

Abstract
Aims: Activation of specific signaling pathways in response to mechanical trauma causes delayed neuronal apoptosis; GSK-3β/β-catenin signaling plays a critical role in the apoptosis of neurons in CNS diseases, SGK was discovered as a regulator of GSK-3β/β-catenin pathway, The goal of this study was to determine if the mechanism of cell death or survival mediated by the SGK/GSK-3β/β-catenin pathway is involved in a rat model of TBI. Main methods: Here, an acute traumatic brain injury model was applied to investigate the expression change and possible roles of SGK, Expression of SGK, and total-GSK-3β, phospho-GSK3β on ser-9, beta-catenin, and caspase-3 were examined by immunohistochemistry and Western blot analysis. Double immunofluorescent staining was used to observe the SGK localizations. Si-RNA was performed to identify whether SGK regulates neuron apoptosis via GSK-3β/β-catenin pathway, ultimately inhibit caspase-3 activation. Key findings: Temporally, SGK expression showed an increase pattern after TBI and reached a peak at day 3. Spatially, SGK was widely expressed in the neuron, rarely in astrocytes and oligodendrocytes; in addition, the expression patterns of active caspase-3 and phospho-GSK3β were parallel with that of SGK, at the same time, the expression of β-catenin shows similarity with SGK. In vitro, to further investigate the function of SGK, a neuronal cell line PC12 was employed to establish an apoptosis model. We analyzed the association of SGK with apoptosis on PC12 cells by western blot, immunofluorescent labeling and siRNA. Significance: the results implied that SGK plays an important role in neuron apoptosis via the regulation of GSK3β/β-catenin signaling pathway; ultimately inhibit caspase-3 activation. Taken together, we inferred traumatic brain injury induced an upregulation of SGK in the central nervous system, which show a protective role in neuron apoptosis.
Keywords: SGK, GSK3β/β-catenin signaling pathway, traumatic brain injury (TBI), neuron apoptosis
Introduction
Traumatic brain injury (TBI) is one of the leading causes of death and disability worldwide, including the developing world [1]. Traumatic brain injury is an insult to the brain caused by an external physical force, resulting in functional disability [2]. Both clinical and experimental studies have shown that the pathophysiology of traumatic brain injury (TBI) is complex and involves both primary and secondary injuries [3]. Primary damage occurs at the moment of insult and includes contusion and laceration, diffuse axonal injury, and intracranial hemorrhage; Secondary damage includes processes that are initiated at the time of insult, but do not appear clinically for hours or even days after injury. It can subsequently trigger astrocyte proliferation, microglia activation and neuronal cell death [4]. Although large amount of clinical and basic researches focus on TBI, there are limited methods for improving the outcome of patients suffering from brain trauma [5,6]. Both anti-apoptotic and pro-apoptotic signaling cascades are activated in secondary tissue injury [7]. In addition to causing direct mechanical injury, trauma initiates secondary cascades of biochemical and cellular changes that substantially contribute to subsequent tissue damage and related neurological deficits.
Activation of specific signaling pathways in response to mechanical trauma causes delayed neuronal apoptosis [8]. One pathway with a prominent role in neurotrauma is the signaling pathway in which the enzyme glycogen synthase kinase 3β (GSK3β) is a key component. Glycogen synthase kinase-3β (GSK3β) activation promotes cell death [9-12] and inhibits cell proliferation, a growing body of evidence suggests that GSK3β is an important modulator in central nervous system diseases, including traumatic brain injury and AD (Alzheimer’s disease) [13], Phosphorylation GSK3β on serine-9 to render it inactive, a mechanism by which neurons become resistant to apoptotic stimuli [14], which makes it becomes the focus for its role in neuron protection, furthermore, Neuroprotective stimuli lead to an inactivation of GSK3β. Prominent in this latter category is the PI3K/Akt pathway. Thus, GSK3β activity appears to correlate inversely with neuronal viability [15]. β-catenin, which is central to the Wnt signaling pathway involved in many stages of development, is a GSK3β substrate. GSK3β-mediated phosphorylation enhances the proteasome-dependent degradation of β-catenin. The inhibition of GSK-3β caused dramatic elevations in the level of β-catenin and stimulated β-catenin-dependent gene transcription that regulate neuronal homeostasis and support neuron cell survival.
GSK-3β/β-catenin pathway can be regulated by many signaling pathways and proteins, Wnt and Akt (also called protein kinase B) are two major signaling pathways that have been shown to regulate GSK-3β activity via distinct mechanisms. Additionally, the recent studies have shown the Serum- and glucocorticoid-regulated kinase (SGK), known as SGK1, was discovered as a regulator of GSK-3β/β-catenin pathway, which is involved in the pathophysiology process of tumor growth, fibrosing disease, ischemia, neurodegeneration, and traumatic brain injury [16]. Previous investigations have demonstrated that SGK phosphorylates GSK3β on serine-9 and then controls β-catenin dynamics, further takes part in the process of tight junction formation in mammary epithelial tumor cells and in the regulation of L-selectin and perforin expression as well as activation induced cell death of T-lymphocytes.
In addition, Many reports have shown that SGK mRNA and protein level upregulated after traumatic brain injury which particularly abundant in the central nervous system and neuron-specific, and play a protective role in the regulation of neuronal function [17-19]. But the inherent role is still little known.
Given the roles of GSK-3β/β-catenin pathway and SGK in central nervous system especially in neuron apoptosis and the mutant relationship between them, accordingly, we speculated whether the SGK is involved in the neuronal survival via GSK-3β/β-catenin signaling and its action through the downstream targets, especially β-catenin, after TBI.
