

Abstract
Hexavalent chromium [Cr (VI)] is a well-known human carcinogen associated with the increased risk of lung cancer. However, the mechanism underlying the Cr (VI)–induced carcinogenesis remains unclear due to the lack of suitable experimental models. In this study, we developed an in vitro model by transforming nontumorigenic human lung epithelial BEAS-2B cells through long-term exposure to Cr (VI). By utilizing this model, we found that miR-143 expression levels were dramatically repressed in Cr (VI)–transformed cells. The repression of miR-143 led to Cr (VI)–induced cell malignant transformation and angiogenesis via upregulation of insulin-like growth factor-1 receptor (IGF-IR) and insulin receptor substrate-1 (IRS1) expression. Moreover, we found that interleukin-8 is the major upregulated angiogenesis factor induced by Cr (VI) through activation of IGF-IR/IRS1 axis followed by activation of downstream ERK/hypoxia-induced factor-1α/NF-κB signaling pathway. These findings establish a causal role and mechanism of miR-143 in regulating Cr (VI)–induced malignant transformation and tumor angiogenesis.
Key Words: chromium (VI), miR-143, lung cancer, IGF-IR/IRS1, tumor angiogenesis.
Lung cancer is the leading cause of cancer mortality worldwide. In 2011, about 28% of cancer deaths in men and 26% in women are caused by lung and bronchus cancer in United States (Siegel et al., 2011). Lung cancer is closely related with environmental carcinogens such as cigarette, air pollution, and heavy metals. Hexavalent chromium [Cr (VI)] compounds are widely used in industry and have been shown to have serious toxic and carcinogenic effects on humans (Cohen et al., 1993). Epidemiological studies suggest a high risk of lung cancer associated with Cr (VI) occupational exposure (Pastides et al., 1994). In addition, environmental chromium is an emerging concern because Cr (VI) compounds are also present in high concentrations in the landfills and environments in United States and other countries. However, the cellular and molecular mechanisms, especially causative factors underlying Cr (VI)–induced carcinogenesis are poorly defined partly due to the lack of suitable experimental models that can closely mimic chronic Cr (VI) exposure in human lung cells (Azad et al., 2010; Balansky et al., 2000; Costa et al., 2010; Levy et al., 1986).
The potential mechanisms of Cr carcinogenesis include sustained oxidative stress, compromised DNA repair system, and microsatellite instability (Holmes et al., 2008; Rodrigues et al., 2009; Wang et al., 2011a). Besides these genotoxic effects, a number of studies show that aberrant gene expressions are critical events for cell malignant transformation induced by Cr (VI) (Medan et al., 2012; Rodrigues et al., 2009). The discovery of microRNAs (miRNAs) provides novel mechanisms of gene regulation. The aberrant expression of miRNAs has been frequently observed in many human cancers including lung cancer (Liu et al., 2011). However, there is little information on the involvement of miRNAs in environmental carcinogenesis. It remains to be determined whether the loss or gain of certain miRNA expression leads to cell malignant transformation in response to chemical carcinogens. A recent study found that miR-200b is involved in arsenic (As) carcinogenesis (Wang et al., 2011b), indicating that miRNAs may have roles in environmental carcinogenesis. miR-143 is generally downregulated in colon cancer, liposarcoma, esophageal squamous cell carcinoma, and prostate cancer, suggesting that it has tumor-suppressive properties (Roa et al., 2010; Ugras et al., 2011; Wach et al., 2012; Wu et al., 2011). In this study, we established an in vitro model by transforming nontumorigenic human lung epithelial BEAS-2B cells through long-term exposure to Cr (VI). We utilized this model to determine the roles of certain miRNAs such as miR-143 in Cr (VI)–induced cell transformation, tumor formation, and tumor angiogenesis.
MATERIALS AND METHODS
Animal experiment.
Male BALB/cA-nu nude mice (4 weeks old) were purchased from Shanghai Experimental Animal Center (Chinese Academy of Sciences, Shanghai, China) and maintained in pathogen-free conditions. BEAS-2B cells, BEAS-Cr cells, BEAS-Cr cells stably expressing miR-143, or BEAS-Cr cells stably expressing miR control were injected sc into the flank of nude mice (2 × 106 cells in 150 µl). Bidimensional tumor volume measurements were obtained with calipers three times a week. Tumor volumes were calculated according to the formula (width2 × length)/2. The mice were euthanized after 28 days, and tumors were weighed.
Antibodies and reagents.
Sodium dichromate (Na2Cr2O7·H2O) was obtained from Sigma (St Louis, MO). Antibodies against insulin-like growth factor-1 receptor (IGF-IR), insulin receptor substrate-1 (IRS1), p-AKT, total AKT, p-ERK, and total ERK were from Cell Signaling Technology (Beverly, MA). Antibodies against NF-κB, c-myc, and CD31 were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against hypoxia-induced factor-1α (HIF-1α) was from BD Bioscience (Franklin Lakes, NJ). siRNA SMARTpools (pool of four individual siRNAs) against IGF-IR, IRS1, interleukin (IL)-8, ERK, NF-κB, HIF-1α, and scrambled control were from Dharmacon (Lafayette, CO). Recombinant human IL-8 was purchased from R&D Systems (Minneapolis, MN).
Cell culture and generation of stable cell lines.
The human bronchial epithelial BEAS-2B cells (purchased from ATCC) were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS). The human umbilical vein endothelial cells (HUVECs) (purchased from ATCC) were cultured in EBM-2 complete medium. Stable cell lines of BEAS-Cr cells overexpressing miR-143 or miR control were generated by infecting with lentivirus carrying miR-143 or a negative control precursor (Open Biosystems, IL) followed by the selection with puromycin. To establish stable cell lines overexpressing IGF-IR or IRS1, the cells were infected with pBABE retrovirus vector alone or with pBABE retrovirus vector carrying IGF-IR or IRS1 cDNA construct without the 3′-UTR (Addgene, MD) followed by the selection with zeocin. To establish BEAS-2B cell line stably expressing IL-8, 293T cells were transfected with lentivirus carrying IL-8 plasmid (GeneCopoeia, Rockville, MD) or empty vector to generate virus soup. Then, BEAS-2B cells were transduced with virus and followed by puromycin selection.
Chronic Cr (VI) exposure.
