

Abstract
Background
Norepinephrine and glutamate are among several neurotransmitters implicated in the neuropathology of major depressive disorder (MDD). Glia deficits have also been demonstrated in people with MDD, and glia are critical modulators of central glutamatergic transmission. We studied glia in men with MDD in the region of the brain (locus coeruleus; LC) where noradrenergic neuronal cell bodies reside and receive glutamatergic input.
Methods
The expression of 3 glutamate-related genes (SLC1A3, SLC1A2, GLUL) concentrated in glia and a glia gene (GFAP) were measured in postmortem tissues from men with MDD and from paired psychiatrically healthy controls. Initial gene expression analysis of RNA isolated from homogenized tissue (n = 9–10 pairs) containing the LC were followed by detailed analysis of gene expressions in astrocytes and oligodendrocytes (n = 6–7 pairs) laser captured from the LC region. We assessed protein changes in GFAP using immunohistochemistry and immunoblotting (n = 7–14 pairs).
Results
Astrocytes, but not oligodendrocytes, demonstrated robust reductions in the expression of SLC1A3 and SLC1A2, whereas GLUL expression was unchanged. GFAP expression was lower in astrocytes, and we confirmed reduced GFAP protein in the LC using immunostaining methods.
Limitations
Reduced expression of protein products of SLC1A3 and SLC1A2 could not be confirmed because of insufficient amounts of LC tissue for these assays. Whether gene expression abnormalities were associated with only MDD and not with suicide could not be confirmed because most of the decedents who had MDD died by suicide.
Conclusion
Major depressive disorder is associated with unhealthy astrocytes in the noradrenergic LC, characterized here by a reduction in astrocyte glutamate transporter expression. These findings suggest that increased glutamatergic activity in the LC occurs in men with MDD.
Introduction
Major depressive disorder (MDD) has a lifetime prevalence of 16% in the United States.1 Antidepressant drugs are the most used intervention for those with a diagnosis of depressive disorders, but the road to remission is long and uncertain, with 40% of patients never reaching full remission and at least 15% not experiencing any symptomatic improvements.2 Elucidating the biological bases of MDD is likely to provide novel targets for the development of more effective drugs, or at the very least, adjunctive treatments for existing antidepressants that increase the chance of remission.
Speculation that norepinephrine plays a role in depressive disorders dates back to the early 1950s, and research since then increasingly supports this. The locus coeruleus (LC) in the pontine brainstem contains the cell bodies of the major source of norepinephrine in the brain and has been the subject of numerous investigations regarding the neuropathology of MDD.3 The human LC is an area with very high densities of radioligand binding of antidepressant drugs to monoamine oxidase,4 the norepinephrine transporter5 and the serotonin transporter.6 Numerous postmortem studies demonstrate abnormal neurochemistry of the noradrenergic LC in people with MDD and in people who died by suicide. A role of norepinephrine in depression and antidepressant drug action is supported by laboratory animal studies demonstrating neurochemical changes in the LC following chronic stress, including changes that are reversed following antidepressant administration, that resemble LC pathology in humans with MDD.3 Collectively, laboratory animal and human findings implicate elevated activity of the noradrenergic LC in depression7 and medication-induced reductions of LC activity as an important biological effect of antidepressant drugs.8
Elevated LC activity in people with MDD could be secondary to elevated excitatory glutamatergic input to the LC. Locus coeruleus activity is modulated by glutamatergic input primarily from 2 areas: the paragigantocellularis nucleus and the prefrontal cortex. Interestingly, elevated glutamate levels have been reported in human serum, the occipital cortex, the prefrontal cortex and plasma of those with a diagnosis of MDD, with a positive correlation found between symptom severity and plasma concentration of glutamate.9–12 To our knowledge, glutamate levels have not been measured in the brainstem of people with MDD. However, chronic stress, which is associated with MDD, increases LC activity at least in part through elevated excitatory glutamatergic input.13 Furthermore, glutamatergic abnormalities in the postmortem LC of people with MDD, including elevated N-methyl-d-aspartate receptor subunit expression and reduced neuronal nitric oxide synthase expression, have been observed.14,15
A growing body of evidence shows a loss of glia or glia dysfunction in people with MDD; results have shown reduced glia, including oligodendrocytes and GFAP-labelled astrocytes, in cerebral cortical areas and the amygdala.16–19 Since the actions of glutamate are terminated largely by its removal from the synaptic junction by the excitatory amino acid transporter 1 (EAAT1) and excitatory amino acid transporter 2 (EAAT2) found on glia, a reduction in glia or glia health could contribute to elevated glutamate in the brain. Recently, Bernard and colleagues20 demonstrated reductions in the levels of expression of several genes known to be enriched in glia in LC tissue from people with MDD compared with matched controls. Among these genes were SLC1A3 (encoding EAAT1), SLC1A2 (encoding EAAT2) and GLUL (encoding glutamine synthase). Reduced uptake of glutamate by LC glia, as a result of reduced SLC1A3 and SLC1A2 expression, could directly contribute to altered glutamatergic signalling in the LC in people with MDD, as has been postulated previously.14,15 However, the study by Bernard and colleagues20 did not determine whether reduced levels of expression of glia-enriched genes were in fact isolated to glia, did not determine which type of glia was affected, and ultimately could not rule out the possibility that reduced glia gene expression was a result of a reduced number of glial cells in the LC region dissected for the gene expression studies. In the present study, we investigated the cellular source of altered glutamate transporter gene expression in the LC of people with MDD using postmortem brain tissues from people with characterized MDD and from psychiatrically healthy controls. We first attempted to reproduce the findings of Bernard and colleagues.20 Then we used laser-capture microdissection to specifically measure gene expression in LC astrocytes and oligodendrocytes. Our results provide evidence of compromised astrocytes in the LC of people with MDD, characterized here by a reduced expression of SLC1A3, SLC1A2 and GFAP.
