

Abstract
Protein aggregates are a common pathological feature of neurodegenerative diseases and several lysosomal diseases, but it is currently unclear what aggregates represent for pathogenesis. Here we report the accumulation of intraneuronal aggregates containing the macroautophagy adapter proteins p62 and NBR1 in the neurodegenerative lysosomal disease late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). CLN2 disease is caused by a deficiency in the lysosomal enzyme tripeptidyl peptidase I, which results in aberrant lysosomal storage of catabolites, including the subunit c of mitochondrial ATP synthase (SCMAS). In an effort to define the role of aggregates in CLN2, we evaluated p62 and NBR1 accumulation in the CNS of Cln2−/− mice. Although increases in p62 and NBR1 often suggest compromised degradative mechanisms, we found normal ubiquitin–proteasome system function and only modest inefficiency in macroautophagy late in disease. Importantly, we identified that SCMAS colocalizes with p62 in extra-lysosomal aggregates in Cln2−/− neurons in vivo. This finding is consistent with SCMAS being released from lysosomes, an event known as lysosomal membrane permeability (LMP). We predicted that LMP and storage release from lysosomes results in the sequestration of this material as cytosolic aggregates by p62 and NBR1. Notably, LMP induction in primary neuronal cultures generates p62-positive aggregates and promotes p62 localization to lysosomal membranes, supporting our in vivo findings. We conclude that LMP is a previously unrecognized pathogenic event in CLN2 disease that stimulates cytosolic aggregate formation. Furthermore, we offer a novel role for p62 in response to LMP that may be relevant for other diseases exhibiting p62 accumulation.
Introduction
Classic late-infantile neuronal ceroid lipofuscinosis (CLN2 disease) is an autosomal recessive neurodegenerative lysosomal disease that presents in children between 2 and 4 years of age with psychomotor retardation, intractable seizures, vision loss, and ultimately premature death. CLN2 disease is caused by mutations in the TPP1 gene, which encodes the lysosomal enzyme tripeptidyl peptidase I (TPP1) (Sleat et al., 1997). TPP1 deficiency leads to lysosomal accumulation of storage material, of which the subunit c of mitochondrial ATP synthase (SCMAS) is the primary component (Palmer et al., 1992; Ezaki et al., 1999). Although lysosomal storage pathology is present in all tissues, the CNS is most severely affected, with neurodegeneration occurring throughout the brain. Importantly, it remains unclear how TPP1 deficiency affects neuronal viability, thus underscoring the need to define pathogenesis for this untreatable disease.
The formation and degradation of protein aggregates is a physiological process that cells use to manage protein quality control (Lamark and Johansen, 2012). In many neurodegenerative states, the formation of aggregates per se is not considered deleterious, as chaperones and adapters function to isolate misfolded proteins and toxic intermediates within neurons (Johnston et al., 1998; Ross and Poirier, 2004; Bjørkøy et al., 2005). However, the persistent presence of aggregates is an indelible marker of compromise of the ubiquitin–proteasome system (UPS) and/or autophagy–lysosomal pathway. It is the dysfunction of these critical mechanisms—through their broader role in protein, organelle, and metabolic homeostasis—that neuronal viability is presumably affected (Hara et al., 2006; Komatsu et al., 2006; Pandey et al., 2007).
Because of the oligomerized nature of aggregates, macroautophagy is the primary mechanism for degrading these structures. Macroautophagy involves the engulfment of cytoplasmic material within double-membrane vesicles called autophagosomes that subsequently fuse with lysosomes for degradation (Mizushima et al., 2002). Two key adapter proteins involved in selective macroautophagy are p62 (p62/SQSTM1) and NBR1 (neighbor of BRCA1 gene 1). p62 and NBR1 function by binding ubiquitinated substrates, promoting aggregation of this cargo, and targeting this material within forming autophagosomes via an interaction with the autophagosomal membrane protein LC3-II (Bjørkøy et al., 2005; Pankiv et al., 2007; Kirkin et al., 2009). Thus, p62 and NBR1 are integral for aggregate formation and processing, and the accumulation of both proteins has been widely associated with compromised macroautophagy (Lamark and Johansen, 2012). Indeed, studies in several lysosomal diseases to date have used p62 accumulation as an indirect marker of macroautophagy impairment (Lieberman et al., 2012).
Here we report a previously unrecognized pathogenic mechanism whereby lysosomal membrane permeability (LMP) stimulates cytosolic aggregate formation in CLN2 disease. Unexpectedly, we found that the storage protein SCMAS colocalizes with p62 in cytosolic aggregates throughout the CNS of Cln2−/− mice. We determined that the release of SCMAS storage from lysosomes results in the sequestration of this material as aggregates by p62 and NBR1. Our studies identify that aggregate formation can be stimulated by LMP independent of macroautophagy and UPS dysfunction. Furthermore, we describe a novel role for p62 in response to LMP that may be relevant in other diseases associated with p62 accumulation.
Materials and Methods
Mice and tissue collection.
All procedures using animals were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. The murine model of CLN2 disease, the TPP1-deficient (Cln2−/−) mouse, has been well established to accurately represent human disease (Sleat et al., 2004). Cln2−/− mice along with wild-type (WT) littermate controls of either sex were used in all experiments and genotyped as described previously (Sleat et al., 2004). Tissue was processed as described previously for immunohistochemistry (Micsenyi et al., 2009). Briefly, mice were deeply anesthetized with ketamine–xylazine and perfused transcardially with saline after which tissue for biochemical analyses was excised and frozen. For immunohistochemistry and electron microscopy (EM), mice were further perfused with 4% paraformaldehyde (PFA), and tissue was immersion fixed overnight in 4% PFA.
Antibodies.
Anti-LC3 antibodies used were a rabbit polyclonal antibody (pAb) (Novus Biologicals) and a mouse monoclonal antibody (mAb) (Nanotools). Anti-p62 antibodies used were a rabbit pAb (Enzo Life Sciences) and a guinea pig pAb (American Research Products). Anti-NBR1 antibody was a mouse mAb (Novus Biologicals). Anti-LAMP2 rat mAb developed by J. T. August was purchased from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences (Iowa City, IA). The anti-SCMAS rabbit pAb was a generous gift from E. F. Neufeld (University of California, Los Angeles, Los Angeles, CA). The anti-ubiquitin antibodies used were a rabbit pAb (Dako) and an anti-polyubiquitinated mouse mAb (Enzo Life Sciences). The cathepsin D rabbit pAb was a generous gift from R. A. Nixon (Nathan Kline Institute, Orangeburg, NY). The anti-calbindin D-28K antibody used was a rabbit pAb (Millipore). Anti-β-actin antibody was a mouse mAb (Sigma-Aldrich). The anti-Beclin-1, anti-mTOR, and anti-phospho-mTOR (Ser2448) antibodies were rabbit pAbs (Cell Signaling Technology). The anti-Tom20 antibody was a rabbit pAb (Santa Cruz Biotechnology). The anti-MAP2 antibodies were a rabbit pAb (Millipore) and a mouse mAb (Sigma-Aldrich). Secondary antibodies used for immunohistochemistry and immunocytochemistry were biotinylated goat anti-rabbit IgG, biotinylated goat anti-mouse IgG, and biotinylated goat anti-guinea pig IgG (Vector Laboratories). Secondary antibodies used for immunofluorescence were Alexa Fluor 350 goat anti-rabbit IgG, Alexa Fluor 488 goat anti-guinea pig IgG, Alexa Fluor 546 goat anti-rabbit IgG (Invitrogen), Alexa Fluor 647 donkey anti-rabbit IgG minimal cross-reactivity, Alexa Fluor 647 donkey anti-rat IgG minimal cross-reactivity, 7-amino-4-methylcoumarin-3-acetic acid goat anti-mouse IgG minimal cross-reactivity, Cy3 donkey anti-rat IgG minimal cross-reactivity, and Cy3 donkey anti-mouse IgG minimal cross-reactivity (Accurate Chemicals). For immunogold EM, secondary gold-conjugated antibodies used were goat anti-guinea pig IgG and goat-anti-rabbit IgG (Electron Microscopy Sciences).
Immunohistochemistry and immunocytochemistry.