BEAS-2B cells were continuously cultured in DMEM containing 1μM Cr (VI). Parallel cultures grown in Cr (VI)–free medium acted as passage-matched controls. After 6 months of exposure, Cr (VI)–treated cells were cultured in normal medium and subjected to cell transformation and tumor growth analysis.
RT-qPCR analysis.
Total RNAs were extracted using Trizol (Life Technologies, Carlsbad, CA). The cDNA synthesis was performed using oligo(dT)18 primers and M-MLV reverse transcriptase (Promega). The amplification was performed by PCR. SYBR-Green RT-qPCR was performed to detect IL-8 and GADPH mRNA levels using Power SYBR Green PCR Master Mix Kit (Applied Biosystems, Carlsbad, CA). Taqman RT-qPCR was performed to detect miRNA expression levels using Taqman miRNA reverse transcription kit and Taqman universal PCR master mix (Applied Biosystems, Austin, TX). Primer sequences for RT-PCR or RT-qPCR were shown as below:
RT-PCR primers
HIF-1α forward: 5′-TCCATGTGACCATGAGGAAA-3′
HIF-1α reverse: 5′-TATCCAGGCTGTGTCGACTG-3′
IL-8 forward: 5′-TAAATCTGGCAACCCTAGTC-3′
IL-8 reverse: 5′-GCGTTCTAACTCATTATTCCGT-3′
GADPH forward: 5′-CCACCCATGGCAAATTCCATGGCA-3′
GADPH reverse: 5′-TCTAGACGGCAGGTCAGGTCCACC-3′
Primers for SYBR-Green RT-qPCR
IL-8 forward: 5′-CACCGGAAGGAACCATCTCA-3′
IL-8 reverse: 5′-AGAGCCACGGCCAGCTT-3′
GAPDH forward: 5′-ATGGGTGTGAACCATGA GAAGTATG-3′
GADPH reverse: 5′GGTGCAGGAGGCATTGCT-3′
Colony isolation.
BEAS-Cr cells were seeded in soft agar for at least 2 weeks. A number of colonies were then picked up from the agarose with a pipette, seeded into a 6-cm dish with complete medium, and cultured to the confluence. All surviving colonies (BEAS-Cr clones) were kept in complete medium (DMEM + 10% FBS) and used for subsequent experiments.
Tube formation assay.
HUVECs were cultured in EBM-2 complete medium and switched to EBM-2 basic medium containing 0.2% FBS for 24h to be used for the tube formation assay. The conditioned media were prepared from different cells by replacing normal culture medium with serum-reduced medium (1% FBS). After culture for 24h, the serum-reduced medium was collected and stored at −20°C for later use. The HUVECs were trypsinized, counted, and resuspended in EBM-2 basic medium and then mixed with equal volume of the conditioned medium and seeded on Matrigel-pretreated 96-well plate at 2 × 104 cells/well. Tube formation was observed under light microscope after culture for 6–12 h and photographed. The total lengths of the tubes for each well were measured using CellSens Standard software.
Chorioallantoic membrane assay.
White Leghorn fertilized chicken eggs were incubated at 37°C under constant humidity. Different cells and control cells were transfected with certain miRNA precursors and treated as specifically indicated. These cells were trypsinized, counted, and resuspended in the serum-free medium. The cell suspensions were mixed with Matrigel at 1:1 ratio and implanted onto the chorioallantoic membranes (CAMs) of chicken eggs at day 9. Tumor angiogenesis responses were analyzed 5 days after the implantation. The tumor/Matrigel plugs were trimmed off CAM and photographed. The number of blood vessels as the index of angiogenesis was obtained by counting the branches of blood vessels in three representative areas (1.5 mm2) by two observers in a double-blind manner.
Immunohistochemistry.
Tumor tissues were sliced and fixed with Bouin’s solution for 24 h and processed by conventional paraffin-embedded method. Dako Envision two-step method of immunohistochemistry was used to stain IGF-IR and IRS1 in human tissues and CD31 in xenograft tumors according to the instruction. For CD31 staining in tumor tissues, sections were prepared from three of the Matrigel plugs in every group, and the microvessel densities were counted in three different fields per section as follows: slides were first scanned under low power (×100) to determine three ‘‘hotspots’’ or areas with the maximum number of microvessels; then, the positive-stained blood vessels in the selected areas were analyzed at ×400 magnification.
miRNA transfection.
The negative control miRNA and miR-143 precursors were obtained from Applied Biosystems. Cells were cultured in six-well plate to reach 60% confluency and transfected using miR-143 or negative control precursor at 30nM using Lipofectamine RNAiMAX reagent (Invitrogen, Grand Island, NY) according to the manufacturer’s instruction. Total proteins and RNAs were prepared from the cells 60–70h after the transfection and were used for subsequent analysis.
miRNA luciferase reporter constructs and luciferase activity assay.
The 3′-UTR-luciferase reporter constructs containing the 3′-UTR of IGF-IR and IRS with wild-type and mutant-binding sites of miR-143 were amplified using PCR method. The PCR products were cloned into the pMiR-luc reporter vector (Ambion), immediately downstream of the luciferase gene. The mutant 3′-UTR constructs were made by introducing four to six mismatch mutations into the putative seed regions of IGF-IR and IRS1. All the constructs containing 3′-UTR inserts were sequenced and verified. The luciferase activity assay was performed as previously described (He et al., 2012).
Statistical analysis.
All the results were obtained from at least three independent experiments. Most results were presented as mean ± SE from independent experiments and analyzed by Student’s t-test and one-way ANOVA. All results were analyzed by SPSS for Windows, version 11.5. Differences were considered significant at a value of p < 0.05.