Methods
Brain tissues
Brain tissues were collected at autopsy, as previously described, at the Cuyahoga County Coroner’s Office in Cleveland, Ohio, in accordance with approval from the University Hospitals Case Medical Center Institutional Review Board for Human Investigation. For the psychiatric assessment of our study sample, the next-of-kin completed the Structured Clinical Interview for DSM-IV psychiatric conditions (SCID) on behalf of the decedents, as described previously.21,22 We also obtained brain tissues from 4 people from the Quebec Suicide Brain Bank at the Douglas Hospital Research Centre, as approved by the Douglas Hospital Research Centre’s Research Ethics Committee. These samples were evaluated in a similar manner as those from the Cuyahoga County collection in that the SCID and SCID-II were completed retrospectively by the next-of-kin.23,24 Finally, we obtained tissues from 8 people from the Brain Tissue Donation Program at the University of Pittsburgh, and these tissues were subjected to similar retrospective psychiatric evaluation techniques and evaluations of medical records.25 For this collection, all procedures were approved by the University of Pittsburgh’s Institutional Review Board and Committee for Oversight of Research Involving the Dead.
In total, brain tissues from 20 psychiatrically healthy control men and 19 men with a diagnosis of MDD were used (Table 1). Blood and urine samples from all decedents were examined for the presence of psychotropic medications and substances of abuse. All controls were void of any depressive disorder diagnosis at the time of death or in the past. Detailed information, including causes of death and comorbidities, on the control and MDD groups is provided in Appendix 1, Table S1, available at cma.ca/jpn. The samples in both groups were matched for age, pH, RNA integrity number (RIN) and nicotine history as closely as was possible with the available tissues (Table 1). Causes of death and comorbidities are not included in Table 1 to protect the identities of the decedents.
Table 1.
Brain tissue information and assays
| Group; ID | Age, yr | Sex | pH | RIN | PMI | Smoker | Toxicology | Assays | Tissue |
|---|---|---|---|---|---|---|---|---|---|
| Controls | |||||||||
| FF1 | 27 | M | 6.88 | 8.4 | 17 | Yes | No drug detected | rtPCR, LCM, IHC, WB | LC, Cx |
| KS6 | 43 | M | 6.58 | 7.4 | 22 | No | No drug detected | rtPCR | LC, Cx |
| KS15 | 43 | M | 5.99 | 6.9 | 17 | No | Carbon monoxide | WB | LC |
| KS21 | 48 | M | 6.98 | 7.4 | 9 | Yes | No drug detected | rtPCR, LCM, IHC, WB | LC, Cx |
| KS27 | 74 | M | 6.62 | 6.7 | 21 | Yes | No drug detected | rtPCR, WB | LC, Cx |
| KS31 | 59 | M | 6.79 | 7.6 | 6 | No, history | Lidocaine | rtPCR, LCM, IHC, WB | LC, Cx |
| KS53 | 30 | M | 6.98 | 7.2 | 19 | Occasional | No drug detected | WB | LC |
| KS57 | 17 | M | 6.71 | 7.4 | 24 | No | Ethanol | rtPCR, LCM, IHC, WB | LC, |
| KS59 | 46 | M | 6.95 | 6.8 | 19 | No | No drug detected | rtPCR, LCM, IHC, WB | LC, Cx |
| KS61 | 31 | M | 6.86 | 3.6 | 28 | No information | No drug detected | WB | LC |
| KS63 | 18 | M | 6.40 | 6.4 | 32 | Unknown | Midazolam | rtPCR, LCM, IHC, WB | LC |
| KS65 | 58 | M | 5.80 | 6.4 | 27 | No | No drug detected | rtPCR, LCM, IHC, WB | LC |
| KS74 | 72 | M | 6.30 | 7.3 | 16 | No | Diltiazem | LCM | LC |
| KS76 | 42 | M | 6.70 | 7.4 | 24 | No | Butalbital | LCM | LC |
| KS78 | 18 | M | 6.60 | 7.9 | 16 | Unknown | No drug detected | LCM | LC |
| KS80 | 18 | M | 7.00 | 8.7 | 15 | No | Diazepam | LCM | LC |
| KS82 | 47 | M | 6.10 | 8.3 | 25 | No | Propoxyphane | WB | LC |
| RR | 37 | M | 6.47 | 7.3 | 17 | No | No drug detected | rtPCR, WB | LC, Cx |
| VV | 54 | M | 6.52 | 7.7 | 19 | Yes | Lidocaine | rtPCR, WB | LC, Cx |
| KS67 | 54 | M | 6.50 | 6.5 | 26 | No | Morphine | IHC | LC |
| Mean | 42 | 6.59 | 7.2 | 20 | |||||
| SEM | 4 | 0.08 | 0.2 | 1 | |||||
| MDD | |||||||||
| GG1 | 30 | M | 6.91 | 8.0 | 18 | Yes | No drug detected | rtPCR, LCM, IHC, WB | LC, Cx |
| KS8 | 42 | M | 6.67 | 6.7 | 44 | No | No drug detected | rtPCR | LC, Cx |
| DD | 52 | M | 6.48 | 5.8 | 18 | No | Carbon monoxide | WB | LC |
| KS12 | 41 | M | 6.24 | 6.7 | 19 | Yes | Chlorpheniramine | rtPCR, LCM, IHC, WB | LC, Cx |
| KS28 | 81 | M | 6.78 | 6.1 | 33 | Yes | No drug detected | rtPCR, WB | LC, Cx |
| KS32 | 60 | M | 6.32 | 6.8 | 20 | Yes | Ethanol | rtPCR, LCM, IHC, WB | LC, Cx |
| KS54 | 40 | M | 6.78 | 6.8 | 22 | Unknown | No drug detected | WB | LC |
| KS58 | 18 | M | 6.58 | 6.8 | 27 | Unknown | Carbon monoxide | rtPCR, LCM, IHC, WB | LC, |
| KS56 | 37 | M | 6.67 | 6.9 | 31 | No | Ethanol | rtPCR, LCM, IHC, WB | LC, Cx |
| KS62 | 26 | M | 6.77 | 7.1 | 7 | Unknown | Unknown | WB | LC |
| KS64 | 20 | M | 6.73 | 6.7 | 20 | No | Diphenhydramine | rtPCR, LCM, IHC, WB | LC |
| KS66 | 48 | M | 6.68 | 6.7 | 17 | No | No drug detected | rtPCR, LCM, IHC, WB | LC |
| KS75 | 77 | M | NA | 6.7 | 20 | No | Carbon monoxide, diazepam, temazepam | LCM | LC |
| KS77 | 38 | M | 6.70 | 8.7 | 19 | No | No drug detected | LCM | LC |
| KS79 | 25 | M | 6.90 | 7.6 | 13 | Yes | No drug detected | LCM | LC |
| KS81 | 18 | M | 7.00 | 8.5 | 10 | No | Ethanol | LCM | LC |
| JJ1 | 54 | M | 6.24 | 6.3 | 23 | No, 30-yr history | Carbon monoxide | WB | LC |
| TT | 38 | M | 6.52 | 7.2 | 24 | No | No drug detected | rtPCR, WB | LC, Cx |
| WW | 65 | M | 6.24 | 6.7 | 30 | Yes | Codeine | rtPCR, WB | LC, Cx |
| Mean | 43 | 6.62 | 7.0 | 22 | |||||
| SEM | 4 | 0.06 | 0.2 | 2 | |||||
Cx = prefrontal cortex (Brodmann area 10); IHC = immunohistochemistry performed on tissue sections; LC = locus coeruleus; LCM = laser-capture microdissected cells used in end-point polymerase chain reaction assays; M = male; MDD = major depressive disorder; NA = not available; PMI = postmortem interval; RIN = RNA integrity number; rtPCR = real-time polymerase chain reaction; SEM = standard error of the mean; WB = Western blot.