Thus the present study was designed to investigate the changes of SGK, phospho-GSK3β/β-catenin and their roles in the regulation of caspase-3 (the apoptosis marker) in a TBI model after 3 days. Moreover, the siRNA was used to confirm the possible roles of SGK regulate neuron apoptosis via GSK3β/β-catenin signaling pathway.
Materials and methods
Models of TBI
All protocols using animals were conducted in accordance with the guidelines published in the NIH Guide for the care and use of laboratory animals and the principles presented in the Guidelines for the use of animals in neuroscience research by the Society for Neuroscience and approved by Nantong University Animal Care Ethics Committee. Male Sprague-Dawley Rats (n=48) with an average body weight of 250 g (220 ± 275 g) were used in this experiment. Unilateral controlled cortical injury was performed as previously described [20,21]. In brief, adult male were anesthetized with Ketamine (90 mg/kg)/xylazine (10 mg/kg), and surgery was performed under aseptic conditions. An anteroposterior surgical incision (5-mm-long, 3-mm-deep, and 1-mm-wide) was made by inserting a microknife into the right cortex.
3 mm lateral from the midline (n=42). Controlled rats (n=6) underwent identical procedures to experimental animals, but did not receive brain injury. Ketoprofen (5 mg/kg) was administered to minimize postsurgical pain and discomfort. The overlying muscles and skin were closed in layers with 4-0 silk sutures and staples, and the animals were allowed to recover on a 30°C heating pad. Animals were individually housed in cages and kept in a temperature-controlled environment (21°C) on a 12-hr light-dark cycle, with access to food and water ad libitum. animals were killed at 12 h, 1 d, 3 d, 5 d, 7 d, 14 d, and 28 d after injury, and sham-operated rats (n=3) were sacrificed at 3 days. All efforts were made to minimize the number of animals used and their suffering.
Cell cultures and treatment
PC12 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% (v/v) fetal bovine serum, 5% donor horse serum and antibiotics at 37°C under 5% CO2 in humidified air. The cells were passed every 3-4 days. In order to study apoptosis, cells were seeded onto a poly-l-lysine-coated 60 mm dishes and incubated in a low concentration of serum (1% horse serum) for 24 hours prior to treatment with H2O2 (300 nmol/L) for different time.
siRNA and transfection
Primer pairs for the SGK (NM_001292567.1) siRNA expression vector was target the sequence: 5’-CAAGGACCUAGCCGCACAA-3’. For transient transfection, the SGK siRNA vector, and the non-specific vector were carried out using lipofectamine 2,000 (Invitrogen) and plus reagent in OptiMEM (Invitrogen) as suggested by the manufacturer. Transfected cells were used for the subsequent experiments 48 h after transfection.
Western blot analysis
Western blots were prepared from normal brain cortex or from injured cortex at 12 h-28 days, to obtain samples for Western blotting, rats were sacrificed at different time points post-operatively (n=3 for each time point), the brain tissue surrounding the wound (extending 2 mm to the lesion site, weighing 70-90 mg) as well as an equal part of the contralateral, unoperated cortex were dissected out and immediately frozen at -70°C until use. To prepare lysates, frozen brain tissue samples were minced with eye scissors in ice. The samples were then homogenized in lysis buffer (1% NP-40, 50 mmol/l Tris, and pH 7.5, 5 mmol/l EDTA, 1% SDS, 1% sodium deoxycholate, 1% Triton X-100, 1 mmol/l PMSF, 10 μg/ml aprotinin, and 1 μg/ml leupeptin) and clarified by centrifuging for 20 min in a microcentrifuge at 4°C. After determination of its protein concentration with the Bradford assay (Bio-Rad), the resulting supernatant (50 μg of protein) was subjected to SDS-polyacrylamide gel electrophoresis (PAGE). The separated proteins were transferred to a polyvinylidene difluoride membrane (Millipore) by a transfer apparatus at 350 mA for 1.5 h. The membrane was then blocked with 5% nonfat milk and incubated with primary antibody against SGK (anti-rabbit, 1:500; Santa Cruz), p-GSK3β, β-catenin (anti-mouse, 1:1,000; Cell Signaling), or GAPDH (anti-rabbit, 1:1,000; Santa Cruz), GFAP (anti-mouse, 1:1,000; Cell Signaling; anti-rabbit, 1:1,000; Santa Cruz). After incubating with an anti-rabbit horseradish peroxidase-conjugated secondary antibody, protein was visualized using an enhanced chemiluminescence system (ECL, Pierce Company, USA).
Immunofluorescence staining
After defined survival times, rats were terminally anesthetized and perfused through the ascending aorta with saline, followed by 4% paraformaldehyde at different survival times (n=3 per time point). The brains were removed and postfixed in the same fixative for 3 hours and then replaced with 20% sucrose for 2-3 days, following 30% sucrose for 2-3 days. After treatment with sucrose solution, the tissues were embedded in OCT compound. Then, 10-μm frozen cross-sections were prepared and examined. All sections were first blocked with 10% normal serum blocking solution-species the same as the secondary antibody, containing 3% (w/v) bovine serum albumin (BSA) and 0.1% Triton X-100 and 0.05% Tween-20 2 h at room temperature in order to avoid unspecific staining. Then the sections were incubated with both rabbit polyclonal primary antibodies for anti-SGK (1:200; Santa Cruz), goat polyclonal primary antibodies for anti-caspase-3 (1:200; Cell Signaling) and mouse monoclonal primary antibodies anti-GFAP (a marker of astrocytes, 1:200; Sigma), anti-NeuN (a marker of neuron, 1:600; Chemicon); Briefly, sections were incubated with both primary antibodies overnight at 4°C, followed by a mixture of FITC- and TRITC-conjugated secondary antibodies for 2 h at 4°C. In sections from each specimen, the primary antibody was omitted to assess for nonspecific binding of the secondary antibody. The stained sections were examined with a Leica fluorescence microscope (Germany).