RESULTS
miR-143 Expression is Downregulated in Cr (VI)—Transformed BEAS-2B Cells
In order to mimic the pathophysiological impact of long-time exposure to chromium, we established an in vitro model by transforming immortalized and nontumorigenic human lung epithelial BEAS-2B cells via exposure to 1µM Cr (VI) for 24 weeks. This dose is relevant to human exposure (Das and Singh, 2011; Woodruff et al., 1998) and it exhibits minimal cellular toxicity (data not shown). The cells cultured in Cr (VI)–free medium acted as passage-matched control. Cr (VI)–exposed BEAS-2B cells showed the characteristics of transformed cells such as increased cell proliferation, anchorage independent growth, and tumor growth in nude mice (hereafter designated as BEAS-Cr) (Figs. 1A–C and Table 1). We performed miRNA microarray analysis to compare the miRNA profiles between BEAS-Cr cells and their parental cells BEAS-2B and found that miR-143 was dramatically suppressed in BEAS-Cr cells (data not shown). Then, we further validated the result by performing Taqman RT-qPCR analysis and showed that miR-143 expression was lowered about 35-fold in BEAS-Cr cells (Fig. 1D). To determine the variations of miR-143 repression among individual Cr (VI)–transformed cells, individual colonies were isolated and cultured separately (BEAS-Cr clones). The repression of miR-143 expression was observed similarly in all of the different clones, suggesting that miR-143 is generally downregulated in BEAS-Cr cells (Fig. 1E). In addition, miR-143 expression levels were much lower in lung cancer cells A549 and H2195 compared with BEAS-2B (Fig. 1F).
Fig. 1.

miR-143 is downregulated in Cr (VI)–transformed BEAS-2B cells and human lung cancer tissues. (A) An equal number of BEAS-2B and BEAS-Cr cells (10,000 cells) were seeded in 12-well plates to analyze cell proliferation by counting with trypan blue exclusion for every 24h. (B) Cells were seeded in 96-well plates and cultured in medium with different concentrations of serum. 3-(4,5-Dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay was performed at 72h to assess the cell proliferation rates. (C) An equal cell number of cells (5000) were seeded in a six-well plate and cultured in soft agar and incubated at 37°C for 2 weeks. Representative images for BEAS-2B and BEAS-Cr are presented (left panel). Number of colonies from soft agar assay was counted for each group shown in lower panel. (D–F) miR-143 expression levels were determined by Taqman RT-qPCR in parental Cr (VI)–transformed BEAS-2B cells (BEAS-Cr) and different BEAS-Cr clones. Relative miRNA expression levels were represented as RQ using 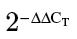 methods. The values were normalized to the U6 expression level and that in BEAS-2B cells. Mean ± SE values were from three separate experiments. **Significantly different compared with control (p < 0.01).
methods. The values were normalized to the U6 expression level and that in BEAS-2B cells. Mean ± SE values were from three separate experiments. **Significantly different compared with control (p < 0.01).
Table 1.
Tumor Growth Assay in Nude Mice
| Number of mice for injection | Number of mice growing tumor | |
|---|---|---|
| Mice receiving BEAS-2B control cells | 8 | 0 |
| Mice receiving BEAS-Cr cells | 7 | 6 (average tumor volume: 375mm3) |
Ectopic Expression of miR-143 Inhibits Cr (VI)–Induced Tumor Angiogenesis
As miR-143 is downregulated in BEAS-Cr cells, they might have biological functions related to cell transformation and tumor growth. Angiogenesis is the essential process for tumor development (Folkman et al., 1971). We evaluated the effect of miR-143 overexpression in tumor-induced angiogenesis both in vitro and in vivo. First, we performed tube formation assay in vitro (Fig. 2A). HUVECs maintained in EBM-2 basic medium were not capable of tube formation. However, it was strongly induced using conditioned medium prepared from BEAS-Cr cells. Further, the ectopic expression of miR-143 in BEAS-Cr impaired the tube formation by 35%. Next, we employed chicken CAM assay to determine angiogenesis responses in vivo. BEAS-Cr cells induced much higher angiogenesis responses compared with their control cells (Fig. 2B). Transient transfection of miR-143 precursor in the cells decreased their ability to induce angiogenesis in the CAM (Fig. 2C). Immunohistochemical detection of CD31 has been extensively used to quantify the angiogenesis responses of xenograft tumors (Wang et al., 2008). In order to evaluate the effect of miR-143 in tumor angiogenesis in nude mice, we established BEAS-Cr cells stably overexpressing miR-143 or negative miR control. Then, we generated xenograft tumors after the implantation of these stable cells for 30 days. Overexpression of miR-143 decreased the tumor volume by 50% compared with negative control cells (Fig. 2D). The number of microvessels indicated by the CD31 staining in miR-143-overexpressing tumors was 50% less than the control (Fig. 2E).
Fig. 2.

Ectopic expression of miR-143 inhibits Cr (VI)–induced tumor angiogenesis. (A) HUVECs were cultured in serum-free medium overnight and resuspended in EBM-2 basic medium. To perform the tube formation assay, HUVECs were incubated in EBM-2 basic medium; conditioned medium prepared from BEAS-2B or BEAS-Cr was transfected with pre–miR control or pre-miR-143, respectively. Tube formation was determined under light microscope after the culture for 12h. Total tube lengths (mm) were presented as mean ± SE from six replicates for each treatment. * and # indicate significant differences compared with BEAS-2B and with BEAS-Cr miR-cont, respectively (p < 0.05). Scale bar: 100 μm. (B and C) BEAS-2B and BEAS-Cr cells were transfected with or without 25nM pre-miR-143 and pre–miR control precursor, respectively. After transfection (24 h), 2 × 106 cells were trypsinized, suspended, and mixed with equal volume of Matrigel and implanted onto the chicken CAMs of 10-day-old chicken embryos. The branches of blood vessels were counted as the relative angiogenesis using 8–10 embryos per treatment 96h after implantation. The data represent as mean ± SE of blood vessel numbers, which were normalized to that of the control. Scale bar: 2 mm. * indicates significant difference compared with that of the control (p < 0.05). (D) Tumor xenograft model in nude mice was established as described in Materials and Methods section. Stable BEAS-Cr cells overexpressing miR-143 or negative miR control were implanted sc into the both flanks of nude mice. Tumor volumes were measured 4 weeks after the cell injection according to the formula (width2 × length)/2 and presented as mean ± SE (n = 10). (E) Mice were euthanized, and tumor sections from the nude mice were used for immunohistochemical staining using antibodies against CD31. Scale bar: 50 μm. Arrow: CD31-positive staining vessels.