Tissue preparation and sectioning
Blocks of tissue containing the right prefrontal cortex (Brodmann area [BA] 10) and the pontine LC were cryopreserved at autopsy and stored at −80°C. Tissues were sectioned using a cryostat microtome (Leica CM3050S). For cytohistochemical staining, tissue was sectioned at 20 μm increments, desiccated overnight and stored at −80°C until use. For real-time quantitative polymerase chain reaction (qPCR) experiments using tissue homogenates, unmounted frozen sections of the prefrontal cortex and LC were punch-dissected with a 5 mm trephine. For cortex tissue, grey matter containing all 6 neocortical layers was isolated in this manner. In the LC, equivalent rostrocaudal levels along the axis of the LC were obtained as previously described.15 Tissue sections for laser capture microdissection (LCM) were prepared as described previously.26 For Western blotting, frozen sections (50 μm) were cut, and tissue containing the LC was punch-dissected (10 punches isolated per tissue donor).
Laser capture microdissection
We identified oligodendrocytes and astrocytes for LCM using a modified Nissl stain and a rapid GFAP immunostain, respectively. Capture was performed on a Veritas Microdissection Instrument model 704 (Life Technologies), and the quality of cell captures was subsequently verified by PCR of cell-specific gene markers, as previously described.27
Polymerase chain reaction
Total RNA was isolated and then reverse transcribed (rt) to complementary DNA from both punch-dissected tissue and laser-captured cells, as previously described.26 We used quantitative rtPCR to assess transcript levels in tissue homogenates, as described previously; briefly, target genes were normalized to the geometric mean of GAPDH, ACTB and UBC.28 All qPCR products were single, individual peaks identical to a synthesized oligonucleotide standard incorporating the complementary sequences to each primer set. We used semiquantitative end-point PCR to quantify messenger RNA levels from laser-captured samples, where levels of expression of target genes were normalized with the geometric mean of expression levels of reference genes, as used in qPCR.27 All semiquantitative end-point PCRs were performed in triplicate, where every replicate was verified using microcapillary electrophoresis (Agilent Bioanalyzer), indicating that each reaction was a single amplified product at the appropriate nucleotide length. All primer sets and genes are listed in Appendix 1, Table S2.
Immunohistochemical analysis
Sections used were immunohistochemically labelled with GFAP, as previously described.27 Two-dimensional analysis was carried out using Image J software to calculate both areal fraction and density of cells. To determine the amount of GFAP immunoreactivity in a given area, we obtained grey-scale images of GFAP-labelled LC tissue. The GFAP immunoreactive staining was determined by assigning a fixed threshold above background (defined on slides lacking primary antibody) and by generating a binary image. We calculated fractional area by dividing the GFAP-positive area (μm2) in the binary image by the total area of the image frame. A total of 10 randomized frames were analyzed in the LC region for each slide, and 3 nonadjacent slides cut at anatomically equivalent levels were analyzed per tissue sample donor (total of 30 images averaged per donor). To estimate the 2-dimensional density of GFAP-positive cells, we used an approach similar to that for the areal fraction. The GFAP-positive cells lying within the sampling area and not touching borders were counted for each of the 10 randomized images per slide (the same 3 nonadjacent slides per donor as those used for area fraction calculations).
Western blotting
Punches were homogenized in ice-cold Tris lysis buffer (20 mM, pH 8) supplemented with ethylenediamine-tetraacetic acid (0.2 mM), NP40 (0.5%), NaCl (150 mM), sodium dodecyl sulfate (0.1%), sodium deoxycholate (0.5%) and protease inhibitor cocktail (Thermo Scientific). Homogenates were sonicated twice, each time for 20 seconds, then divided and stored at −80°C until use. Proteins (2 μg) were separated using a 10% Bis/Tris gel with a Hoefer SE250 gel electrophoresis unit and then transferred to nitrocellulose membranes. Membranes were blocked with 5% instant milk at 22°C, incubated with mouse anti-GFAP (1:1000) for 3 hours at 22°C (Sigma), washed at 22°C 3 times for 10 minutes each time, and incubated with anti-mouse IgG (GE Healthcare) for 1 hour. We identified protein banding using horseradish peroxidase conjugated to the mouse IgG, which was then subjected to a chemiluminescent reaction (PerkinElmer). We quantified bands using MCID 7.0 software (InterFocus Imaging) with standardized parameters for all membranes. Each membrane blot consisted of samples from matched pairs so that experimental conditions were identical. We determined relative protein expression by calculating a ratio of GFAP to α-tubulin immunoreactivity (NB100–690; Novus Biologicals) determined on the same blot. All samples were analyzed in triplicate.
