This system allowed investigation of mitochondrial function in Parkinson's disease neurons, yielding new insights into the relationship between nuclear and mitochondrial function in that disorder, which could eventually be used to screen for drugs that would ameliorate cell type-specific patient symptoms.
The study of mitochondrial disorders in iPS cells may also provide insight into mitochondrial function and mtDNA dynamics in stem cells in general. Human pluripotent stem cells (hPSCs) are reported to rely primarily on glycolysis for ATP production and harbor immature, cristae-poor mitochondria(26–29). Consistent with a relative lack of reliance on OXPHOS, we found that PS-iPS cells carrying high levels of deleted mtDNA could be generated and propagated. However, the kinetics of iPS derivation from fibroblasts with high mutant mtDNA burdens were delayed, and growth of iPS colonies with higher mutant mtDNA was decreased (Figure 4A and Supplementary Table 2). From other patient fibroblasts samples, iPS clones were derived with normal kinetics and efficiency and free of deleted mtDNA, due to low heteroplasmy in the initial fibroblast population but possibly also because of selection against clones carrying the mtDNA deletion (Supplemental Table 2 and Supplementary Figure 5). Furthermore, in two of our PS-iPS lines, we observed a decline in heteroplasmy over time, suggesting that mutant mtDNA are selected against during rapid proliferation over many population doublings in the pluripotent state (Figure 3). Given the inclusion of glucose in fibroblast and iPS culture medium, and the reportedly largely glycolytic state of hPSCs cells(26–29), these findings are somewhat unexpected and suggest that intact OXPHOS machinery may be required for optimal reprogramming of somatic cells, and growth of hPSCs. In keeping with these observations, a recent report shows that the OXPHOS machinery is active in hPSCs but uncoupled from ATP production, leading to speculation that mitochondrial activity is required for as yet undefined homeostatic functions in hPSCs(18). Moreover, our analysis of mitochondrial respiration in PS-iPS cells showed a decrease in hPSCs carrying high levels of mutant mtDNA compared to those without mutant mtDNA, revealing a basal level of oxidative activity that may serve a role in hPSC homeostasis. The availability of isogenic iPS cells and derivatives carrying varying levels of deleted mtDNA, as described here, will provide a valuable platform to study the role of mitochondrial function in reprogramming of somatic cells, and growth and differentiation of hPSCs.
Notably, in one of our PS-iPS cell lines (PS-iPS3), mtDNA heteroplasmy was maintained at high levels over more than fifty passages (approximately 1 year) in culture. Our observations are consistent with independent findings of variable heteroplasmy in iPS cells derived from patients with MELAS, a disorder caused by mtDNA point mutations (T.J. Nelson and colleagues, submitted). We speculate that infrequent epigenetic or genetic changes, including viral integrations, may enable selection of rare iPS clones to tolerate heteroplasmy-induced mitochondrial dysfunction. Because knowledge of such nuclear adaptations to heteroplasmy could inform our understanding of mitochondrial disorders, we are investigating the basis of clone-to-clone variation observed in the dynamics of mutant mtDNA changes during in vitro iPS culture. The availability of self-renewing, highly heteroplasmic PS-iPS cells will also permit screening for chemicals and/or genetic manipulations that facilitate purging of mutant mtDNA.
It is interesting to consider that the observation of decreased heteroplasmy in rapidly-dividing pluripotent stem cells in vitro may be reflective of changes in mutant mtDNA in stem cells in vivo. Pearson syndrome patients generally show spontaneous improvement in hematologic disease and immune function over the course of years, but worsening of neuropathy and myopathy(30, 31). In patients with other mtDNA disorders, mutant mtDNA is found as the predominant species in post-mitotic skeletal muscle fibers, but depleted in mitotic cells such as blood cells and skeletal muscle satellite stem cells(32, 33). Although the mechanisms driving these differences are largely unknown, selection against mutant mtDNA in dividing stem cells has been proposed as an explanation(30). If similar forces are at work in the PS-iPS lines, further in vitro studies may yield insights into methods to promote the extinction of mutant mtDNA species in specific tissues in patients.