miR-143 Targets Both IGF-IR and IRS1 in BEAS-2B Cells
IGF-IR/IRS1 signaling has been closely associated with cell transformation (Baserga, 2006). Compared with its parental cell BEAS-2B, Cr (VI)–transformed cells displayed higher expression levels of both IGF-IR and IRS1 (Fig. 3A, left). Transfection of miR-143 precursor in BEAS-Cr cells inhibited expression of both IGF-IR and IRS1 proteins (Fig. 3A, right). We also investigated several other oncogenes that are potentially regulator or targets of miR-143 (Kent et al., 2010; Zhu et al., 2011), but we did not find any significant change of these oncogene expressions in Cr (VI)–transformed cells (Fig. 3B). To explore whether miR-143 directly regulates IGF-IR or IRS1, we used “Targetsearch” program (He et al., 2012) to search for miR-143 potential targets and found that miR-143 is predicted to bind with 3′-UTR of both IGF-IR and IRS1 mRNAs. We constructed the 3′-UTR reporters of IGF-IR and IRS1 containing the putative miR-143-binding sites and corresponding mutant constructs downstream of the luciferase reporters. Cotransfection of miR-143 precursor with their respective wild-type reporter constructs decreased the luciferase activities in BEAS-2B cells, whereas cotransfection of miR-143 precursor with reporters containing point mutations at putative miR-143-binding sites did not affect the luciferase activities (Fig. 3C). These findings demonstrate that miR-143 directly targets IGF-IR and IRS1.
Fig. 3.

miR-143 targets both IGF-IR and IRS1 in BEAS-2B cells. (A) Total epidermal growth factor receptor, N-Ras, K-Ras, and MDM2 protein levels along with their protein phosphorylation levels were determined by immunoblotting in BEAS-2B cells and BEAS-Cr cells. (B) Basic expression levels of p-IGF-IR, IGF-IR, p-IRS1, and IRS1 were determined by Western blotting in BEAS-Cr cells, BEAS-As cells, and the passage-matched control BEAS-2B cells (left). BEAS-Cr and BEAS-As cells were transiently transfected with 25nM miR-143 or negative miR control precursor. IGF-IR and IRS1 expression levels were determined by Western blotting (right). (C) Top: Sequence alignment of human miR-143 with 3′-UTR of IGF-IR or IRS1. The mutation sites in the 3′-UTR of IGF-IR and IRS1 were shown in the third row for creating the mutant luciferase reporter constructs. The luciferase activities were presented as relative luciferase activities normalized to those of the cells cotransfected with wild-type 3′-UTR reporter and miRNA precursor control. * indicates significant decrease compared with that of control cells (p < 0.05). All tests were performed in triplicate and presented as mean ± SE.
miR-143 Attenuates Tumor Angiogenesis by Directly Targeting IGF-IR/IRS1
To confirm the classical roles of IGF-IR and IRS1 in cell transformation in the context of Cr (VI)–treated cells, siRNA SMARTpools against IGF-IR and IRS1, respectively, were transfected into BEAS-Cr cells for soft agar assay and CAM assay. Knockdown of IGF-IR or IRS1 by their siRNAs inhibited colony formation and angiogenesis (Figs. 4A and B). To determine whether miR-143 decreases angiogenesis responses by targeting IGF-IR and IRS1, we utilized the miR-143-overexpressing BEAS-Cr cells to establish stable cell lines overexpressing IGF-IR or IRS1 without the 3′-UTR (Fig. 4C). As expected, both tube formation assay and CAM assay showed that miR-143-expressing cells suppressed angiogenesis responses, whereas forced expression of IGF-IR or IRS1 ORF completely restored inhibitory effect of miR-143 (Figs. 4D and E). Collectively, these results suggest that miR-143 inhibits angiogenesis by directly targeting IGF-IR and IRS1 expression.
Fig. 4.

miR-143 attenuates tumor angiogenesis by directly targeting IGF-IR/IRS1. (A and B) BEAS-Cr cells were transfected with 50nM of an siRNA scramble control or siRNA SMARTpool against IGF-IR and IRS1, respectively, for 24 h and were subjected to soft agar assay (A) or CAM assay (B) as previously described. Scale bar: 2mm. ** indicates significant difference compared with control group (p < 0.01). (C) BEAS-Cr cells stably expressing miR-143 were established as described in Figure 2. Retrovirus carrying IGF-IR or IRS1 cDNA was used to generate stable BEAS-Cr cells overexpressing miR-143/vector, miR-143/IGF-IR, or miR-143/IRS1. (D) Tube formation assay was performed as described in Figure 2A. Conditioned medium was prepared from BEAS-Cr miR-cont cells, BEAS-Cr miR-143 cells, BEAS-Cr miR-143/IGF-IR cells, and BEAS-Cr miR-143/IRS1 cells, respectively. Total tube lengths for each treatment were analyzed and presented as mean ± SE (mm) from six replicates for each treatment. (E) BEAS-Cr miR-cont cells, BEAS-Cr miR-143 cells, BEAS-Cr miR-143/IGF-IR cells, or BEAS-Cr miR-143/IRS1 cells were implanted onto the CAMs to perform angiogenesis assay as described in Figure 2B. The representative images from each group were shown here. Scale bar: 2mm. The total number of blood vessels in each group was quantified. * and # indicate significant difference compared with miR control and with miR-143 cells, respectively.
miR-143 Inhibits ERK Signaling via IGF-IR/IRS1 in Long-Term Cr (VI)–Treated BEAS-2B
The mitogenic effect of IGF-IR signaling is mainly through downstream AKT and/or ERK pathways. We found that ERK activation was dramatically activated after the long-term exposure to Cr (VI), whereas AKT activation was much less induced in BEAS-Cr cells. To determine the roles of IGF-IR and IRS1 expression in BEAS-Cr cells, siRNA SMARTpools (pool of four individual siRNAs) specifically against IGF-IR or IRS1 were transfected into the BEAS-Cr cells. Knockdown of IGF-IR and IRS1 expression using siRNAs against IGF-IR and IRS1, respectively, markedly decreased ERK phosphorylation and moderately reduced AKT phosphorylation levels (Fig. 5A). Almost all of BEAS-Cr clones as described in Figure 1B showed the elevations of IGF-IR and IRS1 and enhanced activation of ERK signaling compared with their parental BEAS-2B cells (Fig. 5B). Overexpression of miR-143 was able to suppress phospho-ERK expression levels, suggesting that the suppression of miR-143 leads to the activation of ERK signaling (Fig. 5C).