Statistical analysis
All data were subjected to Student t tests for paired comparisons. We paired control and MDD samples according to the matching criteria mentioned previously (pairs were also processed simultaneously through all experimentation). Statistical significance for the multiple comparisons of levels of SLC1A2, SLC1A3, GLUL and GFAP expressions was adjusted by Bonferroni correction to p = 0.012 to reduce chances of a type I error. We considered comparisons of GFAP immunoreactivity levels and cell counts to be significant at p = 0.05, uncorrected. All data were graphed and statistically analyzed using GraphPad Prism version 5.0 (GraphPad Software). All results of our statistical analyses appear in Appendix 1, Tables S3 and S4.
Results
The expression levels of 4 glia-related genes, SCL1A3, SCL1A2, GLUL and GFAP, were first analyzed by real-time PCR of rtRNA isolated from homogenized tissue that was punch-dissected from the LC region from decedents who had MDD and from matched controls (Fig. 1). SCL1A3 expression levels were significantly lower (−43%, p = 0.001) in the MDD than the control group. In contrast, levels of SCL1A2 expression were not significantly different between the MDD and control groups (p = 0.36). GLUL expression was modestly lower (−23%, p = 0.02) in the MDD than in the control group. GFAP expression in the LC was similar between the groups (p = 0.66).
Fig. 1.

Real-time quantitative polymerase chain reaction analysis of SLC1A3 (n = 10), SLC1A2 (n = 9), GLUL (n = 10) and GFAP (n = 9) in punch-dissected locus coeruleus tissue from matched pairs of tissue samples from psychiatrically healthy controls (open symbols) and men with major depressive disorder (closed symbols). Target gene expression was normalized with the geometric mean of 3 reference genes (GAPDH, UBC and ACTB). Relative gene expression levels are shown as fold changes using the method by Livak and Schmittgen.29 *p = 0.001.
Gene expression data collected from tissue homogenates contain an admixture of cell types, including different types of glia, neurons, surrounding neuropil and glial processes, and vascular tissue. A change in the number of a specific type of cell within a dissected brain region could contribute to an observed gene expression change if the change is a gene expressed in a specific cell. With regard to glia themselves, both astrocytes and oligodendrocytes express genes related to glutamate uptake and metabolism.30 Hence, we performed LCM of individual cell types within the LC to examine expression levels of the glia-related genes in the MDD and control groups.
Levels of SLC1A3 expression were robustly lower in astrocytes from decedents with MDD than in samples from matched controls when normalized to reference genes (−53%, p = 0.006, Fig. 2A) or to the number of cells (−58%, p = 0.003, Fig. 2C). Likewise, SLC1A2 expression in astrocytes was significantly lower in MDD than control samples normalized to reference genes (−29%, p = 0.003, Fig. 2A) or to number of cells (−39%, p < 0.001, Fig. 2C). GLUL expression in astrocytes was not significantly different between the groups (Fig. 2B and 2D). GFAP expression normalized to reference genes was modestly reduced (−19%, p = 0.04, Fig. 2B) in astrocytes of MDD tissues, although this did not reach statistical significance. However, normalization of GFAP gene expression to the number of cells captured showed a significantly lower expression in astrocytes from MDD samples than control samples (−27%, p = 0.005, Fig. 2D). In marked contrast to astrocytes, there were no significant differences in the levels of expression of SCLA3, SCLA2 and GLUL in oligodendrocytes between the groups (Fig. 3A–D). GFAP expression was below the limits of detection in oligodendrocytes.
Fig. 2.

Gene expression using quantitative end-point polymerase chain reaction conducted with laser-captured astrocytes from locus coeruleus tissue from matched pairs of psychiatrically healthy controls (open symbols) and men with major depressive disorder (closed symbols). Target gene expression of SCLA3 (n = 7), SCLA2 (n = 7), GLUL (n = 7) and GFAP (n = 11) was normalized using the geometric mean of the gene expression of 3 reference genes (GAPDH, UBC and ACTB; A and B) and normalized to the number of cells captured (C and D). *p < 0.01, **p < 0.001.
Fig. 3.

Gene expression using quantitative end-point polymerase chain reaction conducted with laser-captured oligodendrocytes from locus coeruleus tissue from matched pairs of psychiatrically healthy controls (open symbols) and men with major depressive disorder (closed symbols). Target gene expression of SCLA3 (n = 6), SCLA2 (n = 7) and GLUL (n = 6) was normalized using the geometric mean of the gene expression of 3 reference genes (GAPDH, UBC and ACTB; A and B) and normalized to the number of cells captured (C and D). No significant changes in gene expression were found. GFAP gene expression was undetectable.
It is worth noting through examination of Figures 2 and 3 that levels of SLC1A3 expression were highest in astrocytes, with expression in oligodendrocytes being about 20% of that in astrocytes in the control and MDD groups. SLC1A2 expression was higher in astrocytes than oligodendrocytes, although expression in oligodendrocytes was about 60% of that in astrocytes. The level of GLUL expression in oligodendrocytes was nearly 200% of GLUL expression in astrocytes.
To determine whether reductions in glutamate transporter genes were common to other brain regions, gene expression was measured in homogenates of the prefrontal cortex (BA 10). In contrast to LC homogenates, no statistically significant differences were observed for SLC1A3, SLC1A2, GLUL or GFAP expression in the prefrontal cortex (Fig. 4).
Fig. 4.