Fixed brain sections were cut using a Leica VT-1000S vibratome or fixed tissue was paraffin embedded and mounted on gel-coated slides. For immunoperoxidase staining, sections were blocked in 1% bovine serum albumin (BSA), 1.5% normal goat serum (NGS), and 0.02% saponin in PBS. Primary antibodies were diluted in 1% BSA, 1% NGS, and 0.02% saponin in PBS and incubated overnight at 4°C. Biotinylated secondary antibodies were similarly diluted and applied, followed by incubation with Vectastain avidin and biotinylated horseradish peroxidase macromolecular complex (ABC) kit (Vector Laboratories). Tissue was then stained with DAB Substrate Kit for Peroxidase (Vector Laboratories). Sections were visualized and photographed using an Olympus AX70 upright epifluorescence microscope equipped with MagnaFire CCD camera (Optronics). For immunofluorescence labeling of tissue, sections were blocked using 1% BSA, 5% NGS, 5% normal donkey serum (NDS), and 0.02% saponin in PBS. Primary antibodies were diluted in1% BSA, 2.5% NGS, 2.5% NDS, and 0.02% saponin in PBS and incubated overnight at 4°C. Secondary fluorescent antibodies were similarly diluted and applied to tissue. Sections were mounted with Prolong Antifade reagent with or without DAPI (Invitrogen). For immunocytochemistry, coverslips were fixed in 95% EtOH/5% acetic acid at −20°C for 10 min. Coverslips were blocked with 5% NGS and 5% NDS in PBS. Primary antibodies were diluted in blocking solution and incubated overnight at 4°C. Secondary fluorescent antibodies were diluted in 1% NGS and 1% NDS in PBS. Coverslips were mounted with Prolong Antifade reagent with or without DAPI. For controls of multi-labeling immunofluorescence assays, WT and Cln2−/− tissue and coverslips were labeled with individual primary and all secondary antibodies for each experiment to ensure no secondary antibody cross-reactivity.
Confocal fluorescence microscopy.
Confocal imaging was performed using a Carl Zeiss Meta Duo V2 confocal laser scanning microscope. Imaging laser settings were adjusted using control specimens to ensure autofluorescence and background signal were below immunofluorescence signal for each channel in individual experiments. Single z-plane 8-bit immunofluorescence images were acquired (unless otherwise noted) with a 63× oil-immersion objective [numerical aperture (NA) 1.4] or 20× dry objective (NA 0.40) using all or a combination of 351, 488, 543, and 633 nm laser lines consecutively, with emission bandwidth filters of 435–485, 505–550, 554–629 nm, and 640–704 nm, respectively. Sampling resolution was optimized for the objectives used, and images were constructed from the average of two unidirectional scans. Images were prepared using MetaMorph software (MDS Analytical Technologies) to separate LSM files into single-channel acquisitions and then combine channels for colored merged images. Adobe Photoshop (Adobe Systems) was used to generate figures.
Western blot analyses.
Frozen tissue used for Western blots was processed as described previously (Davidson et al., 2009). Tissue was homogenized in ice-cold lysis buffer [50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Igepal CA-630, 1% sodium deoxycholate, 0.1% SDS supplemented with Complete, Mini Protease Inhibitor Cocktail (Roche)] and centrifuged. The supernatants were then collected as soluble protein fractions, and pellets were washed with lysis buffer, further extracted with 2% SDS in lysis buffer, and sonicated to collect insoluble protein fractions. Protein concentrations were determined using the BCA protein assay kit (Thermo Fisher Scientific). Samples were analyzed by SDS-PAGE under denaturing conditions and transferred to immunoblot PVDF or nitrocellulose membranes (Bio-Rad). Membranes were blocked using 5% nonfat dry milk in 1× TBS with 0.1% Tween 20. Primary antibody incubations were performed overnight at 4°C, and subsequent horseradish peroxidase-conjugated secondary antibodies were applied. Protein detection was performed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), and blots were exposed to film or developed using a KODAK 2000R imaging station. Protein quantification for each sample was performed by densitometric analysis of film using NIH ImageJ software (http://imagej.nih.gov/ij/, 1997–2012), or blots were analyzed using KODAK 2000R imaging software. β-Actin was used as an external loading control for all Western blot analyses. For statistical analysis, all samples were normalized to the WT littermate controls in each experiment, and quantification was represented as the mean ± SEM. There were at least four biological replicates in each experiment, and statistical significance was determined by two-tailed unpaired Student's t tests analyzed by GraphPad Prism software InStat version 5.04 for Windows.
Quantitative real-time PCR.
Cortical tissue specimens were harvested from 13-week-old Cln2−/− mice (n = 4) and corresponding WT littermates (n = 4). RNA was isolated from 30 mg of fresh cortical tissue using RNeasy Plus Mini Kit and RNase-Free DNase set per the instructions of the manufacturer (Qiagen). RNA samples were immediately processed and quantified using the Qubit RNA Broad-Range Assay Kit and Qubit 2.0 Fluorometer (Invitrogen), after which sample concentrations were normalized and reverse transcription of RNA samples was performed to generate single-stranded cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). RT-PCR cycles were as follows: 10 min at 25°C, 120 min at 37°C, and 5 min at 85°C as per the instructions of the manufacturer using a GeneAmp PCR System 9700 (PerkinElmer Applied Biosystems). cDNA was quantified using the Qubit single-stranded cDNA Assay Kit and Qubit Fluorometer (Invitrogen), and samples were normalized. Probes from the TaqMan probe-based gene expression assay were used for singleplex quantitative real-time PCR (qPCR). Assay probes to the target gene Sqstm1 (RefSeq accession number NM_011018.2, 116 bp amplicon length) and the reference gene Gapdh (RefSeq accession number NM_008084.2, 107 bp amplicon length) were used (Applied Biosystems). Each biological replicate was run in quadruplicate for both target and reference gene. qPCR was performed on an ABI Prism 7000 Detection System (Applied Biosystems). qPCR cycles were as follows: 10 min at 95°C, and then 40 cycles of 15 s at 95°C and 1 min at 60°C. The relative quantity (RQ) of Sqstm1 mRNA in Cln2−/− cortical tissue compared with WT was expressed as fold change (RQ ± 95% confidence interval). As additional proof of statistical significance, the p value was determined by the pairwise fixed reallocation randomization test using REST (Relative Expression Software Tool; http://rest.gene-quantification.info/) (Livak and Schmittgen, 2001, Pfaffl et al., 2002).
UPS assay.
Chymotryptic activity of the UPS in cerebral and cerebellar tissue was analyzed using modified protocols described previously (Kisselev and Goldberg, 2005, Berezniuk et al., 2010). Tissue from 6- and 13-week-old WT and Cln2−/− mice (four animals per age and genotype) were compared. Mice were deeply anesthetized with ketamine–xylazine and perfused transcardially with ice-cold 0.9% saline. Half of the cerebrum and cerebellum were immediately excised and mechanically homogenized in extraction buffer (in mm: 50 Tris, pH 7.5, 40 KCl, 5 MgCl2, 0.5 ATP, and 1 DTT). Homogenates were then centrifuged and separated as soluble protein and insoluble pellet fractions. Additional extraction buffer was added to the pellets, and they were resuspended by mechanical trituration. Experiments were conducted using two technical replicates for each condition. Parallel samples were then incubated in the presence or absence of the irreversible UPS inhibitor 20 μm epoxomicin (EMD Chemicals) for 30 min at 37°C. Next, the fluorogenic UPS substrate 100 μm N-succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC) (Sigma-Aldrich) was added to each sample, followed by an additional incubation for 30 min at 37°C. After incubation time, ice-cold extraction buffer was added to stop the reaction, and chymotryptic activity was measured by detecting the fluorescence of free AMC released through the hydrolysis of the fluorogenic substrate (380 nm excitation, 460 nm emission) with a PerkinElmer Life and Analytical Sciences LS-3B spectrofluorometer. The average fluorescence units (FU) from inhibitor-treated samples were subtracted from the average FU from non-inhibitor-treated samples. This average activity value was multiplied by the amount of substrate added to each sample (20 nmol)/maximum FU after total cleavage of this amount of substrate (53.21 FU). Activity was expressed as nanomoles AMC per minutes per milligrams of total protein. Protein concentration was determined using the Bradford protein assay (Bio-Rad).
EM.