Fig. 5.

miR-143 inhibits ERK signaling via IGF-IR/IRS1 in long-term Cr (VI)–treated BEAS-2B. (A) BEAS-Cr cells were transfected with 50nM of an siRNA scramble control or an siRNA SMARTpool against IGF-IR or IRS1 for 72 h. The IGF-IR, IRS1, p-AKT, AKT, p-ERK, and ERK protein levels were assessed by Western blotting. (B) The expression levels of IGF-IR, IRS1, p-AKT, AKT, p-ERK, and ERK in eight BEAS-Cr clones were assessed by Western blotting. The representative protein bands were shown in the top panel, and relative protein expression levels from three independent experiments were shown in the lower panel. (C) BEAS-Cr cells were transiently transfected with 25nM miR-143 or negative miR control precursor and harvested 70h later to analyze p-ERK and ERK expression levels.
IL-8 Mediates Cr (VI)–Induced Angiogenesis via IGF-IR/IRS1/ERK Signaling
To identify angiogenesis factor(s) that may play a critical role in BEAS-Cr cell–inducing and miR-143-inhibiting angiogenesis, we screened potential angiogenic inducers using RayBio human angiogenesis arrayC1000 and found that IL-8 was the most upregulated factors in BEAS-Cr cells, whereas several other factors including VEGF, angiogenin-1, and angiopoietin were increased to much lower degree (data not shown). RT-PCR results showed that IL-8 mRNA expression was greatly elevated in Cr (VI)–transformed cells (Fig. 6A, left). Knockdown of IGF-IR or IRS1 in the cells greatly inhibited IL-8 expression, indicating that IL-8 is a downstream signal of IGF-IR/IRS1 pathway that might be responsible for Cr (VI)–induced angiogenesis (Fig. 6A, right). IL-8 is known to possess proangiogenic properties (Brat et al., 2005). Indeed, HUVECs cultured in basic medium could not form tube. But tube formation was greatly induced when the cells were cultured in medium containing 10 ng/ml recombinant human IL-8 (Fig. 6B). We showed that knockdown of IL-8 using siRNAs decreased the angiogenesis responses in BEAS-Cr cells by 50% (Figs. 6C–E). We then generated BEAS-2B cells stably expressing IL-8 to assess its angiogenic activity (Fig. 6F). Overexpression of IL-8 greatly enhanced tube formation in HUVECs (Fig. 6G). More importantly, overexpression of miR-143 decreased IL-8 expression in BEAS-Cr cells (Figs. 6H and I). A number of studies indicate that the release of IL-8 in human airway epithelial cells is mediated by ERK pathway (Lin et al., 2013; Rath et al., 2013). We demonstrated this link by treating BEAS-Cr cells with ERK inhibitor U0126 or by transfecting the cells with siRNA SMARTpool against ERK. As shown in Figures 6J and 6K, the inhibition of ERK using both the chemical and molecular inhibitors greatly decreased IL-8 expression levels. Taken together, these results demonstrate that IL-8 is the major angiogenesis activator in Cr (VI)–induced angiogenesis, which is mediated via IGF-IR/IRS1/ERK pathway.
Fig. 6.


IL-8 mediates Cr (VI)–induced angiogenesis via IGF-IR/IRS1/ERK signaling. (A) Left panel: RT-PCR analysis was used to evaluate mRNA levels of IL-8 in BEAS-Cr cells and control BEAS-2B cells. Right panel: BEAS-Cr cells were transfected with 50nM of an siRNA scramble control or an siRNA SMARTpool against IGF-IR or IRS1 for 60h. Total RNA samples were prepared and subjected to RT-PCR analysis for IL-8 expression. (B) HUVECs were cultured in Matrigel-coated 96-well plate in basic medium or medium containing 10ng/ml recombinant human IL-8. Tube formation was photographed 6h after the seeding. (C and D) Conditioned media were prepared from BEAS-Cr cells transfected with siRNA control and siIL-8. HUVECs were cultured in conditioned medium. Tube formation was assessed as previously described. (E) BEAS-Cr cells were transfected with 50nM of an siRNA scramble control or a SMARTpool siRNA against IL-8 for 24h. Then, the cells were subjected to CAM assay as described. (F) Establishment of BEAS-2B cells stably expressing IL-8. (G) Tube formation assay was performed using conditioned media prepared from BEAS-2B empty vector cells and BEAS-2B overexpressing IL-8 cells. Scale bar: 100 μm. Data were presented as mean ± SE (n = 6). (H) BEAS-Cr cells were transfected with miR-143 or miR control precursor for 60h. Total RNA samples were prepared for detection of IL-8 expression levels. (I) BEAS-Cr cells were transfected with miR-143 or miR control precursor for 60h. The SYBR-Green RT-qPCR analysis was performed to detect IL-8 mRNA levels. (J) BEAS-Cr cells were treated with or without ERK inhibitor U0126 (5μM) for 24h. Total RNAs were extracted and used to determine IL-8 mRNA levels. (K) BEAS-Cr cells were transfected with siMAPK (50nM) or scrambled siRNA (50nM). Total RNAs were extracted 60h after transfection to detect IL-8 mRNA levels by RT-qPCR. ** indicates significant difference compared with control group (p < 0.01). All experiments were performed in triplicate and presented as mean ± SE. * indicates significant difference compared with control group (p < 0.05).
Induction of IL-8 Expression Is Mediated by HIF-1α and NF-κB
IL-8 can be directly regulated by both HIF-1α and NF-κB (Kim et al., 2006; Mukaida et al., 1994). Thus, we investigated whether HIF-1α and NF-κB are involved in IL-8 activation in BEAS-Cr cells. As shown in Figure 7A, HIF-1α was upregulated in BEAS-Cr cells, whereas knockdown of HIF-1α inhibited both HIF-1α and IL-8 expression levels (Fig. 7B). When cells were cultured under hypoxia, IL-8 levels were significantly higher than those under normoxic condition (Fig. 7C), suggesting that hypoxia plays a role in BEAS-Cr cell–induced IL-8 expression. Similarly, NF-κB p65 expression levels in nuclear fraction were much higher in BEAS-Cr cells compared with its parental cells (Fig. 7D), indicating that NF-κB signaling is activated in Cr (VI)–transformed cells. This result was further confirmed by NF-κB reporter activity assay (Fig. 7E). To test whether NF-κB signaling regulates IL-8 expression, we treated cells with NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) for analyzing IL-8 mRNA levels and showed that PDTC dramatically decreased IL-8 expression in BEAS-Cr cells (Fig. 7F). Moreover, overexpression of miR-143 was sufficient to inhibit both HIF-1α and NF-κB expression (Fig. 7G).