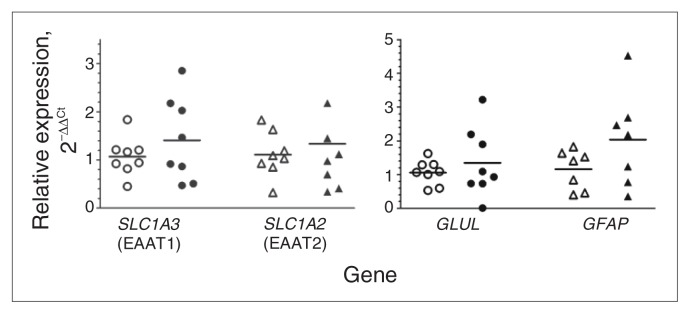
Quantitative polymerase chain reaction analysis of SLC1A3 (n = 8), SLC1A2 (n = 8), GLUL (n = 8) and GFAP (n = 7) using tissue homogenates from the frontal cortex (Brodmann area 10) from pairs of matched psychiatrically healthy controls (open symbols) and men with major depressive disorder (closed symbols). Target gene expression was normalized to the geometric means of gene expression for GAPDH, UBC and ACTB. Gene expression data are displayed as fold changes using the method of Livak and Schmittgen.29 There were no significant differences between any of the groups.
To determine whether levels of glutamate transporter protein were altered in the LC in the MDD group, we attempted Western blotting, but the amount of tissue required for these assays exceeded available tissue. Only frozen blocks of pontine tissue containing the LC were available, and immunostains of frozen postfixed tissue sections for EAAT1 and EAAT2 were very light and of relatively poor quality despite repeated attempts. In contrast, we used GFAP immunoreactivity to identify astrocytes for LCM and staining of Western blots, and tissue sections were of sufficient quality and signal intensity for quantitation. Western blots revealed a strikingly wide range of levels of GFAP immunoreactivity among control and MDD samples (Fig. 5A). Nevertheless, levels of GFAP immunoreactivity were significantly lower (−62%, all p < 0.05) in MDD than in matched control samples. GFAP immmunoreactivity measured in tissue sections also demonstrated significantly lower levels (−52%, all p < 0.05) in MDD than in matched control samples (Figs. 5B, 5D, 5E). Finally, the density of GFAP-labelled astrocytes in MDD samples was significantly lower (−46%, p < 0.05) than that in control samples (Fig. 5C).
Fig. 5.

GFAP immunolabelling in locus coeruleus (LC) tissue from matched pairs of psychiatrically healthy controls (open circles) and men with major depressive disorder (MDD; closed circles). Panel A shows data from Western blots of homogenized punch-dissected LC tissue normalized to tubulin immunoreactivity on the same blots (n = 14). Panel B shows data from pseudoquantitative analyses of images of GFAP immunostained sections (n = 8). Panel C shows estimates of astrocyte densities computed from 2-dimensional analysis of GFAP-stained cell profiles in the same tissue sections used in panel B (n = 8). The lower images are representative GFAP-immunostained sections from a control (D) and MDD (E) decedent and background staining (F) generated by the secondary antibody in the absence of the primary antibody. Panel G shows GFAP-stained cells (magnetization ×60) from the control donor (D). The bars in the lower left corner of panels D and G indicate a scale of 50 μm and 10 μm, respectively. *p < 0.05.
Reference genes and demographic variables
As a result of our careful matching of control and MDD decedents, there were no significant differences between the groups in terms of age, postmortem interval, RIN and pH values (Table 1). In addition, there were no significant differences between the MDD and control groups in the expression levels of the 3 reference genes in RNA isolated from homogenized tissues or from laser-captured astrocytes and oligodendrocytes (data not shown). There were no significant correlations between age and the expression of any of the reference or target genes, or between age and levels of GFAP immunoreactivity. With regard to GFAP immunoreactivity, it should be noted that this study was not designed to specifically address age and GFAP protein levels. That is, tissues from decedents across the range of ages were not run on the same blot. Instead, tissues from paired MDD and age-matched control decedents were run on the same blot. There were no significant correlations between RIN or pH values and the levels of expression of target or reference genes. Primers were designed to generate small amplimers (about 100 nucleotides; Appendix 1, Table S2), reducing the potential impact of RNA degradation on PCR product levels.27,31
Finally, 4 of the 19 decedents with MDD had a history of anti-depressant prescriptions, and 2 of them died as a result of heart disease rather than suicide or overdose of unknown cause. Unfortunately, these small subgroups were not of sufficient size within a given set of experiments to statistically evaluate the effects of antidepressant drug history or nonsuicide on gene or protein expression levels. For comparisons where more than 1 control/MDD pair could be evaluated, the following observations are noted. Individual comparisons of SLC1A3 gene expression in LC tissue homogenates from matched control–MDD pairs in which the decedents with MDD had a history of antidepressants indicated fold changes of 0.42, 0.47 and 0.79 compared to paired controls (mean change of 0.56). The mean change of the remaining control–MDD pairs in which the decedents with MDD had no history of antidepressants was 0.41. Three decedents with MDD whose tissues were used in our Western blotting analysis had previous antidepressant use, and levels of GFAP immunoreactivity in these samples were reduced by 97%, 9% and 89% (mean reduction of 65%) relative to their paired control. The mean reduction of GFAP in decedents with MDD without prior antidepressant drug use was 48%. We used brain tissues from 3 different brain collections, leaving the possibility that population, tissue collection or other differences may have impacted the data. Too few samples were used from the Quebec Suicide Brain Bank to evaluate separately. Four donor pairs from the Pittsburgh collection were used to generate the GFAP gene expression data from LCM-captured cells. The average percentage reduction of GFAP gene expression in astrocytes from MDD samples relative to matched controls was 15% for the Pittsburgh samples and 18% for Cuyahoga County samples.
Discussion
The results of the present study demonstrate pathology of astrocytes in the region of the noradrenergic LC. Evidence of dysfunction of astrocytes in the LC region in individuals with MDD include reduced expression of SLC1A3, SLC1A2 and GFAP; lower GFAP protein levels; and reduced density of GFAP-positive astrocytes. Altered expression of glutamate transporter genes has been reported previously in a number of studies of neurodegenerative and psychiatric diseases,32–34 including MDD.20,35 The present study provides direct evidence of astrocyte pathology in the cell body region of a monoamine neurotransmitter, indicating that glia cell abnormalities reported in more superior/rostral brain regions in MDD16,19 extend to the brainstem and may contribute to pathology of monoamine systems.