Preembedding immunogold silver-enhanced labeling was performed using an adapted protocol described previously (Yi et al., 2001). EM was performed using a Philips CM10 electron microscope. For immunogold EM, 4% PFA-fixed cerebral tissue was sectioned and incubated in 50 mm glycine/PBS for 30 min and then washed with PBS. Tissue was next incubated and blocked with 0.02% saponin in Aurion Blocking Solution and then washed with PBS–0.2% BSA-c (Electron Microscopy Sciences). Primary anti-p62 antibody was diluted in PBS–0.2% BSA-c and incubated overnight at 4°C, followed by extensive washing with PBS–0.2% BSA-c. Tissue was next incubated overnight at 4°C with secondary ultra-small gold-conjugated antibody diluted in PBS–0.2% BSA-c. Sections were then washed extensively with PBS–0.2% BSA-c, washed with phosphate buffer (PB), and finally washed with Enhancement Conditioning Solution (ECS). Tissue was then incubated with R-Gent SE-EM silver enhancement reagent (Electron Microscopy Sciences). The silver enhancement reaction was terminated with 30 mm sodium thiosulfate in ECS for 10 min, followed by washes with ECS and PB, and then postfixed with 2.5% glutaraldehyde in PB for 2 h. Sections were then transferred to 0.5% osmium tetroxide in 0.1 m cacodylate buffer for 30 min, followed by extensive washing with PB. Next, sections were cut into blocks along the cortex, and tissue was serial dehydrated in alcohol and plastic embedded in Epon. An ultramicrotome was used to cut ultrathin sections that were then stained with uranyl acetate and lead citrate.
Primary neuronal cultures.
Primary cortical neuronal cultures from Cln2−/− and WT littermate embryos were generated based on methods reported previously (Dobrenis et al., 1992, 2005). All experiments were performed on mature cultures [at least 21 d in vitro (DIV)]. Briefly, Cln2 heterozygous mice were mated, daily plug checks established day 0 of gestation, and females were killed at gestation day 15 to collect embryos. Brains from embryos were excised and hibernated in medium containing Leibovitz's L-15 medium, CO2-independent medium, and B-27 supplement (Invitrogen), and tissue for genotyping was collected. Next, brains were matched and pooled based on genotype results and litter, and the neocortex of each was further dissected out. Tissue was then mechanically dissociated in the presence of trypsin (Sigma-Aldrich), and cells were plated on poly-d-lysine (Sigma-Aldrich)-coated dishes and coverslips for biochemical and immunocytochemical experiments.
Macroautophagy flux and LMP experiments.
Macroautophagy flux experiments were performed using mature purified primary neuronal cultures and well-established methods (Mizushima and Yoshimori, 2007). LMP experiments and the p62 flux assay were performed using mature mixed primary cortical cultures. Purified primary cortical neurons were generated by treatment of cultures with mitotic inhibitors, 20 μg/ml 5-fluoro-2′-deoxyuridine plus 50 μg/ml uridine (Sigma-Aldrich), at 3 DIV and cultured for the remainder in the absence of FBS. Cultures were maintained until maturity at 21 DIV. Optimized saturating treatments of NH4Cl and leupeptin were determined to block macroautophagy flux without affecting neuronal viability. Cultures were treated for 4 h with 10 mm NH4Cl plus the lysosomal protease inhibitor 50 μm leupeptin to inhibit lysosomal degradation and flux. For LMP induction, mixed primary cortical cultures were grown and maintained until maturity and treated with 2 mm l-leucyl-l-leucine methyl ester (LeuLeuOMe) (Sigma-Aldrich) for 24 h. For the p62 flux assay, mature mixed primary cortical cultures were treated with 2 mm LeuLeuOMe for 20 h, at which point cultures were subsequently treated with vehicle, 10 mm NH4Cl plus 50 μm leupeptin, or 5 μm lactacystin for an additional 4 h. Cells for biochemical studies were harvested in ice-cold lysis buffer [50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Igepal CA-630, 1% sodium deoxycholate, 0.1% SDS supplemented with Complete, Mini Protease Inhibitor Cocktail (Roche)] and centrifuged. Soluble lysates were separated from insoluble pellet fractions and then analyzed by SDS-PAGE and Western blot, and coverslips were fixed and processed for immunocytochemistry. Quantification of neurons containing p62 aggregates in primary neuronal cultures treated with vehicle or 2 mm LeuLeuOMe for 24 h were evaluated in a blinded analysis by immunofluorescence microscopy. Cultures were immunolabeled for p62 and MAP2. Using an Olympus AX70 upright epifluorescence microscope and 60× oil-immersion objective (NA 1.40), at least 200 MAP2-positive neurons were counted per treatment (coverslip) and a minimum of 10 neurons per field of view. Neurons were counted as being positive for p62 aggregates if they contained one or more p62-immunopositive structures with a minimum size of 0.5 μm in diameter. A total of three separate experiments were conducted. Quantification was reported as the mean ± SEM for the percentage of neurons containing p62 aggregates.
Results
p62 and NBR1 are increased and colocalized in focal accumulations in neurons of Cln2−/− mice
We identified widespread focal immunolabeling for p62 and NBR1 in neurons throughout the cerebral cortex, hippocampus, thalamus, hypothalamus, the granule cell layer of the cerebellum, and Purkinje cells (Figure 1A–D and data not shown). Similar focal accumulations were not found in WT controls. Notably, we observed p62 accumulation in cortical neurons in mice as young as 3 weeks of age (data not shown). p62 and NBR1 consistently exhibited perinuclear localization in the basilar region of cortical neurons in which the primary lysosomal storage pathology accumulates in CLN2 and other neuronal ceroid lipofuscinoses (NCLs; Mole et al., 2011). By immunofluorescence and confocal microscopy, we found colocalization of p62 and NBR1 in cortical neurons and Purkinje cells (Fig. 1C,D). Additional experiments showed p62 and NBR1 accumulation in CD68-positive microglial cells, but not GFAP-positive astrocytes (data not shown). Western blot analysis identified significant increases in p62 in both soluble and insoluble protein fractions of Cln2−/− cerebrum and cerebellum (Fig. 1E), whereas NBR1 levels increased in insoluble protein fractions of cerebrum (Fig. 1F). These increases, specifically in insoluble protein levels, are suggestive of oligomerized protein aggregate accumulation commonly associated with many neurodegenerative proteinopathies (Douglas and Dillin, 2010; Lamark and Johansen, 2012). Additionally, by qPCR, we found a small but significant increase in Sqstm1 (p62) mRNA expression in Cln2−/− versus WT neocortical tissue (Fig. 1G). We conclude that the colocalization of p62 and NBR1 is consistent with the redundant function of both proteins as macroautophagy adapters in priming cargo for uptake within autophagosomes (Lamark et al., 2009).
Figure 1.

p62 and NBR1 are increased and colocalized in focal accumulations in neurons of Cln2−/− mice. A, Immunoperoxidase labeling for p62 in Cln2−/− cortical tissue exhibiting a basilar perinuclear focal accumulation in neurons. Scale bars: left, 20 μm; right, 5 μm, also pertains to B. B, Immunoperoxidase labeling for NBR1 in Cln2−/− cortical tissue exhibiting a similar focal localization as p62. C, Subcellular colocalization of p62 and NBR1 (arrows) in a Cln2−/− cortical pyramidal neuron in vivo in a single optical plane by confocal microscopy. DAPI labeling (blue) was used to identify the nucleus. Scale bar, 10 μm. D, Colocalization of p62 and NBR1 in a calbindin-positive Cln2−/− Purkinje cell in vivo in a single optical plane. Scale bar, 10 μm. E, Increased soluble and insoluble p62 protein levels from 13-week-old Cln2−/− (−/−) mice compared with WT (+/+) littermates from both cerebral (CRB) and cerebellar (CRBL) tissue by Western blot. Each lane was loaded with material from an individual mouse. Quantification at right displays the mean ± SEM for 4 biological replicates per genotype. Unpaired Student's t test (two-tailed, *p = 0.0138, **p = 0.0006, ***p < 0.0001, ****p = 0.0031). F, NBR1 protein levels are also elevated in 13-week-old Cln2−/− cerebral insoluble protein fractions compared with WT. Each lane was loaded with material from an individual mouse. Quantification at right displays mean ± SEM for four biological replicates per genotype. Unpaired Student's t test (two-tailed, *p < 0.0001). G, qPCR analysis of relative Sqstm1 mRNA levels in 13-week-old WT and Cln2−/− neocortical tissue identifying a significant (*) 1.134 (13.4%)-fold increase in Sqstm1 mRNA in Cln2−/− compared with WT. RQ values are plotted with RQ minimum and RQ maximum values (95% confidence interval; 1.022–1.258 for Cln2−/− and 0.871–1.148 for WT). We additionally used the pairwise fixed reallocation randomization test using REST (http://rest.gene-quantification.info/) (Livak and Schmittgen, 2001; Pfaffl et al., 2002) and determined p = 0.006, further confirming the statistical significance of increased Sqstm1 mRNA in Cln2−/−.
p62 is extra-lysosomal
To investigate whether p62 is a component of the primary lysosomal storage material and directly accumulating as a result of TPP1 deficiency, we used confocal microscopy and immunofluorescence labeling of tissue for p62, the lysosomal membrane marker LAMP2, and the lysosomal enzyme cathepsin D. We found no evidence that p62 accumulation occurs within LAMP2- or cathepsin D-positive lysosomes (Fig. 2A). Rather, p62 was primarily identified adjacent to lysosomes (Fig. 2A) and in some instances decorating the periphery of enlarged lysosomes (Fig. 2B).
