

Abstract
Estrogens can signal through either estrogen receptor α (ERα) or β (ERβ) to ameliorate experimental autoimmune encephalomyelitis (EAE), the most widely used mouse model of multiple sclerosis (MS). Cellular targets of estrogen-mediated neuroprotection are still being elucidated. Previously, we demonstrated that ERα on astrocytes, but not neurons, was critical for ERα ligand-mediated neuroprotection in EAE, including decreased T-cell and macrophage inflammation and decreased axonal loss. Here, we determined whether ERβ on astrocytes or neurons could mediate neuroprotection in EAE, by selectively removing ERβ from either of these cell types using Cre-loxP gene deletion. Our results demonstrated that, even though ERβ ligand treatment was neuroprotective in EAE, this neuroprotection was not mediated through ERβ on either astrocytes or neurons and did not involve a reduction in levels of CNS inflammation. Given the differential neuroprotective and anti-inflammatory effects mediated via ERα versus ERβ on astrocytes, we looked for molecules within astrocytes that were affected by signaling through ERα, but not ERβ. We found that ERα ligand treatment, but not ERβ ligand treatment, decreased expression of the chemokines CCL2 and CCL7 by astrocytes in EAE. Together, our data show that neuroprotection in EAE mediated via ERβ signaling does not require ERβ on either astrocytes or neurons, whereas neuroprotection in EAE mediated via ERα signaling requires ERα on astrocytes and reduces astrocyte expression of proinflammatory chemokines. These findings reveal important cellular differences in the neuroprotective mechanisms of estrogen signaling through ERα and ERβ in EAE.
Introduction
Estrogens are neuroprotective in numerous animal disease models of the CNS, including Alzheimer's disease, Parkinson's disease, and multiple sclerosis (MS). This neuroprotection ranges from prevention of neuronal loss to proper maintenance of the mitochondria (Lee and McEwen, 2001; Wise, 2002; Brinton, 2008; Spence and Voskuhl, 2012). Whereas therapeutic effects of estrogen treatment in experimental models of diseases have been shown, the cellular targets of estrogen treatment are not yet fully understood.
Experimental autoimmune encephalomyelitis (EAE) is the most widely used mouse model of MS (Gold, 2006; Croxford et al., 2011). Similar to MS, EAE consists of CNS inflammation, demyelination, and axonal loss (Trapp and Nave, 2008). Estrogen treatment exerts well-documented protective effects in EAE (Gold and Voskuhl, 2009; Spence and Voskuhl, 2012). Previous studies showed that treatment with either estrogen receptor α (ERα) ligand or estrogen receptor β (ERβ) ligand ameliorated EAE, as well as prevented demyelination and axonal loss (Morales, 2006; Tiwari-Woodruff et al., 2007; Crawford et al., 2010; Du et al., 2010; Spence et al., 2011). However, ERα ligand and ERβ ligand effects in EAE exhibit important differences. Previous data showed that ERα ligand treatment decreased the amount of immune cell infiltration into the CNS, whereas ERβ ligand treatment did not (Tiwari-Woodruff et al., 2007). However, ERβ ligand treatment was able to qualitatively affect CNS infiltration as well as promote remyelination (Crawford et al., 2009; Du et al., 2010). Together, these observations suggest that the differences between the two ER ligands are not only the result of different pharmacological effects on each receptor, but that each ligand may have different cellular targets.
Regarding cellular targets of estrogen treatments, previous work from our laboratories demonstrated that astrocytes, but not neurons, are the target of ERα ligand's protective effects during EAE (Spence et al., 2011). Here, we determined whether ERβ ligand also ameliorated EAE through direct actions on astrocytes or neurons. To do so, we used a genetic loss-of-function strategy. We selectively deleted ERβ from astrocytes or neurons using a well-characterized Cre-loxP system for astrocyte conditional gene knock-out (astrocyte-CKO) in mice to remove ERβ (astrocyte-CKO-ERβ [aCKO-ERβ]) (Herrmann et al., 2008; Spence et al., 2011) and for neuronal conditional gene knock-out (neuronal-CKO) in mice to remove ERβ (neuronal-CKO-ERβ) (Forss-Petter et al., 1990; Kwon et al., 2006). Our findings demonstrate that ERα expression on astrocytes, but not ERβ expression on astrocytes or neurons, is necessary for estrogen-mediated neuroprotection during EAE. Furthermore, treatment with ERα ligand, but not ERβ ligand, decreased astrocytic levels of chemokines that contribute to CNS inflammation.
Materials and Methods
Animals.
All mice were on a C57BL/6 background achieved by at least 10 generations of back crossing. Astrocyte-ERα-CKO were generated by crossing mice of mGFAP-Cre line 73.12 (Herrmann et al., 2008) with mice carrying an ERα gene in which exon 3 was flanked by loxP sites (ERαflox/flox) were the generous gift from Professor Pierre Chambon (Strasbourg) (Dupont et al., 2000). Astrocyte-ERβ-CKO were generated by crossing mice of mGFAP-Cre line 73.12 (Herrmann et al., 2008) with mice carrying an ERβ gene in which exon 3 was flanked by loxP sites (ERβflox/flox) were the generous gift from Professor Pierre Chambon (Strasbourg) (Dupont et al., 2000). Neuronal-ERβ-CKO were generated by crossing mice of rNSEII-Cre (Forss-Petter et al., 1990; Kwon et al., 2006) with mice carrying an ERβ gene in which exon 3 was flanked by loxP sites (ERβflox/flox) were the generous gift from Professor Pierre Chambon (Strasbourg) (Dupont et al., 2000). Animals were maintained under standard conditions in a 12 h dark/light cycle with access to food and water ad libitum. All procedures were done in accordance to the guidelines of the National Institutes of Health and the Chancellor's Animal Research Committee of the University of California, Los Angeles Office for the Protection of Research Subjects.
Adoptive EAE and hormone manipulations.