Animal studies demonstrating the effects of manipulations of glutamate transporters provide direct evidence that glutamate transporters play a major role in the pathophysiology of depressive behaviour. Blockade of Glt-1 (rodent Slc1a2, homologue of human EAAT2) transporters with dihydrokainic acid in rats induces depressive-like behaviours and impairs spatial memory.36 Whereas Glt-1 knockout mice do not survive, Glast-knockout mice (mouse Slc1a3, homologue of human EAAT1) survive but show signs of social withdrawal and impaired learning.37 If increased synaptic glutamate is a contributing factor to MDD due to etiological dysfunction of glutamate transporters, then medications that increase glutamate uptake in astrocytes would be expected to have beneficial therapeutic effects in the treatment of depression. In fact, treatment with the antibiotic ceftriaxone, which induces the expression of EAAT2 in primary human astrocytes,38 reduces depressive behaviours in animal models.39 Riluzole, which increases glutamate uptake in vitro,40 has been shown in animal models to reverse glial dysfunction induced by chronic stress.41 Collectively, these findings indicate that medications enhancing the glutamate transporter may reverse the effects of reduced glutamate transporter expression in individuals with MDD.
We previously reported evidence of astrocyte pathology in the LC region in individuals with MDD.26 The present study provides further evidence of astrocyte dysfunction in the noradrenergic LC region. Reduced SLC1A3 and SLC1A2 expression on astrocytes would be expected to elevate synaptic glutamate and increase excitatory glutamatergic input to LC neurons found in human MDD. These astrocyte pathologies led us to further explore a more general indicator of astrocyte function, GFAP, and the glial marker glutamine synthase (encoded by GLUL and also expressed by oligodendrocytes). While GLUL expression was similar in control and MDD tissues, reduced GFAP expression was found in astrocytes from MDD samples, along with a modest, yet significant, reduction in GFAP immunoreactivity, as demonstrated by Western blotting and in GFAP-immunostained tissue sections. In addition, we found reduced density of GFAP-immunostained astrocytes in the LC region in the MDD samples compared with matched control samples. Reduced GFAP immunoreactivity in MDD has been reported for anterior cingulate cortex white matter,42 dorsolateral prefrontal cortex deep layers,43,44 the amygdala16 and the cerebellum.45 Interestingly, Wistar-Kyoto rats (a strain often used as a model of depression and stress sensitivity) exhibit lower levels of GFAP immunoreactivity in the cerebral cortex and amygdala and a reduced number of GFAP-immunoreactive astrocytes compared with Sprague-Dawley rats.46 However, to our knowledge, no difference in the number of S100β-labelled astrocytes has been reported between these 2 strains of rat. While we observed a lower density of GFAP-immunoreactive astrocytes in the LC region in MDD samples than control samples, we cannot assume that this reflects fewer astrocytes in individuals with MDD. Reduced GFAP expression and lower GFAP protein levels (as verified in Western blots) would likely result in fewer astrocytes being identified using cell counting methods. This has also been noted by other authors.16 Hence, it is reasonable to suspect that the reduction in GFAP immunoreactivity in the present study may be a regulatory event in existing astrocytes.
While factors such as inflammation and oxidative stress induce GFAP expression, little is known about factors that reduce it. Since inflammation increases GFAP expression, and since MDD has been associated with elevated indices of inflammation,47 a possible interpretation of the present findings is that brainstem astrocytes are generally deficient in their inflammatory response. In fact, the multiple deficiencies in gene expression in these cells in MDD (reduced BMP7,26SLC1A2, SLC1A3, GFAP) are consistent with the postulate that LC astrocytes are unhealthy in individuals with MDD. A possible cause of these gene expression changes in astrocytes in individuals with depression is that they occur after prolonged changes in noradrenergic transmission. We and others have reported several indices of abnormal noradrenergic activity in individuals with MDD.3 Astrocytes express noradrenergic receptors,48 and norepinephrine can regulate glutamate uptake in cultured glia cells.49,50 Hence, elevated noradrenergic activity associated with MDD3 may alter astrocyte function. Of course, an alternative hypothesis could also underlie LC pathology: astrocyte deficits may contribute to noradrenergic neuron dysfunction, since these cells liberate supportive neurotrophic factors.26
Limitations
There are weaknesses in the present study that temper conclusions regarding our interpretation. Overall, small sample numbers may have masked statistical differences. Likewise, the small numbers of comorbid conditions (Appendix 1, Table S1) hampered evaluation of the impact of individual comorbidities on the findings. We were unable to verify that reduced SLC1A3 and SLC1A2 expressions were accompanied by reduced EAAT1 and EAAT2 protein levels because of a lack of sufficient amounts of LC tissue for those assays. Although we were unable to detect gene expression changes in BA 10, these experiments were performed using an admixture of cell types, not with LCM, leaving the possibility that alterations in astrocytes were masked by a lack of change in other cell types in this tissue. It is noteworthy, however, that we observed significant reductions of glutamate transporter gene expression in homogenates of LC tissue before extending the study to laborious laser capture experiments to determine the cellular source of the changes. Also, a lack of change in BA 10 does not imply that other cortical areas do not have glutamate transporter gene expression changes. Probably the most important index of transporter function is uptake, which cannot be performed reliably in postmortem human tissue. We were unable to untangle pathology associated with suicide from that associated with MDD, since most decedents with MDD died by suicide. This shortcoming cannot be addressed currently because of a lack of tissues from people with MDD who died a natural death. Finally, only astrocytes that express GFAP were studied. Hence, we note that the results of this study indicate dysfunction in only a subpopulation of astrocytes in individuals with MDD.