Figure 2.
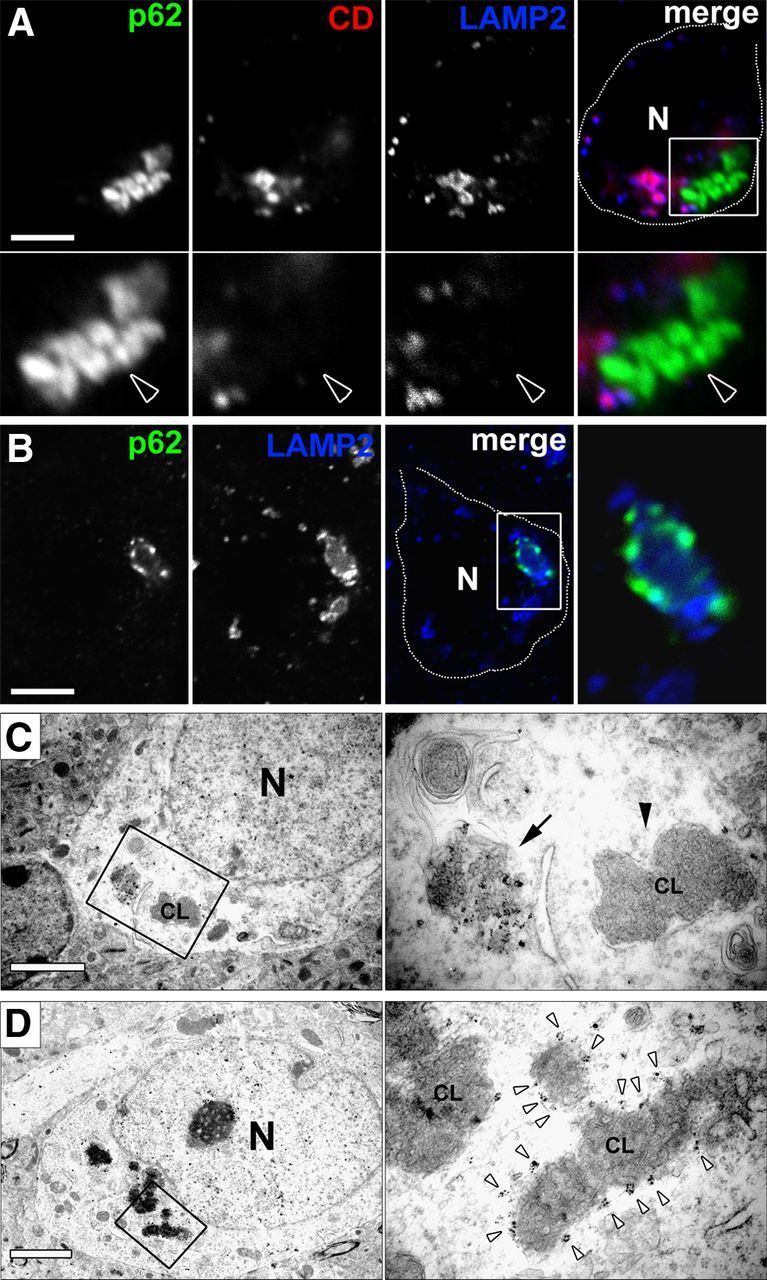
p62 accumulation is extra-lysosomal. A, Immunofluorescence assay shows large p62-positive aggregate (arrowhead) localized adjacent to LAMP2-positive (membrane signal) and cathepsin D-positive (lumenal signal) lysosomes in a Cln2−/− cortical neuron in vivo in a single optical plane by confocal microscopy. Boxed region in merged image is magnified below for each channel. N, Nucleus. Scale bar, 5 μm. B, Single optical plane confocal image of a Cln2−/− cortical neuron in vivo showing p62 immunoreactivity localized to the periphery of a large LAMP2-positive lysosome. Boxed region in merged channel is magnified at right. CD, Cathepsin D; N, nucleus. Scale bar, 5 μm. C, Left EM shows low magnification of a non-membrane-bound p62-positive gold-labeled aggregate and adjacent membrane-bound CL profile in a Cln2−/− cortical neuron. Boxed region in left micrograph is magnified at right with p62 aggregate (denoted by arrow) and membrane-bound CL profile (arrowhead denoted CL). N, Nucleus. Scale bar, 2 μm. D, Left EM shows low magnification of p62-positive gold labeling decorating the periphery of a CL profile in a Cln2−/− cortical neuron. Boxed region in left micrograph is magnified at right showing p62-positive gold (white arrowheads) surrounding the periphery of a CL profile (denoted CL). N, Nucleus. Scale bar, 2 μm. Tissue used in A–D were from 13-week-old Cln2−/− mice.
To evaluate the ultrastructural localization of p62, we used immunogold EM labeling in Cln2−/− cortex. We confirmed that p62 immunogold-positive accumulation was not contained within lysosomes, double-membrane autophagosomes, or other membrane-bound vesicles in neurons (Fig. 2C,D). Consistently, micrographs from Cln2−/− mice at 13 weeks of age showed perinuclear p62-positive non-membrane-bound accumulation in large cytosolic structures. p62-positive labeling of non-membrane bound material was often adjacent to membrane-bound lysosomal storage bodies, known as curvilinear profiles (CL profiles) (Fig. 2C). Notably, p62 localized to cytosolic material that was curvilinear in appearance but lacked a delimiting membrane. This material was morphologically similar to contents within CL profiles but appeared less densely packed. Additionally, we observed evidence for p62-positive gold labeling of the periphery of CL profiles without a clearly demarcated membrane (Fig. 2D). Together with our biochemical analyses, we conclude that p62-positive structures are insoluble cytosolic protein aggregates. The localization of p62 to extra-lysosomal curvilinear material and the periphery of CL profiles suggest that lysosomal storage bodies may succumb to LMP and that p62 responds to this event. The frequent appearance of the large p62-positive perinuclear aggregates may represent a stable and isolated structure at this stage of disease.
p62 colocalizes with ubiquitin; Cln2−/− mice exhibit normal UPS function and only modest macroautophagy impairment
p62-positive aggregates often contain ubiquitin in several neurodegenerative disease pathologies (Salminen et al., 2012), and we identified by immunofluorescence and confocal microscopy that p62 strongly colocalizes with ubiquitin in Cln2−/− cortical neurons in vivo (Fig. 3A). Additionally, increased polyubiquitinated protein was found in insoluble protein fractions of cerebral tissue by Western blot (Fig. 3B). Given this accumulation of ubiquitin and previous evidence of the ability of p62 to target monomeric proteins for degradation through the UPS (Wooten et al., 2006), we evaluated UPS proteolytic function in Cln2−/− cortical tissue to determine whether UPS impairment might account for the presence of aggregates. Using a well-established assay for measuring the chymotryptic activity of the UPS (Kisselev and Goldberg, 2005; Berezniuk et al., 2010), we found no difference between WT and Cln2−/− mice in both cerebral and cerebellar tissue (Fig. 3C), indicating that the UPS catalytic capacity is intact and functional in Cln2−/− and therefore not likely responsible for cytosolic aggregate accumulation.
Figure 3.

p62 colocalizes with ubiquitin in Cln2−/− cortical neurons; chymotryptic activity of the UPS is normal in Cln2−/− brain. A, Single optical plane shows strong p62 colocalization with ubiquitin by immunofluorescence in a Cln2−/− cortical neuron in vivo. Nuclei counterstained with DAPI (blue). Scale bar, 5 μm. B, Western blot showing increased polyubiquitinated proteins from insoluble protein fractions of Cln2−/− cerebral tissue homogenate compared with littermate WT tissue. Actin was used as a loading control. Each lane was loaded with material from an individual mouse. Additional insoluble ubiquitinated proteins that failed to migrate through the gel can be seen in the top of the stacker (arrow). C, Quantification showing no difference (unpaired Student's t test) in UPS chymotryptic activity between WT and Cln2−/− cerebral (CRB) and cerebellar (CRBL) tissue homogenate from 6- and 13-week (Wk)-old mice measured by the release of fluorescent AMC from the UPS substrate Suc-LLVY-AMC (S, soluble protein fraction; P, pellet insoluble fraction).