C57BL/6 donor animals were immunized subcutaneously with myelin oligodendrocyte glycoprotein, amino acids 35–55 (200 μg/animal, American Peptides) emulsified in Complete Freund's Adjuvant, supplemented with Mycobacterium Tuberculosis H37Ra (200 μg/animal, Difco Laboratories), over four sites drained by inguinal and auxiliary lymph nodes in a total volume of 0.1 ml/mouse. Immunized mice had lymph node cells cultured in 24-well plates at a concentration of 3 × 106 cells/ml of complete RPMI medium. Cells were stimulated with 25 μg/ml myelin oligodendrocyte glycoprotein, peptide 35–55, and 20 ng/ml recombinant mouse IL-12 (BD Biosciences and BioLegend) for 72 h. On the third day of culture, lymph node cells were washed with 1× PBS, and each recipient mouse received 3 × 107 cells in 0.3 ml ice-cold PBS by intraperitoneal injection. Recipient female C57 Bl/6 WT and astrocyte-CKO mice had been gonadectomized at 4 weeks of age and had EAE induced by adoptive transfer at 8 weeks of age. Recipient mice were either treated every other day with the ERα ligand, 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol (Tocris Bioscience) at the dose of 10 mg/kg/d or ERβ ligand (DPN) (Tocris Bioscience) at the dose of 8 mg/kg/d or vehicle diluted with 10% molecular-grade ethanol (EM Sciences) and 90% Miglylol 812N liquid oil (Sasol North America) beginning 7 d before adoptive transfer. These doses of 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol and DPN have been previously established (Tiwari-Woodruff et al., 2007). Animals were monitored daily for EAE signs based on a standard EAE 0–5 scale scoring system: 0, healthy; 1, complete loss of tail tonicity; 2, loss of righting reflex; 3, partial paralysis; 4, complete paralysis of one or both hind limbs; and 5, moribund.
Histological preparation.
Female mice were deeply anesthetized in isoflurane and perfused transcardially with ice-cold 1× PBS for 20–30 min, followed by 10% formalin for 10–15 min. Spinal cords were dissected and submerged in 10% formalin overnight at 4°C, followed by 30% sucrose for 24 h. Spinal cords were cut in thirds and embedded in optimal cutting temperature compound (Tissue Tek) and frozen at −80°C; 40-μm-thick free-floating spinal cord cross-sections were obtained with a microtome cryostat (model HM505E) at −20°C. Tissues were collected serially and stored in 0.1 m PBS with 1% sodium azide in 4°C until immunohistochemistry.
Immunohistochemistry.
Before histological staining, 40-μm-thick free-floating sections were thoroughly washed with 0.1 m PBS to remove residual sodium azide. For tissues to be treated with diaminobenzidine (DAB), sections were permeabilized with 0.5% Triton X-100 in 0.1 m TBS and 10% normal goat serum for 60 min at room temperature. The following primary antibodies were used: anti-CD3 at 1:2000 (BD Biosciences PharMingen), anti-neurofilament-NF200 at 1:750 dilutions (Sigma), anti-Iba-1 at 1:10,000 (Wako Chemicals), anti-GFAP at 1:40,000 (Dako), anti-NeuN at 1:250 (Sigma), anti-ERβ at 1:5000 (Millipore), aquaporin 4 at 1:100 (Abcam), anti-MCP-1 (CCL2) at 1:200 (Torrey Pines Biolabs), CCL7 (Sigma) at 1:50, and anti-MBP at 1:750 (Sigma). Tissues were then washed three times for 10 min in 0.1 m TBS. Tissues were labeled with secondary antibodies conjugated to Cy5 or Cy3 (Vector Laboratories and Millipore Bioscience Research Reagents) for 1 h for NF-200, NeuN, MBP, CCL2, CCL5, CCL7, CCL8, aquaporin-4 (AQP4), and GFAP. Tissues were labeled with biotin secondary antibodies for CD3 and Iba-1, followed by ABC/DAB treatment (Vector Laboratories). Fluorescent sections were mounted on slides, allowed to semidry, and coverslipped in fluoromount G (Fisher Scientific). DAB sections were dried overnight and then dehydrated in 70%, 95%, and 100% ethanol, followed by 5 min of Citrasolve and coverslipped with Permount (Fisher Scientific). IgG-control experiments were performed for all primary antibodies, and only nonimmunoreactive tissues under these conditions were analyzed. Immunohistochemistry for ERβ was followed directly as previously described (Giraud et al., 2010).
Quantification.
To quantify immunohistochemical staining results, three dorsal column spinal cord cross-sections at the T1-T5 level from each mouse were captured under microscope at 10× or 40× magnification using the DP70 Image software and a DP70 camera (both from Olympus). All images in each experimental set were captured under the same light intensity and exposure limits. Image analysis was performed using ImageJ Software version 1.30 downloaded from the NIH website (http://rsb.info.nih.gov/ij). Three sections from each animal were then quantified to calculate the mean per animal. Immunohistochemical experiments were combined from three separate clinical trials. To control for variance, each immunohistochemical experiment was run as one large experiment with n = 9–12 per group. Each immunohistochemical experiment was repeated at least twice to confirm data. Myelinated axons were calculated by counting the number of NF200+ axons in a 40× image over the area of the captured tissue section that were fully enclosed by MBP. Axonal densities were calculated by counting the number of NF200+ axons in a 40× image over the area of the captured tissue section. Inflammatory infiltrates were quantified by counting the number of DAB-positive cells in the dorsal column of the thoracic spinal cord at 40× under a light microscope. GFAP was calculated as percentage intensity or area, respectively, from the dorsal column. AQP4, CCL7, and CCL2 were measured as coexpression with GFAP in ImageJ, then divided over the total amount of GFAP in each section and presented as a percentage.
Microscopy.
Stained sections were examined and photographed using a confocal microscope (Leica TCS-SP) or a fluorescence microscope (BX51WI; Olympus) equipped with Plan Fluor objectives connected to a camera (DP70, Olympus). Digital images were collected and analyzed using Leica confocal and DP70 camera software. Images were assembled using Adobe Photoshop (Adobe Systems) and Microsoft PowerPoint. DAB sections were examined at the light level at 40× (Nikon Alphaphot-2 YS2).
Statistical analysis.
Differences in EAE clinical scores were determined by repeated-measures one-way ANOVA. Immunohistochemical data were analyzed by one-way ANOVA. For these analyses, one-way ANOVA, Bonferroni post hoc analysis was performed on F-stat values, and significance was determined at the 95% confidence interval (Prism).
Results
ERβ is specifically deleted from astrocytes in astrocyte-CKO-ERβ mice and in neurons in neuronal CKO-ERβ (nCKO-ERβ) mice
To target CKO-ERα and CKO-ERβ to astrocytes, we used a mouse GFAP (mGFAP)-Cre line previously shown to target Cre activity selectively in astrocytes in the CNS (Herrmann et al., 2008). This mGFAP-Cre line is able to target 98% of all astrocytes with no targeting of other cells in the CNS, such as oligodendrocytes, microglia, and neurons. We then crossed this mouse line with ERα or ERβ-loxP mice, in which exon 3 of ERα or ERβ gene is floxed (Dupont et al., 2000). To target CKO-ERβ to neurons, we used a rat neuronal specific enolase II-Cre line previously shown to target Cre activity selectively in neurons in the CNS (Forss-Petter et al., 1990; Kwon et al., 2006). We then crossed this mouse line with ERβ-loxP mice, in which exon 3 of ERβ gene is floxed (Dupont et al., 2000). To determine the efficacy of ERβ CKO from astrocytes and neurons, we performed immunohistochemistry in WT, astrocyte-CKO-ERα (aCKO-ERα), aCKO-ERβ, and nCKO-ERβ mouse lines with EAE. Our results demonstrated that ERβ was absent in astrocytes in the aCKO-ERβ mouse line, whereas it was present in astrocytes of both the WT and aCKO-ERα mouse line (Fig. 1A). Our results also demonstrated that ERβ was absent in neurons in the nCKO-ERβ mouse line, whereas it was present in astrocytes of both the WT and aCKO-ERβ mouse line the (Fig. 1B). Previous work confirmed that ERα was absent in astrocytes from the aCKO-ERα mouse line (Spence et al., 2011).