Fig. 7.

Induction of IL-8 expression is mediated by HIF-1α and NF-κB. (A) The HIF-1α protein levels were determined by immunoblotting. (B) BEAS-Cr cells were transfected with 50nM of an siRNA scramble control or SMARTpool siRNAs against HIF-1α for 60h. Left panel: HIF-1 protein expression was assessed by immunoblotting. Right panel: total RNAs were extracted and subjected to RT-PCR analysis for HIF-1α and IL-8 expression levels. (C) BEAS-2B and BEAS-Cr cells were cultured in a hypoxic chamber for 24h. Cells cultured under normoxia condition were served as control cells. RNAs were extracted and subjected to SYBR-Green RT-qPCR for IL-8 mRNA expression levels. (D) NF-κB p65 expression was assessed in nuclear and cytosolic fractions of BEAS-2B and BEAS-Cr cells. The nuclear marker c-myc and the cytosolic marker tubulin were used as loading control. (E) Cells were seeded in 12-well plates and transiently transfected with human NF-κB luciferase reporter and β-galactosidase plasmids. The cells were cultured for 48h after transfection, and relative luciferase activities were measured as described in Materials and Methods section. (F) BEAS-Cr cells were treated with or without NF-κB inhibitor PDTC (40μM) for 24h. Total RNAs were extracted and subjected to qRT-PCR analysis for IL-8 expression levels. The experiments were performed in triplicate and presented as mean ± SE. ** indicates significant difference compared with control group (p < 0.01). (G) BEAS-Cr cells were transiently transfected with miR-143 or miR control precursor for 70h. The expression levels of HIF-1α in total protein extracts and NF-κB levels in nuclear fraction were assessed by immunoblotting.
DISCUSSION
Cr (VI)–induced tumorigenesis is thought to be a multistep process involving DNA damage, mutation, and epigenetic modulation (Costa and Klein, 2006). The ultimate outcome of the process is the malignant phenotype with an altered gene expression profile. In this study, we established an in vitro Cr (VI)–transformed cell model by long-term treatment with Cr (VI). The human bronchial epithelial cells BEAS-2B were used to investigate mechanisms of chromium transformation because they share the essential features and behaviors with the normal epithelial cells and are susceptible to carcinogens. Moreover, as BEAS-2B cells retain the ability to undergo squamous differentiation, the cell model reflects more closely squamous carcinoma, which is the major pathological subtype of Cr (VI)–induced human lung cancers (Ishikawa et al., 1994).
A recent study links heavy metal exposure with altered miRNA profiles in leukocytes (Bollati et al., 2010). To determine whether abnormally expressed miRNAs lead to Cr (VI)–induced cell malignant transformation, we performed a genome-wide screening to detect alterations of miRNA profiles in Cr (VI)–transformed cells. We selected miR-143 for further study based on two lines of experimental evidence: (1) miR-143 expression is dramatically repressed in Cr (VI)–transformed cells and lung cancer cells, whereas overexpression of miR-143 is capable of inhibiting colony formation and tumor angiogenesis and (2) miR-143 expression is much lower in human lung cancer tissues compared with the adjacent normal tissues. More importantly, we validated that both IGF-IR and IRS1 are the functional targets of miR-143. IGF-IR and IRS1 are known to be involved in cell transformation and angiogenesis (Baserga, 2006; Jiang et al., 2003) and to play an important role in oncogenic transformation (Wu et al., 2008). Our results showed that both IGF-IR and IRS1 were activated in heavy metal–transformed cells and had functions in cell transformation and angiogenesis. The canonical IGF-IR/IRS1 signaling is activated through the binding of IRS1 with phosphorylated IGF-IR or IR (Lee and White, 2004). The phosphorylated IRS1 serves as the docking site for SH2-containing proteins, resulting in activation of AKT and ERK pathways (Myers et al., 1993; Taniguchi et al., 2006). In this study, ERK is the major downstream pathway induced by Cr (VI) via IGF-IR/IRS1 axis, which can be inhibited by miR-143.
A population-based study reported that serum levels of IL-8 were increased in the arsenic-exposed group (Das et al., 2012), which is consistent with the upregulation of IL-8 in heavy metal–transformed cells. We further identified that IL-8 is the major angiogenic factor involved in Cr (VI)–induced tumor angiogenesis through IGF-IR/IRS1/ERK pathway. IL-8 is known to have proangiogenic properties by enhancing endothelial proliferation, chemotaxis, and protease activation (Brat et al., 2005). Recent study suggests that HIF-1α, a key molecule in tumor angiogenesis (Wang et al., 1995), possesses a mitogen-activated protein kinase (MAPK)-docking domain, indicating that there is direct interaction between HIF-1α and ERK2 MAPK (Karapetsas et al., 2011). Besides, both HIF-1α and NF-κB are able to directly regulate IL-8 transcription via binding the hypoxia response element in IL-8 promoter (Kim et al., 2006). We demonstrated that activation of HIF-1α and NF-κB mediated IL-8 release as one of the downstreams of miR-143, resulting in enhanced angiogenesis.
The mechanism underlying Cr (VI)–induced miR-143 repression is not known yet. A recent study showed that chromium-exposed lung cancer is linked to the progressive methylation of some tumor suppressor genes (Ali et al., 2011). We wondered whether Cr (VI) can induce hypermethylation of promoter region of miR-143, resulting in repression of miR-143. However, our results indicated that DNA methylation levels in control BEAS-2B cells are more extensive than those of Cr (VI)–transformed cells, suggesting that DNA methylation may not be the mechanism underlying miR-143 repression (data not shown). It was reported that chromium (VI) downregulated transcription of MT genes by inhibiting metal-responsive transcription factor-1 (MTF-1) (Majumder et al., 2003). It would be worthwhile to investigate whether MTF-1 and/or other potential transcription factor(s) may be involved in repression of miR-143 expression in the future.