We next analyzed macroautophagy function in Cln2−/− mice, predicting that the primary lysosomal enzyme deficiency may lead to secondary compromise of macroautophagy and inefficient processing of macroautophagy substrates such as p62. Of particular interest was defining where along the macroautophagy pathway impairment might occur because p62 aggregates are extra-lysosomal and not contained within autophagosomes. In mature Cln2−/− purified primary neurons (21 DIV), we found macroautophagy flux—defined as the processing of the autophagosomal membrane protein LC3-II through the macroautophagy–lysosomal pathway—to be equally efficient to that of WT neurons and thus corresponding to an early disease state in vivo (Fig. 4A). Importantly, macroautophagy is efficient in these cultures even while SCMAS storage accumulation is prominent at 21 DIV (Fig. 4B). Evaluation of the autophagosomal membrane protein LC3-II showed only modest increases in Cln2−/− brain tissue compared with WT by end-stage disease (Fig. 4C). We further analyzed the upstream autophagy regulatory proteins mTOR and Beclin 1 and found no changes in the levels of these proteins between Cln2−/− and WT (Fig. 4D,E). This suggests that increased LC3-II levels (associated with an increased number of autophagosomes) in Cln2−/− brains represent modest macroautophagy inefficiency rather than an upregulation in the pathway. It has been argued in other lysosomal diseases that lysosomal storage accumulation can impede autophagosome and lysosome fusion and impair the processing of autophagic material, thus contributing to increased levels of LC3-II and p62 (Settembre et al., 2008; Elrick et al., 2012). Our findings that macroautophagy flux is efficient in mature Cln2−/− primary neuronal cultures indicate that autophagosomes are processed properly even as neurons in these cultures exhibit SCMAS storage. Furthermore, the persistent presence of non-membrane-bound p62-positive protein aggregates in Cln2−/− does not correlate with macroautophagy failure at the step of autophagosome fusion with lysosomes or autophagosome degradation. Rather, the abundance of p62-positive aggregates more appropriately represents a failure to target aggregates within autophagosomes for clearance. In reconciling the modest impairment in autophagosome maturation late in disease with the abundant and early presence of p62-positive protein aggregates in Cln2−/− (as early as 3 weeks), we looked to further define whether aggregate formation is attributable to LMP and independent of macroautophagy.
Figure 4.

Macroautophagy flux is efficient in mature Cln2−/− primary neuronal cultures; autophagosome maturation is modestly inefficient by end-stage disease. A, Macroautophagy flux (Macro Flux) as measured by Western blot for LC3-II levels in the presence or absence of lysosomal proteolysis inhibition (NH4Cl and leupeptin) is efficient, and net macroautophagy (Net Macro) is slightly increased in 21 DIV primary Cln2−/− neuronal cultures compared with WT. Actin was used as a loading control. Each lane represents an individual experimental condition. Quantification of LC3-II/Actin represents the mean ± SEM of four experimental replicates for each condition. Macro flux represents the ratio of LC3-II/Actin mean values for proteolysis inhibitor (NH4Cl and leupeptin)-treated cultures, to LC3-II/Actin mean values for vehicle-treated cultures. Net macro represents the difference of vehicle-treated cultures from proteolysis inhibitor-treated cultures. B, MAP2-positive neuron from 21-d-old Cln2−/− primary neuronal cultures showing SCMAS storage accumulation by immunofluorescence. Boxed region in merged channel is magnified at bottom right. N, Neuron. Scale bar, 10 μm. C, Western blots for LC3-II levels show a slight yet progressive increase from 6 to 13 weeks of age in cerebral and cerebellar tissue from Cln2−/− mice compared with WT. Actin was used as a loading control. Each lane was loaded with material from an individual mouse. Quantified changes in LC3-II at right show statistically significant increases in Cln2−/− cerebellum (CRBL) at 6 and 13 weeks (wk) and in cerebrum (CRB) at 13 weeks compared with WT controls. Quantification is represented as the mean ± SEM for at least four biological replicates per genotype. Statistical significance was determined by unpaired Student's t test (two-tailed, **p < 0.001, ***p < 0.0001). D, No statistically significant changes in levels of p-mTOR (Ser2448) relative to total mTOR/Actin were found in cerebrum (CRB) or cerebellum (CRBL) of Cln2−/− mice compared with WT. Each lane was loaded with material from an individual mouse. Quantification is represented as the mean ± SEM for four biological replicates per genotype. E, Beclin 1 levels in cerebrum (CRB) and cerebellum (CRBL) are comparable between Cln2−/− and WT. Each lane was loaded with material from an individual mouse. Quantification is represented as the mean ± SEM for four biological replicates per genotype.
p62 colocalizes with the primary lysosomal storage protein SCMAS in extra-lysosomal aggregates
SCMAS has been identified as a substrate of TPP1, and therefore its lysosomal accumulation in CLN2 disease appears directly linked to the enzyme deficiency (Ezaki et al., 1999). Given our immuno-EM data suggesting that p62 localizes to curvilinear material, we predicted that if LMP occurs in Cln2−/− neurons, SCMAS should be a component of cytosolic aggregates and therefore colocalized with p62. In evaluating neocortical distribution of SCMAS and p62 in Cln2−/− mice, we observed that SCMAS storage was apparent in neurons throughout all neocortical layers, whereas p62 was found primarily in layers II–IV and layer VI (Fig. 5A). Neurons containing p62 aggregates like those of layer IV exhibited extra-lysosomal SCMAS that colocalized with p62 (Fig. 5B). Importantly, we found that layer V neurons devoid of p62 accumulation contained SCMAS localized entirely within LAMP2-positive lysosomes (Fig. 5C). We also observed evidence of discontinuous LAMP2-positive lysosomal membranes where SCMAS appeared released in the cytosol and colocalized with p62 (Fig. 6A). Higher-resolution images confirmed significant colocalization between p62 and SCMAS in large cytosolic aggregates in Cln2−/− cortical neurons (Fig. 6B). We further considered the neocortical distribution of p62 with regards to the initial characterization of the Cln2−/− mouse model by Sleat et al. (2004). These studies reported that, although neurodegeneration is widespread, a selective pattern of neuronal loss emerges through disease course (Sleat et al., 2004). For example, in Cln2−/− neocortex, neurons of layer IV and layer VI in somatosensory and auditory cortex were found to be particularly vulnerable to degeneration (Sleat et al., 2004). Our analyses show that p62 accumulation is prevalent in neurons of neocortical layers II–IV and layer VI in multiple regions, including posterior parietal cortex (Fig. 5A), as well as somatosensory and auditory cortex (data not shown). To explore the potential relationship between p62 accumulation and neuron fate, we stained cortical tissue for the neurodegenerative marker Fluro-Jade C (FJC), in combination with immunofluorescence labeling for p62. We found that FJC-positive degenerating neurons were devoid of p62, whereas adjacent neurons containing p62-positive aggregates were FJC negative (data not shown). Although p62 accumulates in neuronal populations previously identified to be vulnerable to neurodegeneration, our data suggest that the presence of p62 aggregates alone may not be directly cytotoxic.
Figure 5.

p62 colocalizes with the primary lysosomal storage protein SCMAS in extra-lysosomal aggregates. A, Section of posterior parietal cortex from a 13-week-old Cln2−/− mouse showing the distribution of p62, SCMAS, and LAMP2. p62 accumulation is most prominent in layers II–IV and layer VI, whereas layer V neurons exhibit prominent SCMAS with limited p62 accumulation. Image is a single-plane tile-scan confocal image. Scale bar, 250 μm. B, Single optical plane showing layer IV Cln2−/− cortical neuron in vivo exhibiting p62 accumulation and colocalization with extra-lysosomal SCMAS (arrowheads). Boxed region in merged image is magnified below each individual channel. N, Nuclear region. Scale bar, 5 μm. C, Single optical plane showing layer V Cln2−/− cortical neuron in vivo exhibiting SCMAS storage contained within LAMP2-positive vesicles in the absence of any p62 accumulation by immunofluorescence assay. Boxed region in merged image is magnified below each individual channel. N, Nuclear region. Scale bar, 5 μm.
Figure 6.