Figure 1.

Verification of gene deletion specificity in aCKO-ERα, aCKO-ERβ, and nCKO-ERβ mouse models and EAE disease severity scores showing protective effects of ERα in astrocytes, but not ERβ in astrocytes or neurons. A, Immunohistochemistry shows ERβ colocalized with GFAP in WT and aCKO-ERα, but not aCKO-ERβ mice with EAE. Scale bar, 12 μm. B, Immunohistochemistry shows ERβ colocalized with NeuN in WT and aCKO-ERβ, but not nCKO-ERβ mice with EAE. Scale bar, 11 μm. C, WT, but not aCKO-ERα mice, treated with ERα ligand had significantly better clinical scores compared with WT or aCKO-ERα mice with EAE treated with vehicle. *p < 0.05 versus WT + EAE + ERα ligand (repeated-measures ANOVA with post hoc Bonferroni pairwise analysis). D, WT and aCKO-ERβ mice treated with ERβ ligand had significantly better clinical scores compared with WT or aCKO-ERβ mice with EAE treated with vehicle. *p < 0.05 versus WT + EAE + ERα ligand and aCKO + EAE + ERα ligand (repeated-measures ANOVA with post hoc Bonferroni pairwise analysis). E, WT and nCKO-ERβ mice treated with ERβ ligand had significantly better clinical scores compared with WT or nCKO-ERβ mice with EAE treated with vehicle. *p < 0.05 versus WT + EAE + ERα ligand and nCKO + EAE + ERα ligand (repeated-measures ANOVA with post hoc Bonferroni pairwise analysis). n = 10 per group.
ERβ ligand does not act directly on astrocytes or neurons for clinical disease protection
To determine that astrocytes or neurons were the target of ERβ ligand-mediated protection during EAE, we treated our aCKO-ERβ mice and nCKO-ERβ mice with ERβ ligand during the effector phase of adoptive EAE (Kim et al., 1999). WT mice with EAE treated with vehicle exhibited a level of clinical disease indistinguishable from aCKO-ERβ mice or nCKO-ERβ mice with EAE treated with vehicle, demonstrating that the removal of astrocyte or neuron ERβ did not affect the clinical course of disease (Fig. 1D,E). WT mice with EAE treated with ERβ ligand showed significantly less clinical disease than WT mice with EAE treated with vehicle, confirming that ERβ ligand treatment was able to ameliorate clinical disease (Tiwari-Woodruff et al., 2007; Du et al., 2010). Notably, ERα ligand treatment was not able to ameliorate clinical disease in the aCKO-ERα group, confirming that astrocytes are the target of ERα ligand for clinical disease protection (Fig. 1C) (Spence et al., 2011). In contrast, ERβ ligand treatment was still able to ameliorate clinical disease in the aCKO-ERβ and nCKO-ERβ group, demonstrating that astrocytes and neurons are not the target of ERβ ligand-mediated clinical disease protection (Fig. 1D,E).
ERβ ligand, unlike ERα ligand, does not act on astrocytes to prevent T-cell and macrophage inflammation in the CNS
Given the differential effect of ERα versus ERβ ligand treatment on astrocytes, we used our two aCKO models to examine differences in the downstream effects that each ligand has on astrocytes in EAE. CNS inflammation, comprised principally of T cells and monocytes, are a hallmark of EAE (Tiwari-Woodruff et al., 2007; Voskuhl et al., 2009). Thus, we used immunohistochemistry to quantify the levels of T cells and monocytes in ER ligand-treated EAE aCKO-ERα and aCKO-ERβ mouse lines. Anti-CD3 antibody was used to detect T cells, whereas anti-Iba-1 antibody was used to detect Iba-1 ramified microglia and Iba-1 globoid macrophages as previously described (Voskuhl et al., 2009; Spence et al., 2011). T cells were increased in all mice with EAE regardless of treatment or genotype compared with WT mice without EAE (Figs. 2A, 3A,C). WT mice with EAE treated with ERα ligand had significantly less T-cell inflammation compared with WT mice treated with vehicle, confirming the anti-inflammatory effects of ERα ligand on T-cell inflammation. Furthermore, the effect of ERα ligand on reducing T-cell inflammation was lost in the aCKO-ERα mice treated with ERα ligand, thereby demonstrating that ERα ligand acts upon astrocytes in vivo to decrease T-cell inflammation. In contrast, treatment with ERβ ligand was not able to decrease T cells in any groups with EAE (Figs. 2A, 3C). Furthermore, WT mice with EAE treated with ERα ligand had significantly less Iba-1 globoid macrophage inflammation compared with WT mice treated with vehicle, again confirming the anti-inflammatory effects of ERα ligand on macrophage inflammation. Furthermore, the effect of ERα ligand on reducing macrophages was lost in the aCKO-ERα mice treated with ERα ligand, thereby demonstrating that ERα ligand acts upon astrocytes in vivo to decrease levels of macrophages in the CNS in EAE (Spence et al., 2011). In contrast, treatment with ERβ ligand was unable to decrease macrophages in any groups with EAE (Figs. 2C, 3B,D). Interestingly, when Iba-1 ramified microglia were examined, there were no differences between groups (Fig. 2B, 3B,D). Given that EAE induced an increase in cells with the phenotype of phagocytic macrophages without any effect on cells with the phenotype of microglia suggests that the increase of CNS macrophages in EAE is most likely the result of peripheral infiltrating monocytes, as others have suggested (Ajami et al., 2011).
Figure 2.

Quantification of how ERβ, unlike ERα, in astrocytes does not mediate reduction of CD3 T cells and Iba-1 globoid macrophages in EAE spinal cord. A, CD3 T cells were reduced in WT, but not aCKO-ERα, mice with EAE treated with ERα ligand. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). Treatment with ERβ ligand was unable to reduce CD3 T cells in WT or aCKO-ERβ with EAE. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis). B, Iba-1 ramified microglia exhibited no significant difference in number across all experimental groups. C, Iba-1 globoid macrophages were significantly reduced in WT, but not aCKO-ERα, mice treated with EAE treated with ERα ligand. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). Treatment with ERβ ligand was unable to reduce CD3 T cells in WT or aCKO-ERβ mice with EAE. Scale bar, 12 μm. n = 6 per group. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis).