Conclusion
The present study, showing evidence of astrocyte dysfunction in the noradrenergic LC region in men with MDD, strongly implicates a deficit in the regulation of glutamate action in this region. Excitatory glutamatergic input to the LC is a major modulator of LC activity.51 Stress increases glutamate release in the LC,13,52 and MDD is commonly precipitated by stress. Hence, it seems reasonable to suggest that dysfunctional glutamate uptake by unhealthy astrocytes in the LC could amplify potential detrimental effects of elevated stress-evoked glutamate release in the LC at the level of neurons as well as astrocytes. In fact, findings from postmortem studies of the LC in individuals who had MDD demonstrate a number of characteristics suggesting that people with MDD experience chronic elevated LC activity.3 Many recent studies demonstrate the importance of astrocytes in neurotransmission53 and even demonstrate that destruction of astrocytes can induce depression-like symptoms in rats.54 Future research on the role of glia in depression has great promise to yield new approaches to the management of this disorder.
Acknowledgements
The authors gratefully acknowledge the work of Drs. Bryan L. Roth and George Jurjus, and all other psychiatry and brain autopsy personnel involved in the provision of human brain tissues (Cuyahoga County brain collection). The excellent assistance of the Cuyahoga County Coroner’s Office, Cleveland, Ohio, is greatly appreciated. The authors thank the Quebec Suicide Brain Bank, McGill Group for Suicide Studies, at the Douglas Mental Health Institute for provision of brain tissues. The authors also thank the Brain Tissue Donation Program at the University of Pittsburgh for the provision of brain tissues. This research was supported by MH46692, MH80323, MH68499, RR17701, the American Foundation for Suicide Prevention, the Connecticut Mental Health Center and the Department of Mental Health and Addiction Services.
Footnotes
Competing interests: M.J. Chandley, K. Szebeni, A. Szebeni and G.A. Ordway declare having received grant support from the American Foundation for Suicide Prevention, and grant and travel support from the National Institute of Mental Health. J. Crawford declares having received grant support from the American Foundation for Suicide Prevention and the National Institute of Mental Health. C.A. Stockmeier declares having received grant support and scientific instruments from the National Institute of General Medical Sciences. None declared for G. Turecki and J.J. Miguel-Hidalgo.
Contributors: C.A. Stockmeier, G. Turecki and G.A. Ordway designed the study. M.J. Chandley, K. Szebeni, A. Szebeni, J. Crawford and J.J. Miguel-Hidalgo acquired and analyzed the data. C.A. Stockmeier also acquired data; G.A. Ordway also analyzed data. M.J. Chandley and G.A. Ordway wrote the article. K. Szebeni, A. Szebeni, J. Crawford, C.A. Stockmeier, G. Turecki, J.J. Miguel-Hidalgo and G.A. Ordway reviewed the article. All authors approved its publication.
References
- 1.Kessler RC, Berglund P, Demler O, et al. National Comorbidity Survey Replication. The epidemiology of major depressive disorder: results from the national comorbidity survey replication (NCS-R) JAMA. 2003;289:3095–105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Warden D, Rush AJ, Trivedi MH, et al. The STAR*D project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9:449–59. doi: 10.1007/s11920-007-0061-3. [DOI] [PubMed] [Google Scholar]
- 3.Ordway GA. Neuropathology of central norepinephrine in psychiatric disorders: postmortem research. In: Ordway GA, Schwartz MA, Frazer A, editors. Brain norepinephrine. 1st ed. Cambridge (UK): Cambridge University Press; 2007. pp. 341–62. [Google Scholar]
- 4.Ordway GA, Farley JT, Dilley GE, et al. Quantitative distribution of monoamine oxidase A in brainstem monoamine nuclei is normal in major depression. Brain Res. 1999;847:71–9. doi: 10.1016/s0006-8993(99)02043-0. [DOI] [PubMed] [Google Scholar]
- 5.Klimek V, Stockmeier C, Overholser J, et al. Reduced levels of nor-epinephrine transporters in the locus coeruleus in major depression. J Neurosci. 1997;17:8451–8. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klimek V, Roberson G, Stockmeier CA, et al. Serotonin transporter and MAO-B levels in monoamine nuclei of the human brainstem are normal in major depression. J Psychiatr Res. 2003;37:387–97. doi: 10.1016/s0022-3956(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 7.Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci U S A. 2000;97:325–30. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morilak DA, Barrera G, Echevarria DJ, et al. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1214–24. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62:1310–6. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Kim JS, Schmid-Burgk W, Claus D, et al. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr. 1982;232:299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- 11.Mitani H, Shirayama Y, Yamada T, et al. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1155–8. doi: 10.1016/j.pnpbp.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 12.Sanacora G, Gueorguieva R, Epperson CN, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–13. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 13.Singewald N, Zhou GY, Schneider C. Release of excitatory and inhibitory amino acids from the locus coeruleus of conscious rats by cardiovascular stimuli and various forms of acute stress. Brain Res. 1995;704:42–50. doi: 10.1016/0006-8993(95)01102-1. [DOI] [PubMed] [Google Scholar]
- 14.Karolewicz B, Szebeni K, Stockmeier CA, et al. Low nNOS protein in the locus coeruleus in major depression. J Neurochem. 2004;91:1057–66. doi: 10.1111/j.1471-4159.2004.02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karolewicz B, Stockmeier CA, Ordway GA. Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacology. 2005;30:1557–67. doi: 10.1038/sj.npp.1300781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altshuler LL, Abulseoud OA, Foland-Ross L, et al. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 2010;12:541–9. doi: 10.1111/j.1399-5618.2010.00838.x. [DOI] [PubMed] [Google Scholar]
- 17.Bowley MP, Drevets WC, Ongur D, et al. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry. 2002;52:404–12. doi: 10.1016/s0006-3223(02)01404-x. [DOI] [PubMed] [Google Scholar]
- 18.Ongür D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–5. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajkowska G, Miguel-Hidalgo JJ, Wei J, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–98. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 20.Bernard R, Kerman IA, Thompson RC, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2011;16:634–46. doi: 10.1038/mp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumais A, Lesage AD, Alda M, et al. Risk factors for suicide completion in major depression: a case–control study of impulsive and aggressive behaviors in men. Am J Psychiatry. 2005;162:2116–24. doi: 10.1176/appi.ajp.162.11.2116. [DOI] [PubMed] [Google Scholar]