Immunohistochemistry
After the sections were prepared, they were blocked with 10% goat serum with 0.3% Triton X-100 and 1% BSA for 2 h at room temperature and incubated overnight at 4°C with anti-SGK antibody (rabbit, 1:100; Santa Cruz), followed by incubation in biotinylated secondary antibody (Vector Laboratories, Burlingame, CA). Sections were rinsed again for 5 min (three times) and incubated in the complex avidin-biotin-peroxidase (ABC Kit, Vector Laboratories, Burlingame, CA, USA) for 40 min at 37°C. Staining was visualized with diaminobenzidine (DAB, Vector Laboratories). After reactions, the sections were dehydrated, cleared, and cover slipped. Slides were examined at 10× or 40× magnifications on Leica light microscope (Germany). Cells with strong or moderate brown staining were counted as positive, cells with no staining were counted as negative, whereas cells with weak staining were scored separately.
RNA isolation and reverse transcriptase PCR (RT-PCR) analysis
Total RNA of brain cortex was extracted using Trizol extraction kit according to the manufacturer’s protocol. Total RNA was reverse-transcribed using ThermoScript RT-PCR system (Invitrogen, USA). Primer pairs for SGK: Sense: 5′-CGG AAT TCA CCG TCA AAA CCG AGG CTCG-3′ and Antisense: 5′-GCT CTA GAT CAG AGG AAG GAG TCC ATAGG-3′. Cycling parameters were: 94°C for 45 s, 55°C for 45 s, 72°C for 30 s, and total 30 cycles. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control and was detected using the following primers: Sense, 5′-TGA TGA CAT CAA GAA GGT GGT GAAG-3′; Antisense: 5′-TCC TTG GAG GCC ATG TGG GCCAT-3′. Cycling parameters were: 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and total 28 cycles. After amplification, the products were separated on an agarose (1.5%) gel (cast in the presence of ethidium bromide) and visualized under UV light.
Quantitative analysis
Cells double labeled for SGK, NeuN in the experiment were quantified. Sections were double labeled for SGK with NeuN. To identify the proportion of NeuN positive cells expressing SGK, a minimum of 200 NeuN positive cells were counted adjacent to the wound in each section. Then double labeled cells for SGK and NeuN were recorded. Two or three adjacent sections per animal were sampled.
Statistical analysis
All data were analyzed with Stata 7.0 statistical software. All values are expressed as means ± SEM. The statistical significance of differences between groups was determined by one-way analysis of variance (ANOVA) followed by Turkey’s post-hoc multiple comparison tests. P<0.05 was considered statistically significant. Each experiment consisted of at least three replicates per condition.
Results
SGK is upregulated in animal model of traumatic brain injury
Previous study has shown that SGK is strongly induced in the brains of mice after TBI. In this study, we extended analysis to investigate the time course of SGK. The protein level was relatively lower in normal cortex, then progressively increased from 12 h after TBI, peaked at day 3 (P<0.05), and then gradually decreased to normal level (Figure 1A, 1B). In order to further confirm the protein change, an immunohistochemistry study showed that SGK expression significantly increased in the ipsilateral brain cortex at day 3 after TBI (Figure 2A, 2C, 2F) compared to the untreated brain cortex (Figure 2B, 2D). Error bars represent SEM. Scale bars: 10 μm (Figure 2C-E), 50 μm (Figure 2A, 2B). E was showed as the negative control that the primary antibody was omitted in the staining protocol. Based on the results, we concluded that SGK protein change via transcriptional regulation after TBI.
Figure 1.
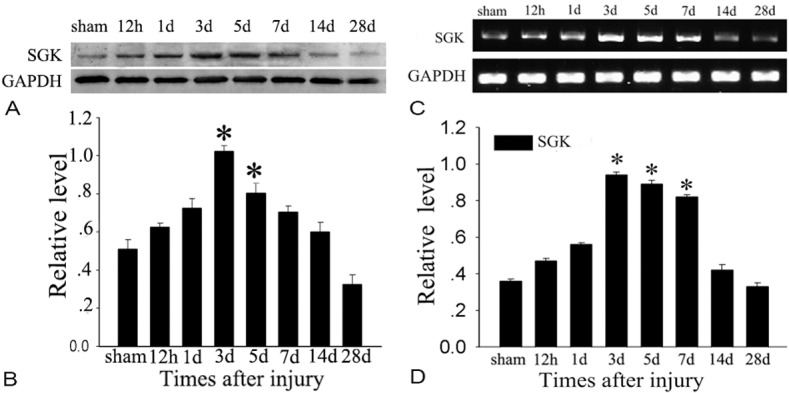
Detection of mRNA level and protein level of SGK change after TBI. Western blot was performed to study the protein level of SGK in the cortex surrounding the wound and sham-unoperated cortex at various survival times after TBI. A: Time courses of SGK expression in ipsilateral brain cortex after TBI. B: Quantification graphs (relative optical density) of the intensity of staining of SGK to GAPDH at each time point. GAPDH was used to confirm equal amount of protein was run on gel. The data are means ± SEM. (n=3, *P<0.01, significantly different from the sham-operated group). The expression of SGK at mRNA level was also detected before and after injury. C: Time courses of mRNA level of SGK expression in ipsilateral brain cortex after TBI. D: The bar chart shows the ratio of SGK mRNA to GAPDH mRNA. The data are means ± SEM. (n=3, *P<0.01, significantly different from the sham-operated group).
Figure 2.