Figure 3.

Representative images of how ERβ, unlike ERα, in astrocytes does not mediate reduction of CD3 T cells and Iba-1 globoid macrophages in EAE spinal cord. A, B, CD3 T cells and Iba-1 globoid macrophages are increased in WT and aCKO-ERα mice with EAE compared with WT mice without EAE. Treatment with ERα ligand prevents this increase in WT, but not aCKO-ERα, mice with EAE. C, D, CD3 T cells and Iba-1 globoid macrophages are increased in WT and aCKO-ERβ mice with EAE compared with WT mice without EAE. Treatment with ERβ ligand did not prevent this increase in either WT or aCKO-ERβ mice. Scale bars: A, C, 41 μm; D, 27 μm.
ERβ ligand, unlike ERα ligand, does not act on astrocytes to prevent demyelination, astrogliosis, and axonal loss in the CNS
We next examined classical neuropathology of EAE by quantifying differences in myelin, gliosis, and axons in the spinal cord (Morales, 2006; Tiwari-Woodruff et al., 2007; Voskuhl et al., 2009; Du et al., 2010; Spence et al., 2011). To quantify myelin, we counted the number of axons using anti-neurofilament 200 (NF200) that were fully encompassed by a ring of MBP. Both ERα ligand and ERβ ligand treatment of WT mice with EAE reduced demyelination compared with WT mice with EAE treated with vehicle, confirming the protective effect of both of these ligand on myelin loss (Tiwari-Woodruff et al., 2007; Du et al., 2010) (Figs. 4, 5A,D). Interestingly, ERβ ligand treatment was able to prevent demyelination in aCKO-ERβ mice, suggesting that ERβ ligand acts upon another cell other than astrocytes to protect against myelin loss. In contrast, ERα ligand treatment was not able to prevent demyelination in aCKO-ERα mice, suggesting that ERα ligand acts upon astrocytes in vivo to prevent myelin loss (Figs. 4, 5A,D). Axonal loss is known to correlate with clinical disease scores (Wujek et al., 2002). Thus, we counted the number of axons using NF200. Both ERα ligand and ERβ ligand treatment of WT mice with EAE prevented axonal loss compared with WT mice with EAE treated with vehicle, confirming the protective effect of both of these ligand on axonal loss (Tiwari-Woodruff et al., 2007; Du et al., 2010; Spence et al., 2011) (Figs. 4B, 5B,E). Interestingly, ERβ ligand treatment was able to prevent axonal loss in aCKO-ERβ mice, suggesting that ERβ ligand acts upon another cell other than astrocytes to protect against axonal loss. In contrast, ERα ligand treatment was not able to prevent axonal loss in aCKO-ERα mice, suggesting that ERα ligand acts upon astrocytes in vivo to prevent axonal loss (Figs. 4B, 5B,E). To quantify reactive astrogliosis, GFAP expression was assessed. WT mice with EAE treated with ERα ligand demonstrated a significant decrease in reactive gliosis compared with WT mice with EAE treated with vehicle. However, ERα ligand treatment was not able to decrease reactive astrogliosis in aCKO-ERα mice, suggesting that ERα ligand acts via ERα on astrocytes in vivo to decrease reactive astrogliosis (Figs. 4C, 5C). In contrast, ERβ ligand treatment had no effect on astrogliosis in any of the groups with EAE (Figs. 4C, 5F), suggesting that ERβ ligand, unlike ERα ligand, is not able to decrease reactive astrogliosis in the CNS.
Figure 4.

Quantification of how ERβ, unlike ERα, in astrocytes does not protect against demyelination, axonal loss, and reactive astrogliosis. A, Myelinated NF200 axons fewer significantly reduced in WT mice with EAE, and treatment with ERα ligand prevented demyelination in WT, but not aCKO-ERα, mice. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). Treatment with ERβ ligand was able to reduce demyelination in WT and aCKO-ERβ mice. *p < 0.05 versus WT + No EAE, WT + EAE + ERβ ligand, aCKO + EAE + ERβ ligand (ANOVA with post hoc Bonferroni pairwise analysis). Scale bar, 15 μm. B, Numbers of NF200 axons were significantly reduced in WT mice with EAE, and treatment with ERα ligand prevented axonal loss in WT, but not aCKO-ERα, mice. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). Treatment with ERβ ligand was able to prevent axonal loss in WT and aCKO-ERβ mice. *p < 0.05 versus WT + No EAE, WT + EAE + ERβ ligand, aCKO + EAE + ERβ ligand (ANOVA with post hoc Bonferroni pairwise analysis). Scale bar, 40 μm. C, Reactive gliosis staining was significantly increased in WT mice with EAE, and treatment with ERα ligand prevented this increase in WT, but not aCKO-ERα, mice with EAE. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). Treatment with ERβ ligand was unable to prevent reactive gliosis in WT or aCKO-ERβ mice with EAE. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis). Scale bar, 122 μm. n = 6 per group.
Figure 5.