The IGF-IR pathway has been implicated in the pathogenesis of non–small cell lung cancer (NSCLC). Preclinical studies have shown that several IGF-IR inhibitors suppress the proliferation and survival of human cancer cells (Yap et al., 2011). The findings provide a new potential approach for IGF-IR target therapy because miR-143 negatively regulates both IGF-IR and IRS1. Moreover, as approximately 50% of patients with NSCLC will develop systemic metastasis that requires tumor angiogenesis (D’Amico, 2004), the antiangiogenic effect of miR-143 may benefit the treatment of NSCLC patients. In conclusion, this study provides new mechanism of heavy metal–induced lung carcinogenesis. The repression of miR-143 plays a causal role in Cr (VI)–induced cell transformation and tumor angiogenesis through directly targeting IGF-IR/IRS1/ERK/IL-8 pathway.
FUNDING
National Natural Science Foundation of China (81071642, 30871296); National Cancer Institute; National Institutes of Health (R01CA109460).
ACKNOWLEDGMENTS
The authors disclose no potential conflicts of interest.
REFERENCES
- Ali A. H., Kondo K., Namura T., Senba Y., Takizawa H., Nakagawa Y., Toba H., Kenzaki K., Sakiyama S., Tangoku A. (2011). Aberrant DNA methylation of some tumor suppressor genes in lung cancers from workers with chromate exposure. Mol. Carcinog. 50, 89–99 [DOI] [PubMed] [Google Scholar]
- Azad N., Iyer A., Vallyathan V., Wang L., Castranova V., Stehlik C., Rojanasakul Y. (2010). Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann. N. Y. Acad. Sci. 1203, 1–6 [DOI] [PubMed] [Google Scholar]
- Balansky R. M., D’Agostini F., Izzotti A., De Flora S. (2000). Less than additive interaction between cigarette smoke and chromium(VI) in inducing clastogenic damage in rodents. Carcinogenesis. 21, 1677–1682 [DOI] [PubMed] [Google Scholar]
- Baserga R. (2006). Building a cathedral. Cancer Biol. Ther. 5, 240–242 [DOI] [PubMed] [Google Scholar]
- Bollati V., Marinelli B., Apostoli P., Bonzini M., Nordio F., Hoxha M., Pegoraro V., Motta V., Tarantini L., Cantone L., et al. (2010). Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ. Health Perspect. 118, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat D. J., Bellail A. C., Van Meir E. G. (2005). The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro. Oncol. 7, 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. D., Kargacin B., Klein C. B., Costa M. (1993). Mechanisms of chromium carcinogenicity and toxicity. Crit. Rev. Toxicol. 23, 255–281 [DOI] [PubMed] [Google Scholar]
- Costa A. N., Moreno V., Prieto M. J., Urbano A. M., Alpoim M. C. (2010). Induction of morphological changes in BEAS-2B human bronchial epithelial cells following chronic sub-cytotoxic and mildly cytotoxic hexavalent chromium exposures. Mol. Carcinog. 49, 582–591 [DOI] [PubMed] [Google Scholar]
- Costa M., Klein C. B. (2006). Toxicity and carcinogenicity of chromium compounds in humans. Crit. Rev. Toxicol. 36, 155–163 [DOI] [PubMed] [Google Scholar]
- D’Amico T. A. (2004). Angiogenesis in non-small cell lung cancer. Semin. Thorac. Cardiovasc. Surg. 16, 13–18 [DOI] [PubMed] [Google Scholar]
- Das A. P., Singh S. (2011). Occupational health assessment of chromite toxicity among Indian miners. Indian J. Occup. Environ. Med. 15, 6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N., Paul S., Chatterjee D., Banerjee N., Majumder N. S., Sarma N., Sau T. J., Basu S., Banerjee S., Majumder P., et al. (2012). Arsenic exposure through drinking water increases the risk of liver and cardiovascular diseases in the population of West Bengal, India. BMC Public Health. 12, 639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J., Merler E., Abernathy C., Williams G. (1971). Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 133, 275–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Xu Q., Jing Y., Agani F., Qian X., Carpenter R., Li Q., Wang X. R., Peiper S. S., Lu Z., et al. (2012). Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 13, 1116–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A. L. Wise S. S. Wise J. P.,Sr (2008). Carcinogenicity of hexavalent chromium. Indian J. Med. Res. 128, 353–372 [PubMed] [Google Scholar]
- Ishikawa Y., Nakagawa K., Satoh Y., Kitagawa T., Sugano H., Hirano T., Tsuchiya E. (1994). Characteristics of chromate workers’ cancers, chromium lung deposition and precancerous bronchial lesions: An autopsy study. Br. J. Cancer. 70, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z. Y., He Z., King B. L., Kuroki T., Opland D. M., Suzuma K., Suzuma I., Ueki K., Kulkarni R. N., Kahn C. R., et al. (2003). Characterization of multiple signaling pathways of insulin in the regulation of vascular endothelial growth factor expression in vascular cells and angiogenesis. J. Biol. Chem. 278, 31964–31971 [DOI] [PubMed] [Google Scholar]
- Karapetsas A., Giannakakis A., Pavlaki M., Panayiotidis M., Sandaltzopoulos R., Galanis A. (2011). Biochemical and molecular analysis of the interaction between ERK2 MAP kinase and hypoxia inducible factor-1α. Int. J. Biochem. Cell Biol. 43, 1582–1590 [DOI] [PubMed] [Google Scholar]
- Kent O. A., Chivukula R. R., Mullendore M., Wentzel E. A., Feldmann G., Lee K. H., Liu S., Leach S. D., Maitra A., Mendell J. T. (2010). Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 24, 2754–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. S., Rajagopal V., Gonsalves C., Johnson C., Kalra V. K. (2006). A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. 177, 7211–7224 [DOI] [PubMed] [Google Scholar]
- Lee Y. H., White M. F. (2004). Insulin receptor substrate proteins and diabetes. Arch. Pharm. Res. 27, 361–370 [DOI] [PubMed] [Google Scholar]