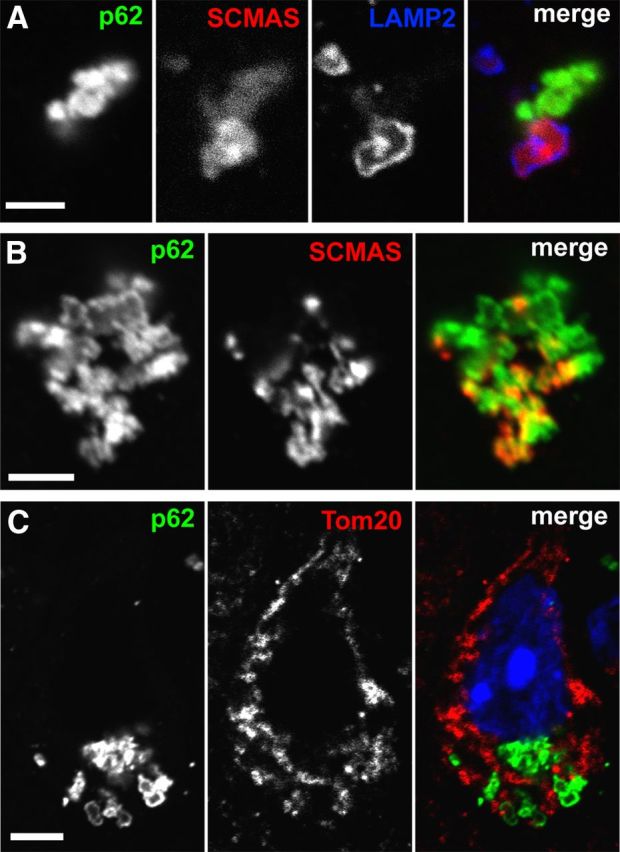
SCMAS storage colocalizes with p62 after being released from lysosomes. A, Single plane image showing a discontinuous large LAMP2-positive lysosomal membrane containing SCMAS and adjacent extra-lysosomal SCMAS that is colocalized with p62 in a Cln2−/− cortical neuron in vivo at 13 weeks. Scale bar, 1.5 μm. B, High-magnification single plane image of significant colocalization between p62 and SCMAS in a large aggregate in a Cln2−/− cortical neuron in vivo at 13 weeks. Scale bar, 1.5 μm. C, p62 does not colocalize with the mitochondrial marker Tom20 in a single-plane image in a Cln2−/− cortical neuron in vivo at 13 weeks. DAPI (blue) counterstained the nucleus. Scale bar, 5 μm.
SCMAS is a transmembrane proton transporter functioning as part of the mitochondrial ATP synthase complex and normally localized within the inner mitochondrial membrane. In CLN2 and several other lysosomal storage diseases, SCMAS is found to accumulate within lysosomes (Elleder et al., 1997). The primary turnover of mitochondria occurs through macroautophagy, also known as mitophagy (Youle and Narendra, 2011). In addition to autophagic delivery of mitochondria to lysosomes, recent evidence suggests that, during oxidative stress, mitochondria-derived vesicles (MDVs) can shuttle mitochondrial proteins and cargo to lysosomes independent of mitophagy (Soubannier et al., 2012). In CLN2 disease, the delivery of mitochondria to lysosomes results in SCMAS storage as a result of lysosomal TPP1 deficiency. Notably, no other mitochondrial proteins have been identified to accumulate in CLN2 storage material (Palmer et al., 1992). Given our findings of p62 colocalization with SCMAS, we evaluated whether extra-lysosomal p62- and SCMAS-positive cytosolic aggregates could represent mitochondria or mitochondrial components that have not yet reached lysosomes, thus indicative of mitophagy or MDV trafficking impairment. Despite a lack of ultrastructural evidence for p62 localizing with mitochondria by immuno-EM, and the fact that p62-positive aggregates are non-membrane bound (in contrast to MDVs), we evaluated p62 in relation to additional mitochondrial markers. By confocal microscopy, we found no colocalization between p62 and the outer mitochondrial protein Tom20, a common protein component of MDVs (Soubannier et al., 2012) (Fig. 6C). Furthermore, we found no colocalization of p62 and an additional component of the mitochondrial ATP synthase complex, the subunit B of mitochondrial ATP synthase (data not shown). Together, our results strongly suggest that the accumulation of p62 and colocalization with extra-lysosomal SCMAS occurs after SCMAS has been released from lysosomes following LMP.
Induction of LMP in primary neuronal cultures stimulates p62 aggregate formation
To test the hypothesis that LMP stimulates p62-positive aggregate formation, we treated primary neuronal cultures with the lysosomotropic agent LeuLeuOMe, which has been well characterized to induce LMP (Thiele and Lipsky, 1990; Ivanova et al., 2008). LeuLeuOMe is endocytosed and targeted to lysosomes in which it accumulates and is cleaved, producing polymerized products that permeabilize lysosomal membranes (Thiele and Lipsky, 1990; Ivanova et al., 2008). Although p62-positive aggregates are prevalent throughout the CNS of Cln2−/− mice, Cln2−/− primary neuronal cultures exhibit little p62 accumulation. Importantly, treatment of WT and Cln2−/− primary neuronal cultures with LeuLeuOMe to induce LMP resulted in a significant increase of p62-positve aggregates (Fig. 7A,D). Additionally by Western blot, LeuLeuOMe treatment of WT and Cln2−/− cultures led to increases in soluble and insoluble levels of p62, as well as increases in insoluble NBR1 and ubiquitinated proteins (Fig. 7B,C). These changes in protein levels with LMP induction in WT and Cln2−/− cultures correlate with analyses from Cln2−/− brain tissue (Fig. 1); specifically, p62 increases in both soluble and insoluble protein fractions, and NBR1 and ubiquitin show significant increases in insoluble protein fractions. Most notably, p62 was found localized to the periphery of LAMP2-positive lysosomes in both WT and Cln2−/−-treated neurons, often completely surrounding the organelle (Fig. 7E), and thus mirroring our in vivo findings.
Figure 7.

Induction of LMP in primary neuronal cultures with LeuLeuOMe stimulates aggregate formation. A, Single-plane images labeled for p62 in WT (top row) and Cln2−/− (bottom row) primary neuronal cultures showing increased appearance of p62 with LeuLeuOMe treatment (right column) compared with vehicle-treated cultures (left column). Scale bar, 25 μm. B, C, Western blot analyses of soluble and insoluble protein fractions for p62, ubiquitin, and NBR1 in WT (B) and Cln2−/− (C) primary neuronal cultures treated with vehicle (−) or 2 mm LeuLeuOMe (+). LMP induction results in p62 increases in soluble and insoluble protein fractions and increased insoluble NBR1 and polyubiquitinated proteins. Actin was used as a loading control. D, Quantification of percentage of MAP2-positive neurons with p62-aggregates after 24 h vehicle- or LeuLeuOMe-treatment in WT and Cln2−/− cultures. LeuLeuOMe treatment results in significant increases in p62 aggregates in both WT and Cln2−/−. Statistical significance was determined comparing treated versus untreated cultures for each genotype. Quantification is represented as the mean ± SEM of three separate experiments (two-tailed, *p = 0.0025, **p = 0.0005). E, Neuron from Cln2−/− primary cultures treated with LeuLeuOMe exhibiting p62-positive accumulation in which p62 is found decorating the periphery of LAMP2-positive structures (arrowheads) in a single-plane confocal image. Boxed region in merged image is magnified below each channel. MAP2-positive labeling (data not shown) confirmed this as a neuron. N, Nuclear region. Scale bar, 5 μm.
Additional evaluation of p62 after LeuLeuOMe treatment of Cln2−/− cultures identified p62 surrounding LAMP2-positive vesicles containing SCMAS storage, whereas vehicle-treated cultures exhibited no p62 accumulation (Fig. 8A,B). Notably, LeuLeuOMe treatment also resulted in p62 localization to the periphery of extra-lysosomal cathepsin D (Fig. 8C). This suggests that, in addition to SCMAS storage, LMP stimulation results in a p62 response to other lysosomal lumenal components that may be released in the cytosol. We next evaluated p62 turnover or flux through the lysosomal and UPS systems in Cln2−/− cultures to determine whether increased levels of p62 after LeuLeuOMe treatment were specific for LMP induction or simply a blockage of p62 degradation (Fig. 8D). We found that soluble p62 levels increased with LeuLeuOMe treatment only, but not in the presence of lysosomal proteolysis inhibition (NH4Cl plus leupeptin treatment) or UPS inhibition (lactacystin treatment). Lysosomal and UPS inhibition leads to increased insoluble p62 but has no effect on the soluble p62 protein pool. Importantly, the accumulation of soluble p62 with LeuLeuOMe treatment indicates that this pool of p62 is specifically increasing because of LMP induction and not because of decreased p62 degradation. Furthermore, the concomitant treatment of cultures with LeuLeuOMe along with lysosomal proteolysis inhibition or UPS inhibition led to additional increases in insoluble p62 levels, indicating that LeuLeuOMe treatment on its own is not blocking p62 degradation, and notably that LMP is causing an increase in total p62 protein. Thus, we conclude that increased levels of soluble and a large portion of insoluble p62 after LeuLeuOMe treatment represents a specific response to LMP induction. Together and relative to CLN2 disease, we conclude that LMP results in the release of lysosomal contents including SCMAS storage material in the cytosol of neurons. This in turn stimulates a response by the macroautophagy adapter proteins p62 and NBR1 that function to sequester this material as cytosolic protein aggregates.