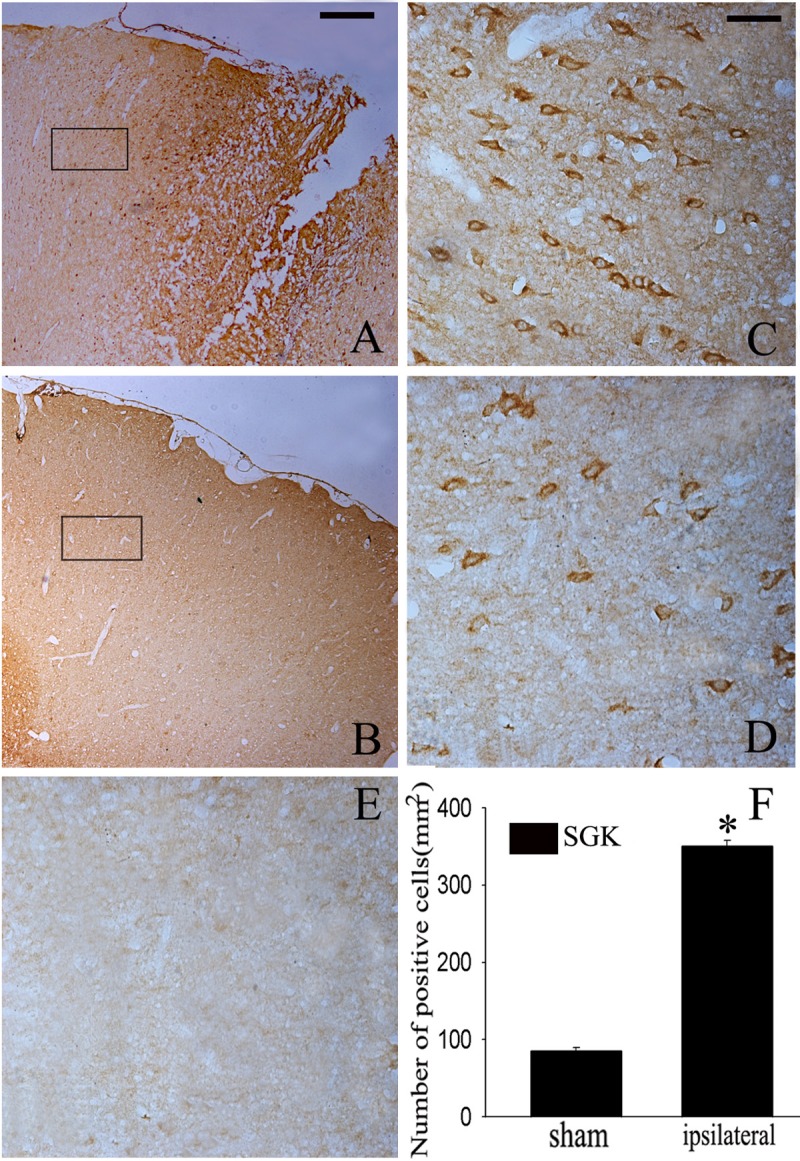
Representative Microphotographs for SGK immunohistochemistry in the rat brain cortex after TBI. SGK staining was mainly observed in the cortex surrounding the wound (A, C). SGK-IR was also detected in the sham-controlled (B, D), Quantitative analysis of SGK positive cells in brain cortex 3 day after injury (F). SGK staining was largely increased in the ipsilateral brain cortex at SGK day 3 after TBI. *indicates significant difference at P<0.05 compared with sham group. Error bars represent SEM. Scale bar: left column, 50 μm (A, B), right columns, and 10 μm (C-E).
TBI-induced SGK mRNA upregulation correlates with an increase in SGK protein level
The translation of differential gene expression into protein changes is one prerequisite for a role of this regulation in the pathogenesis of a disease. Therefore, we analyzed the level of SGK protein in untreated mice and mice treated with the acute brain injury. In this study, the SGK protein level also underwent a similar expression pattern as SGK mRNA level following rat brain injury (Figure 1C, 1D).
The detection of SGK with different cellular markers in the adult rat brain cortex after TBI
To further determine the cell types expressing SGK around the lesion site, we used double labeling immunofluorescent staining with cell-specific markers: NeuN (a marker of neurons), GFAP (a marker of astrocytes), CNPase (a marker of oligodendrocytes). The localization of SGK (red) in neurons (green) was confirmed by co-staining with anti-NeuN (Figure 3A-C). However, the absences of SGK (red) in astrocytes (green) and oligodendrocytes (green) were demonstrated by co-staining with anti-GFAP (Figure 3E-G) and anti-CNPase (Figure 3I-K), suggesting that SGK is not expressed within this cell population in the brain cortex. To investigate the proportion of NeuN-positive cells expressing SGK, a minimum of 200 NeuN-positive cells were counted between sham and day 3 after injury. SGK expression was increased significantly in neurons (P<0.05) at day 3 after injury compared with sham brain cortex (Figure 3O). This data suggest that the increase of SGK play a crucial role in neuron.
Figure 3.