Summary
In conclusion, our study describes the creation of iPS cells that carry varying burdens of mutated mtDNA from a patient with Pearson syndrome. These isogenic lines provide a new tool for in vitro studies of mitochondrial dysfunction. iPS cells offer a valuable complement to cybrid techniques because disease-carrying and disease-free iPS lines can be directly differentiated into precise tissue types to illuminate the pathophysiology of mtDNA disorders, and possibly to screen drugs that will support impaired cell-type specific functions. Mutation-free iPS cells are also a potential source of healthy, autologous tissue-specific progenitors for patients with mitochondrial diseases, although significant hurdles remain in the translation of iPS based cellular therapy. By exploiting their capacity for self-renewal and pluripotency, iPS cells will be a valuable tool to understand tissue-specific pathophysiology and innovate treatments for patients with Pearson syndrome and other mitochondrial genetic disorders.
Supplementary Material
Acknowledgements
The work was supported by grants from the National Heart, Lung, and Blood Institute, The National Institute of Diabetes and Digestive and Kidney Diseases, The National Institutes of Health, and Manton Center for Orphan Disease Research (G.Q.D. and S.A.).
Footnotes
- Anne B. C. Cherry: Conception and design, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
- Katelyn E. Gagne: Conception and design, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
- Erin M. McLoughlin: Conception and design, Collection and/or assembly of data
- Anna Baccei: Collection and/or assembly of data
- Bryan Gorman: Data analysis and interpretation
- Odelya Hartung: Collection and/or assembly of data
- Justine D. Miller: Collection and/or assembly of data
- Jin Zhang: Collection and/or assembly of data, Data analysis and interpretation
- Rebecca L. Zon: Collection and/or assembly of data
- Tan A. Ince: Data analysis and interpretation
- Ellis J. Neufeld: Provision of study material or patients
- Paul H. Lerou: Collection and/or assembly of data
- Mark D. Fleming: Collection and/or assembly of data
- George Q. Daley: Conception and design, Data analysis and interpretation, Final approval of manuscript
- Suneet Agarwal: Conception and design, Provision of study material or patients, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Conflict of interest: The authors disclose no potential conflicts of interest relevant to the work.
References
- 1.DiMauro S. Mitochondrial DNA medicine. Biosci Rep. 2007;27:5–9. doi: 10.1007/s10540-007-9032-5. [DOI] [PubMed] [Google Scholar]
- 2.Grossman LI, Shoubridge EA. Mitochondrial genetics and human disease. Bioessays. 1996;18:983–91. doi: 10.1002/bies.950181208. [DOI] [PubMed] [Google Scholar]
- 3.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–9. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 4.Schon EA. Mitochondrial genetics and disease. Trends Biochem Sci. 2000;25:555–60. doi: 10.1016/s0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–30. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 6.Swerdlow RH. Mitochondrial DNA--related mitochondrial dysfunction in neurodegenerative diseases. Arch Pathol Lab Med. 2002;126:271–80. doi: 10.5858/2002-126-0271-MDRMDI. [DOI] [PubMed] [Google Scholar]
- 7.Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–93. [PubMed] [Google Scholar]
- 8.Shoubridge EA. Mitochondrial DNA segregation in the developing embryo. Hum Reprod. 2000;15(Suppl 2):229–34. doi: 10.1093/humrep/15.suppl_2.229. [DOI] [PubMed] [Google Scholar]
- 9.Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95:976–84. doi: 10.1016/s0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- 10.Rotig A, Colonna M, Bonnefont JP, Blanche S, Fischer A, Saudubray JM, Munnich A. Mitochondrial DNA deletion in Pearson's marrow/pancreas syndrome. Lancet. 1989;1:902–3. doi: 10.1016/s0140-6736(89)92897-3. [DOI] [PubMed] [Google Scholar]