Figure 8.

LeuLeuOMe-stimulated LMP results in a specific response by p62 in primary neuronal cultures. A, Neuron of vehicle-treated Cln2−/− cultures exhibiting no p62-positive accumulation in a single optical plane by confocal microscopy. MAP2-positive labeling (data not shown) confirmed this as a neuron. N, Nuclear region. Scale bar, 5 μm. B, Neuron of LeuLeuOMe-treated Cln2−/− cultures exhibiting two p62-positive aggregates in a single optical plane by confocal microscopy. One large aggregate is adjacent to storage lysosomes (arrow) and another shows p62 decorating a LAMP2-positive lysosome containing SCMAS storage (arrowhead). Boxed region in merged image is magnified below each channel. MAP2-positive labeling (data not shown) confirmed this as a neuron. N, Nucleus. Scale bar, 5 μm. C, Neuron of LeuLeuOMe-treated Cln2−/− cultures exhibiting p62 localized around extra-lysosomal cathepsin D (CD). Image is a single confocal plane image taken from a z-series. Boxed region in merged image is magnified below each channel. MAP2-positive labeling (data not shown) confirmed this as a neuron. N, Nuclear region. Scale bar, 5 μm. D, Analysis of p62 flux in Cln2−/− primary neuronal cultures analyzed by Western blot demonstrating that LeuLeuOMe treatment increases total p62 protein levels independent of proteolytic blockage by lysosomal inhibition (NH4Cl plus leupeptin) or UPS inhibition (lactacystin). Actin was used as a loading control. Quantification of p62/Actin for soluble and insoluble protein fractions is represented as the mean ± SEM for two separate experiments. p62 flux quantification represents the ratio of p62/Actin mean values for lysosomal or UPS proteolysis inhibition (NH4Cl and leupeptin, or lactacystin, respectively) to p62/Actin mean values for vehicle-treated cultures in the presence or absence of LeuLeuOMe.
Discussion
Here we show for the first time that LMP can stimulate protein aggregate formation in neurons and that this is the driving pathogenic event for aggregate formation in CLN2 disease. Because p62 is preferentially degraded by macroautophagy, its accumulation is commonly used as an indirect marker of macroautophagy dysfunction (Bjørkøy et al., 2005; Lamark and Johansen, 2012; Lieberman et al., 2012). Notably, in several lysosomal disease studies, p62 accumulation and its potential role in disease pathogenesis has yet to be comprehensibly evaluated. Our findings show that LMP, independent of macroautophagy and UPS impairment, stimulates p62-positive cytosolic aggregate formation.
Evidence of modest inefficiency of macroautophagy (LC3-II increases) in the brain of Cln2−/− mice late in disease is consistent with a failure in autophagosome maturation. However, by immuno-EM, p62 does not accumulate within autophagosomes or other membrane-bound structures, suggesting that the accumulation of p62 is not attributable to inefficient autophagosome maturation. The fact that p62-positive aggregates are non-membrane-bound implicates failure at the initiation step of macroautophagy, specifically in the proper targeting of cargo within autophagosomes. Notably, it has been found that the composition of protein aggregates can influence their preferential targeting for degradation (Wong et al., 2008). Additionally, cargo recognition failure has been proposed in Huntington's disease, in which, despite evidence that macroautophagy flux is intact, autophagosomes fail to properly engulf mutant huntingtin protein and damaged mitochondria (Martinez-Vicente et al., 2010). Regardless of the reason for the persistent presence of protein aggregates in Cln2−/− brain, given the deficiency in TPP1 enzyme, proper targeting of these insoluble aggregates (which we have identified to contain SCMAS) to lysosomes would only serve to exacerbate lysosomal storage and to compound lysosomal dysfunction. Lack of evidence for changes in canonical macroautophagy signaling through mTOR and Beclin 1 indicate that there is no attempt to upregulate this system in response to aggregate accumulation. Therefore, it is probable that protein aggregates are isolated in neurons in Cln2−/−, similar to the formation of aggresomes identified in other neurodegenerative diseases and model systems in which proteolysis is compromised (Kopito, 2000).
LMP has been reported in the MPTP model of Parkinson's disease, in which loss of lysosomal membrane integrity is believed to result in a depletion of lysosomes (Dehay et al., 2010). Given that autophagosomes must fuse with lysosomes for degradation, Dehay et al. (2010) concluded that LMP directly leads to the aberrant accumulation of autophagosomes associated with this model. Notably, this study did not evaluate aggregate formation or a potential link to LMP. Previous lysosomal disease studies have argued that lysosomal storage and compromised lysosomal function directly inhibit autophagosome fusion and processing, resulting in autophagosome accumulation (Lieberman et al., 2012). We propose that the modestly increased levels of autophagosomes in the brain of Cln2−/− mice are potentially attributable to both storage accumulation and LMP hindering fusion events. Importantly, however, we conclude that p62-positive cytosolic aggregate formation is a direct result of LMP, whereas the persistent presence of these aggregates over time likely represents impaired macroautophagy initiation.
Our findings show that LMP is occurring in the CNS of Cln2−/− mice in specific neuronal populations defined by the presence of p62. In addition to a distinct neocortical distribution, p62 aggregates are prevalent within Purkinje cells, which have also been reported to be susceptible to degeneration (Sleat et al., 2004). Although the relationship between p62 and LMP and neuronal viability requires additional studies, we speculate that the sequestration of released lysosomal contents by p62 may represent a beneficial physiological response to this potentially deleterious event. Several studies have proposed a neuroprotective role for aggregates in other neurodegenerative disease states (Saudou et al., 1998; Cummings et al., 1999; Arrasate et al., 2004). For example, in Huntington's disease, interference with p62 function results in reduced cell viability (Bjørkøy et al., 2005). This study showed that p62 forms a shell around huntingtin protein aggregates ameliorating cytotoxicity. This p62-shell formation is strikingly similar to our results in Cln2−/− in which p62 decorates the periphery of lysosomal storage bodies as well as extra-lysosomal SCMAS. Furthermore, our in vitro studies confirm that p62 plays a role in responding to LMP by surrounding storage-laden lysosomes and cytosolic cathepsin D in a shell-like formation. Our finding that p62 sequesters released lysosomal content following LMP is consistent with the known function of p62 (Lamark and Johansen, 2012).
The response to LMP by p62 and other adapter molecules (of which we have identified NBR1 and ubiquitin) predictably involves a sequential series of steps. We speculate that the soluble versus insoluble protein fractions of p62 may provide insight regarding this process. Specifically, the initial response to lysosomal membrane destabilization might correspond with the soluble p62 protein pool in which p62 is found localized to the periphery of lysosomes. As the lysosomal membrane further deteriorates, p62 sequesters released lumenal components, including storage. Because of its ability to self-oligomerize via its PB1 domain (phox and bem 1) (Noda et al., 2003), p62 promotes the stability of the sequestered material as insoluble aggregates present at end-stage disease in the Cln2−/− brain. Interestingly, in the Cln3Δex7/8 mouse model of CLN3 disease, we have also identified the accumulation and localization of p62 to the periphery of storage-laden lysosomes in cortical neurons (data not shown). This pattern is strikingly similar to our data in Cln2−/− brains (Fig. 2B,D). To date, the presence of p62 aggregates has been observed in several mouse models of the NCLs, including CLN1, CLN2, CLN3, CLN10 (M.C.M. and S.U.W., unpublished observation), and CLN6 (Thelen et al., 2012). The accumulation of p62 in these genetically distinct diseases suggests a remarkable pathophysiological convergence that warrants closer evaluation. Given our current findings, we propose that defining the accumulation of p62 at the subcellular level and in relation to known pathology is critical for determining the significance of this protein in disease.