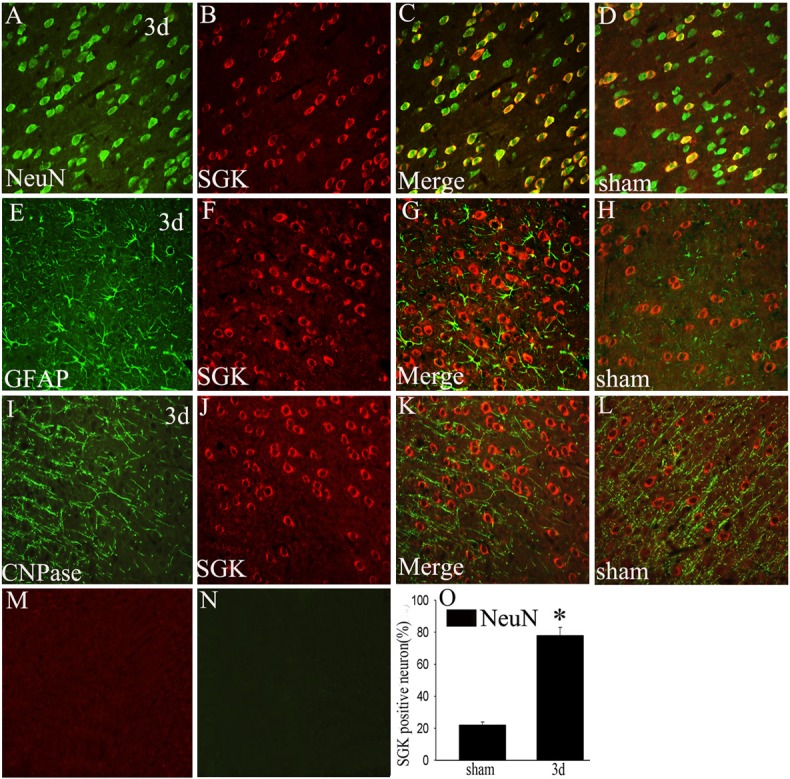
Co-localization of SGK and different phenotype-specific markers in brain cortex. In the adult rat brain cortex within 5 mm distance from the lesion site at day 3 after TBI, horizontal sections labeled with SGK (red, B, F, J) and different cell markers, such as neuron marker (green, A, NeuN), astrocyte marker (green, E, GFAP), oligodendrocyte marker (green, I, CNPase), The yellow color visualized in the merged images represented co-localization of SGK with different phenotype-specific markers (C, G, K), co-localizations of SGK with different phenotype-specific markers in the sham-operated group are shown in the brain cortex (D, H, L). M, N: Negative controls, (O) Quantitative analysis of NeuN-positive cells expressing SGK (%) in the sham-operated group and day 3 after injury. *indicate significant difference at P<0.01 compared with sham-operated group. Error bars represent SEM. Scale bars: 20 μm (A-L).
Dynamic change of GSK3β phosphorylation and β-catenin expression after TBI
Because SGK functions through phosphorylation and inhibition of GSK3β, we further examined the phosphorylation of GSK3β. Western blot analysis showed that phospho-GSK3β expression was significantly increased at 1, 3, 5 and 7 days in the cerebral cortex. GSK3β also influences a number of transcription factors that regulate the expression of anti-apoptotic proteins, such as β-catenin. These results indicate that phosphorylation of GSK3β and β-catenin is accelerated after TBI, and the timing and distribution change is similar to SGK (Figure 4).
Figure 4.
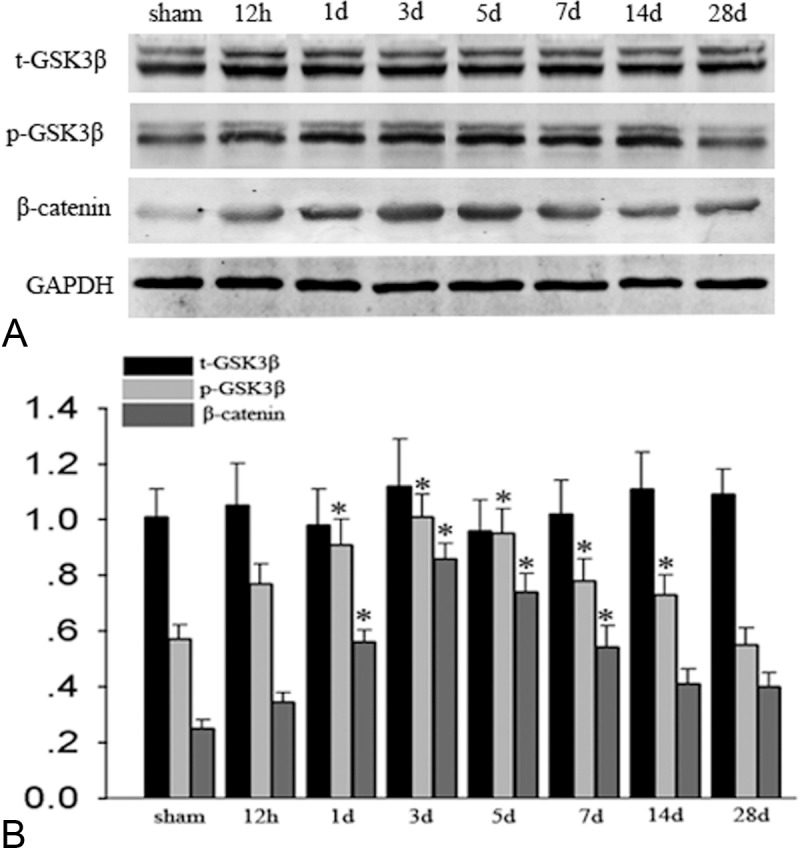
Phosphorylation of GSK3β on serine-9 and of β-catenin regulation after TBI. A: Western blot analysis showed that phospho-GSK3β (serine-9) expression increased significantly at 1 day and peaked at 3 days in the cerebral cortex. There was no prominent change in total GSK3β (t-GSK3β) (P>0.05). Results of the β-catenin analysis are shown here, it gradually increased at 1 day and also peaked at 3 days after TBI, GAPDH are shown as an internal control. B: The bar chart shows the ratio of protein level to GAPDH, *indicate significant difference (P<0.01).
Detection of SGK mRNA and protein level, GSK3β phosphorylation, β-catenin change in H2O2-induced PC12 cells apoptosis model
In vivo, we have demonstrated that phosphorylation of GSK3β and β-catenin is accelerated after TBI and the timing and distribution change is similar to SGK. Furthermore, we investigated spatial and temporal change in vitro. A H2O2-induced PC12 cells apoptosis model described as previously was employed here, the SGK mRNA and protein level increased at 6 h and peaked 9 h, additionally, and the phosphorylation GSK3β on serine-9 and β-catenin also show a similarity with it. These results indicate that the expression profile of which in vitro is consistent in vivo (Figure 5).