- Levy L. S., Martin P. A., Bidstrup P. L. (1986). Investigation of the potential carcinogenicity of a range of chromium containing materials on rat lung. Br. J. Ind. Med. 43, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. H., Yu M. C., Chiang C. C., Bien M. Y., Chien M. H., Chen B. C. (2013). Thrombin-induced NF-κB activation and IL-8/CXCL8 release is mediated by c-Src-dependent Shc, Raf-1, and ERK pathways in lung epithelial cells. Cell. Signal. 25, 1166–1175 [DOI] [PubMed] [Google Scholar]
- Liu X., Sempere L. F., Guo Y., Korc M., Kauppinen S., Freemantle S. J., Dmitrovsky E. (2011). Involvement of microRNAs in lung cancer biology and therapy. Transl. Res. 157, 200–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S., Ghoshal K., Summers D., Bai S., Datta J., Jacob S. T. (2003). Chromium(VI) down-regulates heavy metal-induced metallothionein gene transcription by modifying transactivation potential of the key transcription factor, metal-responsive transcription factor 1. J. Biol. Chem. 278, 26216–26226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medan D., Luanpitpong S., Azad N., Wang L., Jiang B. H., Davis M. E., Barnett J. B., Guo L., Rojanasakul Y. (2012). Multifunctional role of Bcl-2 in malignant transformation and tumorigenesis of Cr(VI)-transformed lung cells. PLoS One. 7, e37045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaida N., Okamoto S., Ishikawa Y., Matsushima K. (1994). Molecular mechanism of interleukin-8 gene expression. J. Leukoc. Biol. 56, 554–558 [PubMed] [Google Scholar]
- Myers M. G., Jr., Sun X. J., Cheatham B., Jachna B. R., Glasheen E. M., Backer J. M., White M. F. (1993). IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3’-kinase. Endocrinology. 132, 1421–1430 [DOI] [PubMed] [Google Scholar]
- Pastides H., Austin R., Lemeshow S., Klar J., Mundt K. A. (1994). A retrospective-cohort study of occupational exposure to hexavalent chromium. Am. J. Ind. Med. 25, 663–675 [DOI] [PubMed] [Google Scholar]
- Rath S., Ziesemer S., Witte A., Konkel A., Muller C., Hildebrandt P., Volker U., Hildebrandt J. P. (2013). S. aureus hemolysin A-induced IL-8 and IL-6 release from human airway epithelial cells is mediated by activation of p38- and Erk-MAP kinases and additional, cell-type specific signalling mechanisms. [published online ahead of print January 24, 2013] Cell Microbiol. [DOI] [PubMed] [Google Scholar]
- Roa W., Brunet B., Guo L., Amanie J., Fairchild A., Gabos Z., Nijjar T., Scrimger R., Yee D., Xing J. (2010). Identification of a new microRNA expression profile as a potential cancer screening tool. Clin. Invest. Med. 33, E124 [DOI] [PubMed] [Google Scholar]
- Rodrigues C. F., Urbano A. M., Matoso E., Carreira I., Almeida A., Santos P., Botelho F., Carvalho L., Alves M., Monteiro C., et al. (2009). Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat. Res. 670, 42–52 [DOI] [PubMed] [Google Scholar]
- Siegel R., Ward E., Brawley O., Jemal A. (2011). Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA. Cancer J. Clin. 61, 212–236 [DOI] [PubMed] [Google Scholar]
- Taniguchi C. M., Emanuelli B., Kahn C. R. (2006). Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- Ugras S., Brill E., Jacobsen A., Hafner M., Socci N. D., Decarolis P. L., Khanin R., O’Connor R., Mihailovic A., Taylor B. S., et al. (2011). Small RNA sequencing and functional characterization reveals MicroRNA-143 tumor suppressor activity in liposarcoma. Cancer Res. 71, 5659–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach S., Nolte E., Szczyrba J., Stöhr R., Hartmann A., Ørntoft T., Dyrskjøt L., Eltze E., Wieland W., Keck B., et al. (2012). MicroRNA profiles of prostate carcinoma detected by multiplatform microRNA screening. Int. J. Cancer. 130, 611–621 [DOI] [PubMed] [Google Scholar]
- Wang D., Stockard C. R., Harkins L., Lott P., Salih C., Yuan K., Buchsbaum D., Hashim A., Zayzafoon M., Hardy R. W., et al. (2008). Immunohistochemistry in the evaluation of neovascularization in tumor xenografts. Biotech. Histochem. 83, 179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. L., Jiang B. H., Rue E. A., Semenza G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A. 92, 5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Son Y. O., Chang Q., Sun L., Hitron J. A., Budhraja A., Zhang Z., Ke Z., Chen F., Luo J., et al. (2011a). NADPH oxidase activation is required in reactive oxygen species generation and cell transformation induced by hexavalent chromium. Toxicol. Sci. 123, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zhao Y., Smith E., Goodall G. J., Drew P. A., Brabletz T., Yang C. (2011b). Reversal and prevention of arsenic-induced human bronchial epithelial cell malignant transformation by microRNA-200b. Toxicol. Sci. 121, 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff T. J., Axelrad D. A., Caldwell J., Morello-Frosch R., Rosenbaum A. (1998). Public health implications of 1990 air toxics concentrations across the United States. Environ. Health Perspect. 106, 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A., Chen J., Baserga R. (2008). The role of insulin receptor substrate-1 in the oncogenicity of simian virus 40 T antigen. Cell Cycle. 7, 1999–2002 [DOI] [PubMed] [Google Scholar]
- Wu B. L., Xu L. Y., Du Z. P., Liao L. D., Zhang H. F., Huang Q., Fang G. Q., Li E. M. (2011). MiRNA profile in esophageal squamous cell carcinoma: Downregulation of miR-143 and miR-145. World J. Gastroenterol. 17, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap T. A., Olmos D., Molife L. R., de Bono J. S. (2011). Targeting the insulin-like growth factor signaling pathway: Figitumumab and other novel anticancer strategies. Expert Opin. Investig. Drugs. 20, 1293–1304 [DOI] [PubMed] [Google Scholar]
- Zhu H., Dougherty U., Robinson V., Mustafi R., Pekow J., Kupfer S., Li Y. C., Hart J., Goss K., Fichera A., et al. (2011). EGFR signals downregulate tumor suppressors miR-143 and miR-145 in Western diet-promoted murine colon cancer: Role of G1 regulators. Mol. Cancer Res. 9, 960–975 [DOI] [PMC free article] [PubMed] [Google Scholar]