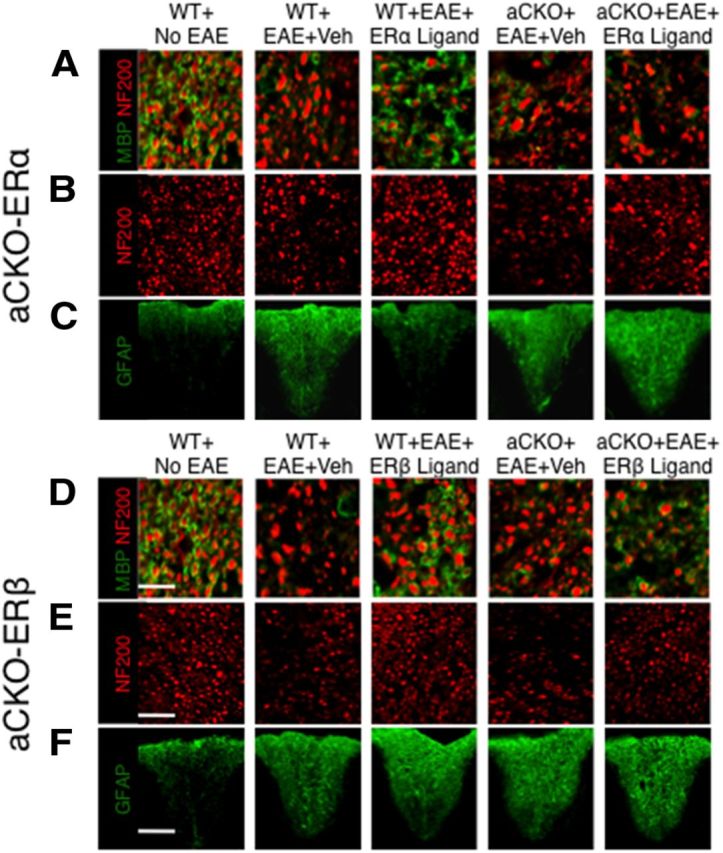
Representative images of how ERβ, unlike ERα, in astrocytes does not protect against demyelination, axonal loss, and gliosis in EAE spinal cord. A, D, Myelinated NF200 axons were decreased in WT mice with EAE. Treatment with ERα ligand reduced demyelination in WT, but not aCKO-ERα, mice with EAE. Treatment with ERβ ligand reduced demyelination in both WT and aCKO-ERβ mice with EAE. Scale bar, 22 μm. B, E, NF200 axons exhibited patchy reductions in numbers in WT mice with EAE. Treatment with ERα ligand reduced axonal loss in WT, but not aCKO-ERα, mice with EAE. Treatment with ERβ ligand reduced axonal loss in both WT and aCKO-ERβ mice with EAE. Scale bar, 50 μm. C, F, Reactive gliosis staining was increased in WT mice with EAE. Treatment with ERα ligand decreased reactive gliosis in WT, but not aCKO-ERα, mice with EAE. Treatment with ERβ ligand was unable to decrease reactive gliosis in WT and aCKO-ERβ mice with EAE. Scale bar, 130 μm.
ERα ligand treatment decreases CCL2 and CCL7, but not AQP4, expression in astrocytes in EAE
Because we had shown a distinction between ERα and ERβ ligand's ability to affect T-cell and macrophage inflammation in the CNS, we used this selectivity to ascertain which small inflammatory molecules within astrocytes that ERα ligand, but not ERβ ligand, might regulate. One molecule of interest was chemokine (C-C motif) ligand 2 (CCL2), previously known as monocyte chemotactic protein 1 (MCP-1). CCL2 is expressed in astrocytes and is known to correlate with a higher disease severity in various CNS injury models (Brambilla, 2005; Conductier et al., 2010; Carrillo-de Sauvage et al., 2012; Hamby et al., 2012). In MS tissue, CCL2 is expressed in astrocytes surrounding active and chronic lesions (McManus et al., 1998; Simpson et al., 1998; Van Der Voorn et al., 2010). In animal models, CCL2, and its receptor chemokine (C-C motif) receptor 2 (CCR2), KO mice are resistant to EAE (Izikson et al., 2000; Huang et al., 2001). This resistance could be the result of a decrease in macrophage recruitment into the CNS in these CCL2 KO mice (Huang et al., 2001). Here, we demonstrated that CCL2 is expressed within reactive astrocytes and immune cells in the spinal cord of EAE mice (Fig. 6A–C). Treatment with ERα ligand in vivo was able to reduce CCL2 expression in reactive astrocytes in WT mice compared with vehicle. Interestingly, ERα ligand treatment lost its ability to reduce CCL2 expression in reactive astrocytes in aCKO-ERα mice with EAE, thereby demonstrating that ERα ligand does indeed act on astrocytes in vivo to reduce CCL2 expression (Fig. 6D). In contrast, in vivo treatment with ERβ ligand was not able to reduce CCL2 expression in reactive astrocytes in any of the groups with EAE (Fig. 6E). Together, this demonstrates the selectivity of ligation of ERα, but not ERβ, on astrocytes in decreasing CCL2 expression during EAE.
Figure 6.

ERα is required on astrocytes to reduce CCL2 expression within reactive astrocytes. A, CCL2 (red), GFAP (green), and DAPI (blue) are expressed in EAE spinal cord in a WT mouse with EAE. Scale bar, 120 μm. B, CCL2 is coexpressed with GFAP. Scale bar, 27 μm. C, CCL2 is coexpressed with infiltrating immune cells stained with DAPI. Scale bar, 27 μm. D, Treatment with ERα ligand was able to reduce coexpression of CCL2 with GFAP in WT, but not aCKO-ERα, mice with EAE. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). E, Treatment with ERβ ligand was not able to reduce coexpression of CCL2 with GFAP in WT or aCKO-ERβ mice with EAE. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis). n = 5 per group.
Given our CCL2 data, we investigated other chemokines expressed by astrocytes that could be differentially affected by ERα versus ERβ ligation during EAE. Our next molecule of interest was CCL7, previously known as MCP-3. CCL7 is expressed in astrocytes, and previous research showed that CCL7 expression directly correlated with CCL2 expression in astrocyte cultures (Thompson and Van Eldik, 2009; Hamby et al., 2012). In MS postmortem tissue, CCL7 expression is expressed in astrocytes and surrounds MS lesions (McManus et al., 1998). Furthermore, CCL7 expression has also been detected in the spinal cords of EAE animals (Adzemovic et al., 2012). Therefore, we examined CCL7 expression within reactive astrocytes and saw a similar expression pattern to that of CCL2 in astrocytes (Fig. 7A,B). However, unlike CCL2, CCL7 expression was only found in reactive astrocytes and was not detected in infiltrating immune cells (Fig. 7C). Treatment with ERα ligand in vivo was able to reduce CCL7 expression in reactive astrocytes in WT mice compared with vehicle. However, ERα ligand treatment lost its ability to reduce CCL7 expression in reactive astrocytes in aCKO-ERα mice with EAE, thereby demonstrating that ERα ligand does indeed act on astrocytes in vivo to reduce CCL7 expression (Fig. 7D). In contrast, treatment with ERβ ligand was not able to reduce CCL7 expression in reactive astrocytes in any of the groups with EAE (Fig. 7E), suggesting that CCL7 expression in astrocytes can be decreased by ERα, but not ERβ, ligand treatment in vivo. We also looked for expression of CCL5 and CCL8 but did not observe expression of these chemokines within astrocytes (data not shown).
Figure 7.

ERα is required on astrocytes to reduce CCL7 expression within reactive astrocytes. A, CCL7 (red), GFAP (green), and DAPI (blue) are expressed in EAE spinal cord in a WT mouse with EAE. Scale bar, 120 μm. B, CCL7 is coexpressed with GFAP. Scale bar, 27 μm. C, CCL7 is not coexpressed in infiltrating immune cells (DAPI). Scale bar, 27 μm. D, Treatment with ERα ligand was able to reduce coexpression of CCL7 with GFAP in WT, but not aCKO-ERα, mice with EAE. *p < 0.05 versus WT + No EAE and WT + EAE + ERα ligand (ANOVA with post hoc Bonferroni pairwise analysis). E, Treatment with ERβ ligand was not able to reduce coexpression of CCL7 with GFAP in WT or aCKO-ERβ mice with EAE. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis). n = 5 per group.