Figure 5.
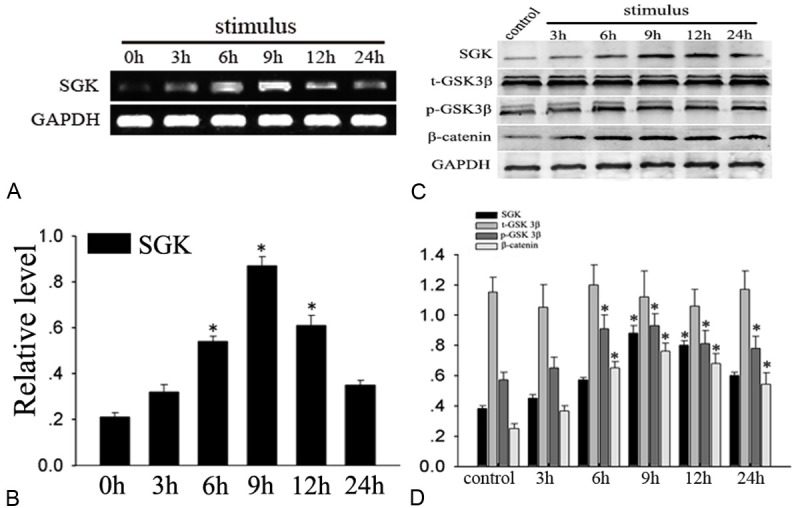
Detection of SGK mRNA and protein, total-GSK3β, phospho-GSK3β, β-catenin level in H2O2-induced apoptosis model in PC12. RT-PCR analyses of SGK mRNA expression, it is upregulated at 6 h and peaked at 9 h (*P<0.01), (C) Western blot analysis showed that SGK, phospho-GSK3β (p-GSK3β) and β-catenin expression significantly increased. There was no prominent change in total GSK3β (t-GSK3β) (P>0.05). B, D: Quantitative analysis, *indicate significant difference (P<0.01).
Downregulation of SGK increases cell death after TBI
Downregulation of a gene is a powerful tool for understanding its biological functions. First, we detected the mRNA and protein of SGK after siRNA, the results showed Interference effect is very significant, we also investigated the functional effects of RNAi-mediated silencing of SGK on neuron death processes. Among apoptosis pathway, caspase-3 is a key effecter. In this experiment, following administration of siRNA-SGK, phospho-GSK3β and β-catenin was significantly reduced in the siRNA group which markedly different from that in the non-specific group (P<0.01). However, caspase-3 is markedly upregulated, which was dramatically difference from that in the non-specific group (P<0.01). Furthermore, double immunofluorescence was applied to explore the relationship between SGK and neuron apoptosis, as shown in Figure 6. After H2O2 stimulation, SGK and caspase-3 were significantly expressed compared with control group, however, in the siRNA group, SGK can rarely be detected, caspase-3 were higher than H2O2 stimulated group. Additionally, DAPI staining indicated that silencing SGK promoted nuclear condensation and perinuclear apoptotic bodies after H2O2 treatment in PC12 cells than H2O2 stimulated group and control group. Based on our data, we conclude that SGK plays a protective role in neuron apoptosis (Figure 6).
Figure 6.
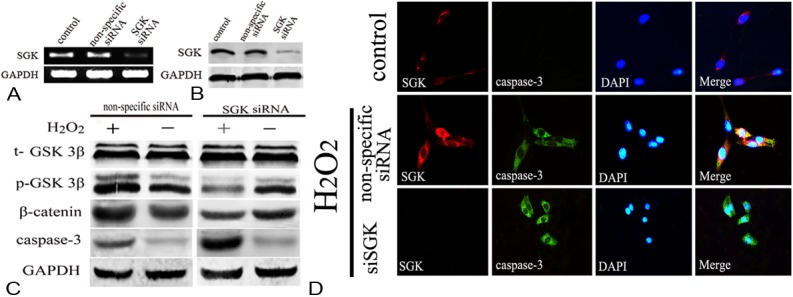
Down regulation of SGK and the relationship between of SGK/GSK3β signaling pathway and apoptosis. A, B: After knocking down SGK, the mRNA and protein expression significantly decreased (P<0.01). C: Down regulation of SGK, the p-GSK3β and β-catenin expression significantly decreased compared to the nonspecific group, however, the caspase-3 show a significant increase (P<0.01). D: Double-immunofluorescent staining for SGK (red) and caspase-3 (green) 9 hours after H2O2 stimulation. Nuclei were counterstained with DAPI (blue).
Discussion
In the present study, we evaluated the relation between SGK and GSK3β/β-catenin signaling pathway after acute brain injury. Interestingly, SGK, phosphorylation of GSK3β (serine-9), caspase-3 and β-catenin were correlated with the severity of brain injury. In the cerebral cortex, where acute brain injury was the most severe, those proteins were accelerated in the early phase and peaked at day 3 after TBI, gradually decreased to normal level, in addition, Immunofluorescent Staining suggests SGK just show co-localization with neuron, not in astrocytes and microgliocyte. After knocking down SGK, we founded that SGK shows a protective role in neuron apoptosis. These results suggest that the timing and distribution of SGK might largely depend on the severity of the stress caused by TBI and play an important role in neuron apoptosis.