- 22.Stockmeier CA, Mahajan GJ, Konick LC, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 2004;56:640–50. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conner KR, Conwell Y, Duberstein PR. The validity of proxy-based data in suicide research: a study of patients 50 years of age and older who attempted suicide. II. Life events, social support and suicidal behavior. Acta Psychiatr Scand. 2001;104:452–7. doi: 10.1034/j.1600-0447.2001.00091.x. [DOI] [PubMed] [Google Scholar]
- 24.Kelly TM, Mann JJ. Validity of DSM-III-R diagnosis by psychological autopsy: a comparison with clinician ante-mortem diagnosis. Acta Psychiatr Scand. 1996;94:337–43. doi: 10.1111/j.1600-0447.1996.tb09869.x. [DOI] [PubMed] [Google Scholar]
- 25.Austin MC, Whitehead RE, Edgar CL, et al. Localized decrease in serotonin transporter-immunoreactive axons in the prefrontal cortex of depressed subjects committing suicide. Neuroscience. 2002;114:807–15. doi: 10.1016/s0306-4522(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 26.Ordway GA, Szebeni A, Chandley MJ, et al. Low gene expression of bone morphogenetic protein 7 in brainstem astrocytes in major depression. Int J Neuropsychopharmacol. 2012;15:855–68. doi: 10.1017/S1461145711001350. [DOI] [PubMed] [Google Scholar]
- 27.Ordway GA, Szebeni A, Duffourc MM, et al. Gene expression analyses of neurons, astrocytes, and oligodendrocytes isolated by laser capture microdissection from human brain: detrimental effects of laboratory humidity. J Neurosci Res. 2009;87:2430–8. doi: 10.1002/jnr.22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang L, Szebeni K, Szebeni A, et al. Dopamine receptor gene expression in human amygdaloid nuclei: elevated D4 receptor mRNA in major depression. Brain Res. 2008;1207:214–24. doi: 10.1016/j.brainres.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Pitt D, Nagelmeier IE, Wilson HC, et al. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology. 2003;61:1113–20. doi: 10.1212/01.wnl.0000090564.88719.37. [DOI] [PubMed] [Google Scholar]
- 31.Fleige S, Walf V, Huch S, et al. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol Lett. 2006;28:1601–13. doi: 10.1007/s10529-006-9127-2. [DOI] [PubMed] [Google Scholar]
- 32.Jacob CP, Koutsilieri E, Bartl J, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 33.Jen JC, Wan J, Palos TP, et al. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–34. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- 34.Rao JS, Kellom M, Reese EA, et al. Dysregulated glutamate and dopamine transporters in postmortem frontal cortex from bipolar and schizophrenic patients. J Affect Disord. 2012;136:63–71. doi: 10.1016/j.jad.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Miguel-Hidalgo JJ, Waltzer R, Whittom AA, et al. Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J Affect Disord. 2010;127:230–40. doi: 10.1016/j.jad.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bechtholt-Gompf AJ, Walther HV, Adams MA, et al. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35:2049–59. doi: 10.1038/npp.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karlsson RM, Tanaka K, Saksida LM, et al. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology. 2009;34:1578–89. doi: 10.1038/npp.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SG, Su ZZ, Emdad L, et al. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–23. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–2. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 40.Fumagalli E, Funicello M, Rauen T, et al. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–6. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 41.Banasr M, Chowdhury GM, Terwilliger R, et al. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2010;15:501–11. doi: 10.1038/mp.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gittins RA, Harrison PJ. A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J Affect Disord. 2011;133:328–32. doi: 10.1016/j.jad.2011.03.042. [DOI] [PubMed] [Google Scholar]
- 43.Davis S, Thomas A, Perry R, et al. Glial fibrillary acidic protein in late life major depressive disorder: an immunocytochemical study. J Neurol Neurosurg Psychiatry. 2002;73:556–60. doi: 10.1136/jnnp.73.5.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Si X, Miguel-Hidalgo JJ, O’Dwyer G, et al. Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004;29:2088–96. doi: 10.1038/sj.npp.1300525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fatemi SH, Laurence JA, Araghi-Niknam M, et al. Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophr Res. 2004;69:317–23. doi: 10.1016/j.schres.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 46.Gosselin RD, Gibney S, O’Malley D, et al. Region specific decrease in glial fibrillary acidic protein immunoreactivity in the brain of a rat model of depression. Neuroscience. 2009;159:915–25. doi: 10.1016/j.neuroscience.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–41. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hösli L, Hosli E, Zehntner C, et al. Evidence for the existence of alpha- and beta-adrenoceptors on cultured glial cells — an electro-physiological study. Neuroscience. 1982;7:2867–72. doi: 10.1016/0306-4522(82)90109-9. [DOI] [PubMed] [Google Scholar]
- 49.Fahrig T. Receptor subtype involved and mechanism of norepinephrine-induced stimulation of glutamate uptake into primary cultures of rat brain astrocytes. Glia. 1993;7:212–8. doi: 10.1002/glia.440070304. [DOI] [PubMed] [Google Scholar]
- 50.Hansson E, Ronnback L. Adrenergic receptor regulation of amino acid neurotransmitter uptake in astrocytes. Brain Res Bull. 1992;29:297–301. doi: 10.1016/0361-9230(92)90060-b. [DOI] [PubMed] [Google Scholar]
- 51.Aston-Jones G, Ennis M, Pieribone VA, et al. The brain nucleus locus coeruleus: restricted afferent control of a broad efferent network. Science. 1986;234:734–7. doi: 10.1126/science.3775363. [DOI] [PubMed] [Google Scholar]
- 52.Valentino RJ, Rudoy C, Saunders A, et al. Corticotropin-releasing factor is preferentially colocalized with excitatory rather than inhibitory amino acids in axon terminals in the peri-locus coeruleus region. Neuroscience. 2001;106:375–84. doi: 10.1016/s0306-4522(01)00279-2. [DOI] [PubMed] [Google Scholar]
- 53.Perea G, Araque A. GLIA modulates synaptic transmission. Brain Res Rev. 2010;63:93–102. doi: 10.1016/j.brainresrev.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64:863–70. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]