Another possible small molecule expressed by astrocytes that could be responsible for the differential effects of ERα versus ERβ ligand treatment on inflammation is AQP4. AQP4 is a water channel expressed by astrocytes and thought to play a critical role in the maintenance of the blood–brain barrier (Verkman et al., 2006, 2011; Verkman, 2009). CNS diseases, such as spinal cord injury, are known to alter the expression of AQP4 (Kimura et al., 2010; Nesic et al., 2010). In EAE, AQP4 expression is upregulated and redistributed, whereas AQP4 KO mice are resistant to EAE (Li et al., 2009; Miyamoto et al., 2009; Wolburg-Buchholz et al., 2009). Interestingly, treatment with estradiol had been previously shown to decrease AQP4 expression in astrocytes in vitro (Rutkowsky et al., 2011). Thus, we quantified the expression of AQP4 in reactive astrocytes. Our results demonstrated that AQP4 is indeed expressed within reactive astrocytes in the spinal cord of EAE mice (Fig. 8A,B). However, AQP4 expression in astrocytes was increased in all mice with EAE regardless of genotype or treatment, suggesting that AQP4 was not altered with ERα or ERβ ligand treatment in either WT or aCKO mice (Fig. 8C,D). Together, these data revealed that the effects of ERα ligand treatment on astrocytes in vivo during EAE are selective in that they affect the expression of specific chemokines, such as CCL2 and CCL7, but not other immunomodulatory molecules, such as AQP4.
Figure 8.

AQP4 expression within reactive astrocytes is increased in all groups with EAE, regardless of genotype or treatment. AQP4 (red), GFAP (green), and DAPI (blue) are expressed in EAE spinal cord in a WT mouse with EAE. Scale bar, 145 μm. B, AQP4 is coexpressed with GFAP. Scale bar, 8 μm. C, D, AQP4 coexpression with GFAP is increased in all groups with EAE, regardless of genotype or treatment. *p < 0.05 versus WT + No EAE (ANOVA with post hoc Bonferroni pairwise analysis). n = 5 per group.
Discussion
Together, our findings reveal important differences in the cellular mechanisms that underlie the neuroprotective effects of estrogen signaling through either ERα or ERβ in EAE. We show that neuroprotection in EAE-mediated via ERβ signaling does not require ERβ on astrocytes or neurons, whereas neuroprotection in EAE mediated via ERα signaling requires ERα on astrocytes and reduces astrocyte expression of chemokines that contribute to CNS inflammation. These findings have important implications for understanding the different possible means by which estrogens can ameliorate EAE and MS, and for strategies to exploit the potential of selective ERα or ERβ ligands in treatment strategies for MS and other neuroinflammatory conditions.
Previous studies from various laboratories have shown that estrogens exert neuroprotective effects in EAE that consist of a decrease in clinical disease, a reduction of CNS inflammation, and a decrease in axonal loss (Elloso, 2005; Morales, 2006; Tiwari-Woodruff et al., 2007; Polanczyk et al., 2010; Spence et al., 2011). Our findings here extend these observations by showing that estrogens can exert neuroprotection in EAE through markedly different cellular mechanisms when signaling through either ERα or ERβ. Consistent with previous studies, we found that both ERα ligand and ERβ ligand decreased clinical disease, demyelination, and axonal loss in EAE and that ERα ligand, but not ERβ ligand, significantly reduced inflammation (Morales, 2006; Tiwari-Woodruff et al., 2007; Crawford et al., 2009; Du et al., 2010; Spence et al., 2011). Notably, we found that the neuroprotective effects of ERα ligand were completely lost when ERα was selectively deleted from astrocytes, whereas selective deletion of ERβ from astrocytes or neurons had no effect on the neuroprotective effects of ERβ ligand. Thus, the neuroprotective effects of ERα ligand are mediated through astrocytes, whereas the neuroprotective effects of ERβ ligand are mediated by some other cell type, perhaps by acting on microglia, dendritic cells, or oligodendrocytes (Crawford et al., 2010; Du et al., 2010; Saijo et al., 2011). In addition, we show that only signaling through ERα, but not ERβ, decreased inflammation in EAE and that this effect required ERα on astrocytes and that ERα ligand reduced astrocyte levels of inflammatory chemokines, such as CCL2 and CCL7. Together, these findings demonstrate that markedly different cellular mechanisms underlie the neuroprotective effects of signaling through ERα or ERβ.
Implications for our results are not limited to the MS model (Morissette et al., 2008). In animal models of ischemia, estradiol is known to reduce total infarct size. However, this protective effect is lost in a global ERα KO, but not in a global ERβ KO, mice treated with estradiol, demonstrating that ERα, but not ERβ, expression is necessary for estradiol neuroprotection during ischemia (Dubal et al., 2001; Dubal, 2006). In another study, MPTP was given to WT as well as both global ERα KO and global ERβ KO. In the absence of exogenous estrogen administration, the level of striatal dopamine loss was greater in global ERα KO compared with the WT and ERβ KO, suggesting that ERα is more important than ERβ in the MPTP model. However, when exogenous estradiol was given, both the ERα KO and ERβ KO groups demonstrated a decrease in dopamine, suggesting that, whereas ERα is more important at physiological levels, ERβ can play a role at therapeutic levels (Morissette et al., 2007). Although the exact mechanisms of ERα versus ERβ neuroprotection are unknown, researchers have started to uncover differential signaling events between ERα and ERβ in the brain. In one such study, ERα ligand-treated mice showed an enhanced protection against an MPTP-induced decrease in striatal dopamine concentration compared with ERβ ligand-treated mice. In addition, ERα ligand-treated mice demonstrated an enhanced protection against an MPTP-induced increase in IGF-1R levels compared with ERβ ligand-treated mice, which showed no effect. Interestingly, independent of MPTP administration, ERα ligand increased the phosphorylation of neuroprotective kinases GSK3 and Akt at higher levels than ERβ ligand (D'Astous et al., 2006). In another example, in hippocampal primary neurons, ERα ligand was not as effective as ERβ ligand in increasing the concentration of a glutamate-induced intracellular calcium rise. Furthermore, although both ligands were able to increase ERK phosphorylation in these neurons, the magnitude and timing of this increase were unique to each ligand (Zhao and Brinton, 2007). In another hippocampal study, treatment with ERβ ligand, but not ERα ligand, increased the synaptic proteins PSD-95, synaptophysin, and the AMPA-receptor subunit GluR1. This increase in synaptic proteins correlated with an improved performance in hippocampus-dependent memory tasks. These positive effects of ERβ ligand treatment were lost in the global ERβ KO model (Liu et al., 2008). Together, these findings are all consistent with the notion that ERα or ERβ signaling mediates neuroprotection via different cellular mechanisms, similar to our observations reported here.