GSK3, a serine/threonine protein kinase, is involved in a variety of fundamental physiological functions such as cell membrane signal-to-gene transcription/protein translation, neuronal polarity and cell survival or apoptosis [22,23]. There were two closely related isoforms, GSK3α and GSK3β [24,25], the GSK3β is neuron-specific in CNS and has the potential to be phosphorylated at Ser9 and Tyr216 [26]. Phosphorylation of GSK-3β at Ser9 was increased at the injury region of cerebral cortical after TBI [14,27], which induces the inactivation of GSK3β, and plays a critical role in the neuroprotection via inhibit the degradation of downstream substrates such as β-catenin [22], likewise, in our study, immunoreactivity of phospho-GSK3β at Ser9 increased after TBI, peaked at 3 days and sustained after 14 days, however, no change in expression of total GSK3β was observed in this model , which is consistent with results seen in brain injury in rats. Although mechanisms downstream of the GSK3β pathway are largely unknown, GSK3β mediated phosphorylation of β-catenin seems to enhance neuronal cell death. β-catenin, originally identified as a component of Wnt signaling pathway and cell-cell adherences junctions, plays an important role in cell proliferation, differentiation, polarity, morphogenesis and development [28]. An important function of β-catenin to the current study is its phosphorylation by GSK-3β, which leads to β-catenin’s stabilization and translocation to the nucleus where it interacts with the TCF/LEF transcription factors to induce gene expression, which encodes proteins that support cell survival [29]. Furthermore, over-expressing β-catenin reduced apoptosis similarly to the protection provided by inhibitors of GSK3 [30], Consistent with these findings, in this study, we founded the expression of β-catenin up-regulated at the tissue and cellular level, which shows an similar expression profile with phospho-GSK3β, this data suggesting that phosphorylation of GSK3β can regulate β-catenin expression after TBI, which show similarity with previous reports.
GSK3β/β-catenin pathway can be regulated by lots of serine/threonine protein kinases, such as Akt, SGK [31,32]. The serine-threonine kinase, Akt, plays an important role in the cell death/survival pathway [33]. Zhao S et al. have illustrated activation of Akt/GSK-3β/β-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats [27]. Interestingly, SGK contains a catalytic domain, which is most similar to Akt (also known as protein kinase B or PKB) [34,35], was originally identified as serum- and glucocorticoid-inducible kinase, and plays an important role in central nervous system diseases [17,36], previous study has shown that SGK mRNA was upregulated after brain injury [18], but it is how to play a role in the brain injury is still not clear. Additionally, Akt and SGK always cooperate in controlling GSK3β phosphorylation on serine-9 and β-catenin dynamics [37,38], in the light of the role of Akt/GSK3β/β-catenin signaling pathway in survival of neurons after traumatic brain injury in rats and the similarity of catalytic domain between them. So we speculate that SGK also plays an important role in CNS injury. In our study, we first detected the expression profile of SGK in TBI model at mRNA and protein level by RT-PCR, Western blot and immunohistology, we founded SGK mRNA and protein level were increased and peaked at day 3 after TBI, the results of double labeling indicates SGK might play a role in the neurons. In addition, we also checked the phospho-GSK3β on ser-9 and phosphorylation of β-catenin, we discovered phospho-GSK3β on ser-9 and the expression pattern of β-catenin show similarity with SGK, this phenomenon explained that SGK induces phospho-GSK3β on ser-9 upregulation and β-catenin stabilization, in vitro, a H2O2-induced neuronal-like PC12 cell apoptosis model was performed to confirm it. Accumulating evidence suggests that the GSK3β/β-catenin signaling pathway plays a crucial role in neuronal survival in several models of neurodegenerative diseases. The most well-defined regulatory mechanism is by up phosphorylation of serine-9 in GSK-3β [39], and correlates with an increase expression of the β-catenin [40], finally contributes to an inactivation of caspase-dependent pathway which induces neuron apoptosis in neurodegeneration disease [41]. We previously have certified the up-regulation of caspase-3 after brain injury [42]. In this experiment, we also tested the caspase-3 (an apoptosis marker of caspase-dependent pathway) expression, it shown the same expression profile with SGK and pho-GSK3β on ser-9, but whether the SGK is a pro-apoptotic and anti-apoptotic function after TBI? siRNA was employed to confirmed it, interestingly, after interfering the SGK expression, we discovered phospho-GSK3β on ser-9 decreased, phosphorylation of β-catenin and caspase-3 upregulated, moreover, immunofluorescent staining suggested knocking down SGK along with the rise of caspase-3, Additionally, DAPI staining indicated that silencing SGK promoted nuclear condensation and perinuclear apoptotic bodies after H2O2 treatment in PC12 cells than H2O2 stimulated group and control group. Based on our data, we concluded that SGK upregulation can be induced by traumatic brain injury at mRNA and protein level, which shows a protective role on neuron apoptosis via GSK3β/β-catenin signaling relates to caspase-3 inhibition.
This study aimed to define the role of SGK-dependent regulation of GSK3β/β-catenin pathway in the control of neuron apoptosis after TBI. Collectively, SGK is upregulated at mRNA and protein level after TBI and induced the phosphorylation of GSK3β on ser-9 and stabilized the β-catenin, siRNA-SGK leads to caspase-3 increase and show an anti-apoptotic role on neuron after brain injury. Our results have shown that SGK and GSK3β (Ser9) was accelerated in the injured cortex, and involved in neuronal survival after TBI. Moreover, neuroprotection of β-catenin against TBI was partly mediated by enhanced and persistent activation of the SGK/GSK3β signaling pathway. These findings suggest that activation of SGK is able to control GSK3β/β-catenin signaling pathway and finally inactivate the caspase-3 in neuron apoptosis.
Acknowledgements
This work was supported by the Natural Science Foundation of China (81070992); and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure of conflict of interest
The authors declare no conflict of interest associated with this work.