Few studies to date have explored LMP in the context of lysosomal or neurodegenerative disease pathogenesis. Susceptibility to LMP has been proposed in neurons of the Niemann–Pick type C1 mouse model (Amritraj et al., 2009). Other instances in which LMP has been implicated include studies in lymphocytes from mucopolysaccharidosis type I mice and patient fibroblasts from Niemann–Pick type A and B disease (Kirkegaard et al., 2010; Pereira et al., 2010). Although the mechanisms involved in LMP remain unclear, several inducers of this event have been identified, including the lysosomal accumulation of pathogenic proteins (Johansson et al., 2010). For example, the accumulation of β-amyloid in lysosomes has been found to stimulate LMP possibly through its ability to oligomerize and promote detergent-like properties (Johansson et al., 2010). Because SCMAS is a proton pore in its native state, it is feasible that LMP could occur by SCMAS insertion into lysosomal membranes. However, our findings suggest that the lysosomal accumulation of SCMAS on its own is not sufficient to stimulate LMP. As noted previously, neurons in culture exhibit significant SCMAS storage yet present no evidence for LMP. Additionally, layer V pyramidal neurons in Cln2−/− mice contain significant SCMAS storage localized to LAMP2-positive lysosomes but lack evidence of LMP. Given that LMP has been reported in other genetically heterogeneous lysosomal diseases, the trigger for this event is likely stimulated through a yet to be identified common pathogenic mechanism that is downstream of the primary disease defect. Importantly, targeting LMP may be beneficial as a therapeutic strategy in lysosomal diseases. In Niemann–Pick type A and B disease, Kirkegaard et al. (2010) identified a critical role for Hsp70 in stabilizing lysosomal membranes of patient fibroblasts. This finding suggests that inducing Hsp70 expression can prevent LMP and promote cell viability, providing a clear rationale for testing drugs in Cln2−/− mice that accomplish this aim (Kieran et al., 2004; Kalmar et al., 2008).
Although this is the first identification of cytosolic protein aggregates in CLN2 disease, histochemical and ultrastructural studies in human CLN2 tissue have demonstrated that biochemically and morphologically distinct forms of storage—believed to share the same pathogenesis—occur concomitantly in disease (Elleder, 1978; Elleder and Tyynela, 1998). Furthermore, a biochemical study analyzing storage isolated from Cln2−/− mice identified material associated with both a lysosomal subcellular fraction and an insoluble protein fraction (Xu et al., 2010). Notably, p62 was found to be the predominant component associated with the insoluble fractions. Our studies provide mechanistic insight for these previous observations and a more complete picture of CLN2 disease pathogenesis. Given our current findings, the accumulation of p62 and its colocalization with storage offers a novel marker to evaluate drug treatment studies aimed at preventing LMP. In addition to CLN2 disease, future efforts to define whether LMP occurs in other disorders characterized by p62 aggregate formation will be needed. Such studies will not only expand our understanding of the neurobiology of these disorders but will also provide clues for elucidating broader neurodegenerative processes and help identify whether LMP is a common therapeutic target in lysosomal disease.
Footnotes
This work was supported by National Institute of Child Health and Human Development Grants 5R01 HD045561 and 5P30 HD071593 (S.U.W.) and NCL-Stiftung National Contest for Life (M.C.M.). J.S. is supported by National Institute of Neurological Disorders and Stroke Award 1F05 NS074790. We thank P. Lobel and D. E. Sleat for the Cln2−/−mice, I. Berezniuk and L. D. Fricker for help with the UPS assay, A. Molero for help with the qPCR, N. F. Ali for technical support, and A. M. Cuervo for her helpful comments.
The authors declare no competing financial interests.
References
- Amritraj A, Peake K, Kodam A, Salio C, Merighi A, Vance JE, Kar S. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am J Pathol. 2009;175:2540–2556. doi: 10.2353/ajpath.2009.081096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Berezniuk I, Sironi J, Callaway MB, Castro LM, Hirata IY, Ferro ES, Fricker LD. CCP1/Nna1 functions in protein turnover in mouse brain: implications for cell death in Purkinje cell degeneration mice. FASEB J. 2010;24:1813–1823. doi: 10.1096/fj.09-147942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–892. doi: 10.1016/S0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Bov é J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrenis K, Joseph A, Rattazzi MC. Neuronal lysosomal enzyme replacement using fragment C of tetanus toxin. Proc Natl Acad Sci U S A. 1992;89:2297–2301. doi: 10.1073/pnas.89.6.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrenis K, Chang HY, Pina-Benabou MH, Woodroffe A, Lee SC, Rozental R, Spray DC, Scemes E. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J Neurosci Res. 2005;82:306–315. doi: 10.1002/jnr.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190:719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleder M. A histochemical and ultrastructural study of stored material in neuronal ceroid lipofuscinosis. Virchows Arch B Cell Pathol. 1978;28:167–178. doi: 10.1007/BF02889067. [DOI] [PubMed] [Google Scholar]
- Elleder M, Tyynela J. Incidence of neuronal perikaryal spheroids in neuronal ceroid lipofuscinoses (Batten disease) Clin Neuropathol. 1998;17:184–189. [PubMed] [Google Scholar]
- Elleder M, Sokolov á J, Hrebícek M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol. 1997;93:379–390. doi: 10.1007/s004010050629. [DOI] [PubMed] [Google Scholar]
- Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet. 2012;21:4876–4887. doi: 10.1093/hmg/dds324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaki J, Tanida I, Kanehagi N, Kominami E. A lysosomal proteinase, the late infantile neuronal ceroid lipofuscinosis gene (CLN2) product, is essential for degradation of a hydrophobic protein, the subunit c of ATP synthase. J Neurochem. 1999;72:2573–2582. doi: 10.1046/j.1471-4159.1999.0722573.x. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Ivanova S, Repnik U, Bojic L, Petelin A, Turk V, Turk B. Lysosomes in apoptosis. Methods Enzymol. 2008;442:183–199. doi: 10.1016/S0076-6879(08)01409-2. [DOI] [PubMed] [Google Scholar]
- Johansson AC, Appelqvist H, Nilsson C, Kågedal K, Roberg K, Ollinger K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010;15:527–540. doi: 10.1007/s10495-009-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem. 2008;107:339–350. doi: 10.1111/j.1471-4159.2008.05595.x. [DOI] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jäättel ä M. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Øvervatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsenyi MC, Dobrenis K, Stephney G, Pickel J, Vanier MT, Slaugenhaupt SA, Walkley SU. Neuropathology of the Mcoln1(−/−) knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol. 2009;68:125–135. doi: 10.1097/NEN.0b013e3181942cf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- Mole SE, Williams RE, Goebel HH. The neuronal ceroid lipofuscinoses (Batten disease) Oxford: Oxford UP; 2011. [Google Scholar]
- Noda Y, Kohjima M, Izaki T, Ota K, Yoshinaga S, Inagaki F, Ito T, Sumimoto H. Molecular recognition in dimerization between PB1 domains. J Biol Chem. 2003;278:43516–43524. doi: 10.1074/jbc.M306330200. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease) Am J Med Genet. 1992;42:561–567. doi: 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pereira VG, Gazarini ML, Rodrigues LC, da Silva FH, Han SW, Martins AM, Tersariol IL, D'Almeida V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: rupture of calcium and proton homeostasis. J Cell Physiol. 2010;223:335–342. doi: 10.1002/jcp.22039. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med [Suppl] 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer's disease. Prog Neurobiol. 2012;96:87–95. doi: 10.1016/j.pneurobio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/S0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Thelen M, Daμμe M, Schweizer M, Hagel C, Wong AM, Cooper JD, Braulke T, Galliciotti G. Disruption of the autophagy-lysosome pathway is involved in neuropathology of the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS One. 2012;7:e35493. doi: 10.1371/journal.pone.0035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, Lipsky PE. Mechanism of l-leucyl-l-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A. 1990;87:83–87. doi: 10.1073/pnas.87.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL, Dawson TM, Cuervo AM, Lim KL. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–2582. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62's role in neurodegenerative disease. J Biomed Biotechnol. 2006;2006:62079. doi: 10.1155/JBB/2006/62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Sleat DE, Jadot M, Lobel P. Glial fibrillary acidic protein is elevated in the lysosomal storage disease classical late-infantile neuronal ceroid lipofuscinosis, but is not a component of the storage material. Biochem J. 2010;428:355–362. doi: 10.1042/BJ20100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Leunissen J, Shi G, Gutekunst C, Hersch S. A novel procedure for pre-embedding double immunogold-silver labeling at the ultrastructural level. J Histochem Cytochem. 2001;49:279–284. doi: 10.1177/002215540104900301. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]