- 11.Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–8. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 15.van de Corput MP, van den Ouweland JM, Dirks RW, Hart LM, Bruining GJ, Maassen JA, Raap AK. Detection of mitochondrial DNA deletions in human skin fibroblasts of patients with pearson's syndrome by two-color fluorescence in situ hybridization. J Histochem Cytochem. 1997;45:55–61. doi: 10.1177/002215549704500108. [DOI] [PubMed] [Google Scholar]
- 16.Willis JH, Capaldi RA, Huigsloot M, Rodenburg RJ, Smeitink J, Marusich MF. Isolated deficiencies of OXPHOS complexes I and IV are identified accurately and quickly by simple enzyme activity immunocapture assays. Biochim Biophys Acta. 2009;1787:533–8. doi: 10.1016/j.bbabio.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc. 2008;3:1180–6. doi: 10.1038/nprot.2008.92. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. Embo J. 2011;30:4860–73. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerdan C, Hong SH, Bhatia M. Formation and hematopoietic differentiation of human embryoid bodies by suspension and hanging drop cultures. Curr Protoc Stem Cell Biol. 2007;Chapter 1(Unit 1D 2) doi: 10.1002/9780470151808.sc01d02s3. [DOI] [PubMed] [Google Scholar]
- 20.Bacman SR, Moraes CT. Transmitochondrial technology in animal cells. Methods Cell Biol. 2007;80:503–24. doi: 10.1016/S0091-679X(06)80025-7. [DOI] [PubMed] [Google Scholar]
- 21.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–28. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- 22.Cock HR, Tabrizi SJ, Cooper JM, Schapira AH. The influence of nuclear background on the biochemical expression of 3460 Leber's hereditary optic neuropathy. Ann Neurol. 1998;44:187–93. doi: 10.1002/ana.410440208. [DOI] [PubMed] [Google Scholar]
- 23.Dunbar DR, Moonie PA, Jacobs HT, Holt IJ. Different cellular backgrounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc Natl Acad Sci U S A. 1995;92:6562–6. doi: 10.1073/pnas.92.14.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Ouweland JM, de Klerk JB, van de Corput MP, Dirks RW, Raap AK, Scholte HR, et al. Characterization of a novel mitochondrial DNA deletion in a patient with a variant of the Pearson marrow-pancreas syndrome. Eur J Hum Genet. 2000;8:195–203. doi: 10.1038/sj.ejhg.5200444. [DOI] [PubMed] [Google Scholar]
- 25.Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, Hyslop SJ, et al. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur J Biochem. 1998;257:192–201. doi: 10.1046/j.1432-1327.1998.2570192.x. [DOI] [PubMed] [Google Scholar]
- 26.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–71. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–33. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]
- 28.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CAt, Ramalho-Santos J, et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011;6:e20914. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–77. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajasimha HK, Chinnery PF, Samuels DC. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A-->G mutation in blood. Am J Hum Genet. 2008;82:333–43. doi: 10.1016/j.ajhg.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simonsz HJ, Barlocher K, Rotig A. Kearns-Sayre's syndrome developing in a boy who survived Pearson's syndrome caused by mitochondrial DNA deletion. Doc Ophthalmol. 1992;82:73–9. doi: 10.1007/BF00156996. [DOI] [PubMed] [Google Scholar]
- 32.Fu K, Hartlen R, Johns T, Genge A, Karpati G, Shoubridge EA. A novel heteroplasmic tRNALeu(CUN) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum Mol Genet. 1996;5:1835–40. doi: 10.1093/hmg/5.11.1835. [DOI] [PubMed] [Google Scholar]
- 33.Weber K, Wilson JN, Taylor L, Brierley E, Johnson MA, Turnbull DM, Bindoff LA. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet. 1997;60:373–80. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.