Astrocytes have multiple functions and play complex roles in CNS function and disease (Sofroniew, 2005; Barres, 2008; Sofroniew and Vinters, 2009; Freeman, 2010), including MS and EAE (Liedtke et al., 1998; Voskuhl et al., 2009; Chastain et al., 2011). Astrocytes have been shown to produce a multitude of proinflammatory and anti-inflammatory molecules that can alter CNS inflammation (Sofroniew and Vinters, 2009; Argaw et al., 2012; Hamby et al., 2012). Given estrogens' ability to alter proinflammatory and anti-inflammatory molecules in astrocytes (Arevalo et al., 2010; Cerciat et al., 2010; Giraud et al., 2010); and given our observed differences in effects on inflammation between ERα and ERβ ligand-treated EAE mice, we investigated inflammatory molecules expressed by astrocytes that could be responsible for ERα ligand-mediated disease protection during EAE. Previous research showed that estrogen treatment in vivo correlated with a decrease in CCL2 expression in astrocytes during EAE, but whether estrogen acted directly or indirectly on astrocytes remained unknown. Further in vitro analysis, using astrocyte cultures, showed that this effect most likely occurred through ERα (Giraud et al., 2010). Our results extend these results by demonstrating for the first time in vivo that CCL2 and CCL7 are decreased upon ligation of ERα, but not ERβ, on astrocytes in EAE. Another astrocyte molecule reported to influence inflammation is AQP4. Previous in vivo data reported that estrogen was able to decrease AQP4 expression in an animal model of ischemia (Rutkowsky et al., 2011). Interestingly, our in vivo data showed that neither ERα ligand nor ERβ ligand significantly altered AQP4 expression on astrocytes. These differences could result from the fact that we used ER-specific ligands as opposed to estradiol, which can act on both nuclear and membrane ERα and ERβ. Alternatively, there may be disease-related differences because the previous reports used an animal model of ischemia, whereas we focused on an animal model of MS. To fully elucidate the role that astrocyte expression of CCL2 and CCL7 play in inflammation, studies using an astrocyte CKO of CCL2 (Ge et al., 2009) or CCL7 or both CCL2 and CCL7 are warranted.
The neuroprotective effects of estrogens, as well as their ligands that bind specifically to ERα or ERβ, are under investigation for potential therapeutic application in various clinical conditions, including MS. Our findings here demonstrate distinctly different cellular mechanisms for neuroprotective effects mediated via signaling through either ERα or ERβ in EAE, the most widely used mouse model of MS. The anti-inflammatory and neuroprotective effects of ERα ligand are mediated via ERα on astrocytes, whereas the neuroprotective effects of ERβ ligand occur without reducing inflammation and are independent of ERβ on astrocytes or neurons and are therefore exerted via other cell types. These findings suggest that the effects of ERα ligand and ERβ ligand treatment in MS are likely to vary and be influenced by the nature and phase of the disease, and that the different ligands may exhibit markedly different efficacies depending on the timing of their delivery. Such factors warrant further investigation.
Footnotes
This work was supported by National Institutes of Health Grant RO1 K24 NS 062117 to R.R.V., Grant NMSS RG 4364 to R.R.V., and Grant T32 HD 7228-31 to R.D.S., Howard Hughes Medical Institute (A.J.W.), Adelson Medical Research Foundation (R.R.V. and M.V.S.), Skirball Foundation, Hilton Foundation, Sherak Family Foundation, and KaroBio Research Foundation (R.R.V.). We thank members of the R.R.V. and M.V.S. laboratories as well as Dr. Tiwari-Woodruff for discussion and assistance; and Dr. Pierre Chambon (Strasbourg) for sharing the ERα and ERβ floxed mice.
The authors declare no competing financial interests.
References
- Adzemovic MZ, Öckinger J, Zeitelhofer M, Hochmeister S, Beyeen AD, Paulson A, Gillett A, Thessen Hedreul M, Covacu R, Lassmann H, Olsson T, Jagodic M. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS One. 2012;7:e39794. doi: 10.1371/journal.pone.0039794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Arevalo MA, Santos-Galindo M, Bellini MJ, Azcoitia I, Garcia-Segura LM. Actions of estrogens on glial cells: implications for neuroprotection. Biochim Biophys Acta. 2010;1800:1106–1112. doi: 10.1016/j.bbagen.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood–brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;6:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor B reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease. Adv Drug Delivery Rev. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-de Sauvage MA, Gómez A, Ros CM, Ros-Bernal F, Martín ED, Perez-Vallés A, Gallego-Sanchez JM, Fernández-Villalba E, Barcia C, Sr, Barcia C, Jr, Herrero MT. CCL2-expressing astrocytes mediate the extravasation of T lymphocytes in the brain: evidence from patients with glioma and experimental models in vivo. PLoS One. 2012;7:e30762. doi: 10.1371/journal.pone.0030762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerciat M, Unkila M, Garcia-Segura LM, Arevalo MA. Selective estrogen receptor modulators decrease the production of interleukin-6 and interferon-γ-inducible protein-10 by astrocytes exposed to inflammatory challenge in vitro. Glia. 2010;58:93–102. doi: 10.1002/glia.20904. [DOI] [PubMed] [Google Scholar]
- Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 2011;1812:265–274. doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Crawford DK, Mangiardi M, Xia X, López-Valdés HE, Tiwari-Woodruff SK. Functional recovery of callosal axons following demyelination: a critical window. Neuroscience. 2009;164:1407–1421. doi: 10.1016/j.neuroscience.2009.09.069. [DOI] [PubMed] [Google Scholar]
- Crawford DK, Mangiardi M, Song B, Patel R, Du S, Sofroniew MV, Voskuhl RR, Tiwari-Woodruff SK. Oestrogen receptor ligand: a novel treatment to enhance endogenous functional remyelination. Brain. 2010;133:2999–3016. doi: 10.1093/brain/awq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxford AL, Kurschus FC, Waisman A. Mouse models for multiple sclerosis: historical facts and future implications. Biochim Biophys Acta. 2011;1812:177–183. doi: 10.1016/j.bbadis.2010.06.010. [DOI] [PubMed] [Google Scholar]
- D'Astous M, Mendez P, Morissette M, Garcia-Segura LM, Di Paolo T. Implication of the phosphatidylinositol-3 kinase/protein kinase B signaling pathway in the neuroprotective effect of estradiol in the striatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Mol Pharmacol. 2006;69:1492–1498. doi: 10.1124/mol.105.018671. [DOI] [PubMed] [Google Scholar]
- Du S, Sandoval F, Trinh P, Umeda E, Voskuhl R. Estrogen receptor-β ligand treatment modulates dendritic cells in the target organ during autoimmune demyelinating disease. Eur J Immunol. 2011;41:140–150. doi: 10.1002/eji.201040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Rau SW, Shughrue PJ, Zhu H, Yu J, Cashion AB, Suzuki S, Gerhold LM, Bottner MB, Dubal SB, Merchanthaler I, Kindy MS, Wise PM. Differential modulation of estrogen receptors (ERs) in ischemic brain injury: a role for ER in estradiol-mediated protection against delayed cell death. Endocrinology. 2006;147:3076–3084. doi: 10.1210/en.2005-1177. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol. 2005;185:243–252. doi: 10.1677/joe.1.06063. [DOI] [PubMed] [Google Scholar]
- Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330:774–778. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Murugesan N, Pachter JS. Astrocyte- and endothelial-targeted CCL2 conditional knockout mice: critical tools for studying the pathogenesis of neuroinflammation. J Mol Neurosci. 2009;39:269–283. doi: 10.1007/s12031-009-9197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud SN, Caron CM, Pham-Dinh D, Kitabgi P, Nicot AB. Estradiol inhibits ongoing autoimmune neuroinflammation and NF B-dependent CCL2 expression in reactive astrocytes. Proc Natl Acad Sci U S A. 2010;107:8416–8421. doi: 10.1073/pnas.0910627107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J Neurol Sci. 2009;286:99–103. doi: 10.1016/j.jns.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci. 2012;32:14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52:1230–1238. doi: 10.1212/WNL.52.6.1230. [DOI] [PubMed] [Google Scholar]
- Kimura A, Hsu M, Seldin M, Verkman AS, Scharfman HE, Binder DK. Protective role of aquaporin-4 water channels after contusion spinal cord injury. Ann Neurol. 2010;67:794–801. doi: 10.1002/ana.22023. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhou J, Li Y, Kim KW, Hensley LL, Baker SJ, Parada LF. Neuron-specific enolase-Cre mouse line with Cre activity in specific neuronal populations. Genesis. 2006;44:130–135. doi: 10.1002/gene.20197. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McEwen BS. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annu Rev Pharmacol Toxicol. 2001;41:569–591. doi: 10.1146/annurev.pharmtox.41.1.569. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang H, Verkman AS. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009;10:94. doi: 10.1186/1471-2202-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS. Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. J Pathol. 1998;152:251–259. [PMC free article] [PubMed] [Google Scholar]
- Liu F, Day M, Muñiz LC, Bitran D, Arias R, Revilla-Sanchez R, Grauer S, Zhang G, Kelley C, Pulito V, Sung A, Mervis RF, Navarra R, Hirst WD, Reinhart PH, Marquis KL, Moss SJ, Pangalos MN, Brandon NJ. Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat Neurosci. 2008;11:334–343. doi: 10.1038/nn2057. [DOI] [PubMed] [Google Scholar]
- McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–29. doi: 10.1016/S0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Nagaosa N, Motoyama M, Kataoka K, Kusunoki S. Upregulation of water channel aquaporin-4 in experimental autoimmune encephalomyeritis. J Neurol Sci. 2009;276:103–107. doi: 10.1016/j.jns.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Morales LB, Loo KK, Liu HB, Peterson C, Tiwari-Woodruff S, Voskuhl RR. Treatment with an estrogen receptor ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci. 2006;26:6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette M, Jourdain S, Al Sweidi S, Menniti FS, Ramirez AD, Di Paolo T. Role of estrogen receptors in neuroprotection by estradiol against MPTP toxicity. Neuropharmacology. 2007;52:1509–1520. doi: 10.1016/j.neuropharm.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Morissette M, Le Saux M, D'Astous M, Jourdain S, Al Sweidi S, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di Paolo T. Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol. 2008;108:327–338. doi: 10.1016/j.jsbmb.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Nesic O, Guest JD, Zivadinovic D, Narayana PA, Herrera JJ, Grill RJ, Mokkapati VU, Gelman BB, Lee J. Aquaporins in spinal cord injury: the janus face of aquaporin 4. Neuroscience. 2010;168:1019–1035. doi: 10.1016/j.neuroscience.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, Teuscher C, Vandenbark AA, Offner H. The protective effect of 17β-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-α. J Pathol. 2010;163:1599–1605. doi: 10.1016/S0002-9440(10)63516-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowsky JM, Wallace BK, Wise PM, O'Donnell ME. Effects of estradiol on ischemic factor-induced astrocyte swelling and AQP4 protein abundance. Am J Physiol Cell Physiol. 2011;301:C204–C212. doi: 10.1152/ajpcell.00399.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/S0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front Neuroendocrinol. 2012;33:105–115. doi: 10.1016/j.yfrne.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, Cao Y, Bondar G, Lam J, Ao Y, Sandoval F, Suriany S, Sofroniew MV, Voskuhl RR. Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc Natl Acad Sci U S A. 2011;108:8867–8872. doi: 10.1073/pnas.1103833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-7 through NFκB and MAPK dependent pathways in rat astrocytes. Brain Res. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci U S A. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Van Der Voorn P, Tekstra J, Beelen RHJ, Tensen CP, Van Der Valk P, De Groot CJA. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. J Pathol. 2010;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins: translating bench research to human disease. J Exp Biol. 2009;212:1707–1715. doi: 10.1242/jeb.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta. 2006;1758:1085–1093. doi: 10.1016/j.bbamem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Verkman AS, Ratelade J, Rossi A, Zhang H, Tradtrantip L. Aquaporin-4: orthogonal array assembly, CNS functions, and role in neuromyelitis optica. Acta Pharmacol Sin. 2011;32:702–710. doi: 10.1038/aps.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise PM. Estrogens and neuroprotection. Trends Endocrinol Metab. 2002;13:229–230. doi: 10.1016/S1043-2760(02)00611-2. [DOI] [PubMed] [Google Scholar]
- Wolburg-Buchholz K, Mack AF, Steiner E, Pfeiffer F, Engelhardt B, Wolburg H. Loss of astrocyte polarity marks blood–brain barrier impairment during experimental autoimmune encephalomyelitis. Acta Neuropathol. 2009;118:219–233. doi: 10.1007/s00401-009-0558-4. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Estrogen receptor α and β differentially regulate intracellular Ca2+ dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]