References
- 1.Reilly P. The impact of neurotrauma on society: an international perspective. Prog Brain Res. 2007;161:3–9. doi: 10.1016/S0079-6123(06)61001-7. [DOI] [PubMed] [Google Scholar]
- 2.Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol. 2012;3:60. doi: 10.3389/fphar.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochanek PM, Clark RS, Ruppel RA, Adelson PD, Bell MJ, Whalen MJ, Robertson CL, Satchell MA, Seidberg NA, Marion DW, Jenkins LW. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: Lessons learned from the bedside. Pediatr Crit Care Med. 2000;1:4–19. doi: 10.1097/00130478-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartings JA, Bullock MR, Okonkwo DO, Murray LS, Murray GD, Fabricius M, Maas AI, Woitzik J, Sakowitz O, Mathern B, Roozenbeek B, Lingsma H, Dreier JP, Puccio AM, Shutter LA, Pahl C, Strong AJ. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. 2011;10:1058–1064. doi: 10.1016/S1474-4422(11)70243-5. [DOI] [PubMed] [Google Scholar]
- 6.Andriessen TM, Horn J, Franschman G, van der Naalt J, Haitsma I, Jacobs B, Steyerberg EW, Vos PE. Epidemiology, severity classification, and outcome of moderate and severe traumatic brain injury: a prospective multicenter study. J Neurotrauma. 2011;28:2019–2031. doi: 10.1089/neu.2011.2034. [DOI] [PubMed] [Google Scholar]
- 7.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 8.Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 2008;28:1845–1859. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- 10.Jin Y, Sui HJ, Dong Y, Ding Q, Qu WH, Yu SX, Jin YX. Atorvastatin enhances neurite outgrowth in cortical neurons in vitro via up-regulating the Akt/mTOR and Akt/GSK-3beta signaling pathways. Acta Pharmacol Sin. 2012;33:861–872. doi: 10.1038/aps.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- 12.Tong N, Sanchez JF, Maggirwar SB, Ramirez SH, Guo H, Dewhurst S, Gelbard HA. Activation of glycogen synthase kinase 3 beta (GSK-3beta) by platelet activating factor mediates migration and cell death in cerebellar granule neurons. Eur J Neurosci. 2001;13:1913–1922. doi: 10.1046/j.0953-816x.2001.01572.x. [DOI] [PubMed] [Google Scholar]
- 13.Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 2009;33:193–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endo H, Nito C, Kamada H, Yu F, Chan PH. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke. 2006;37:2140–2146. doi: 10.1161/01.STR.0000229888.55078.72. [DOI] [PubMed] [Google Scholar]
- 15.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 16.BelAiba RS, Djordjevic T, Bonello S, Artunc F, Lang F, Hess J, Gorlach A. The serum- and glucocorticoid-inducible kinase Sgk-1 is involved in pulmonary vascular remodeling: role in redox-sensitive regulation of tissue factor by thrombin. Circ Res. 2006;98:828–836. doi: 10.1161/01.RES.0000210539.54861.27. [DOI] [PubMed] [Google Scholar]
- 17.Lang F, Strutz-Seebohm N, Seebohm G, Lang UE. Significance of SGK1 in the regulation of neuronal function. J Physiol. 2010;588:3349–3354. doi: 10.1113/jphysiol.2010.190926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imaizumi K, Tsuda M, Wanaka A, Tohyama M, Takagi T. Differential expression of sgk mRNA, a member of the Ser/Thr protein kinase gene family, in rat brain after CNS injury. Brain Res Mol Brain Res. 1994;26:189–196. doi: 10.1016/0169-328x(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 19.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Logan A, Frautschy SA, Gonzalez AM, Baird A. A time course for the focal elevation of synthesis of basic fibroblast growth factor and one of its high-affinity receptors (flg) following a localized cortical brain injury. J Neurosci. 1992;12:3828–3837. doi: 10.1523/JNEUROSCI.12-10-03828.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Wang Y, Cheng C, Chen Y, Shi S, Qin J, Xiao F, Zhou D, Lu M, Lu Q, Shen A. A relationship between p27(kip1) and Skp2 after adult brain injury: implications for glial proliferation. J Neurotrauma. 2010;27:361–371. doi: 10.1089/neu.2008.0581. [DOI] [PubMed] [Google Scholar]
- 22.Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- 23.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 25.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci U S A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S, Fu J, Liu X, Wang T, Zhang J, Zhao Y. Activation of Akt/GSK-3beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurol Res. 2012;34:400–407. doi: 10.1179/1743132812Y.0000000025. [DOI] [PubMed] [Google Scholar]
- 28.Zhu D, Kang Q, Huang PY, He TC, Xie P. Neurogenesis-related genes expression profiling of mouse fibroblastic stem cells induced by Wnt signaling. Neurol Res. 2009;31:200–203. doi: 10.1179/174313209X393915. [DOI] [PubMed] [Google Scholar]
- 29.Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 30.Yuan J, Zhang J, Wong BW, Si X, Wong J, Yang D, Luo H. Inhibition of glycogen synthase kinase 3beta suppresses coxsackievirus-induced cytopathic effect and apoptosis via stabilization of beta-catenin. Cell Death Differ. 2005;12:1097–1106. doi: 10.1038/sj.cdd.4401652. [DOI] [PubMed] [Google Scholar]
- 31.Bhavsar SK, Merches K, Bobbala D, Lang F. AKT/SGK-sensitive phosphorylation of GSK3 in the regulation of L-selectin and perforin expression as well as activation induced cell death of T-lymphocytes. Biochem Biophys Res Commun. 2012;425:6–12. doi: 10.1016/j.bbrc.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 32.Sakoda H, Gotoh Y, Katagiri H, Kurokawa M, Ono H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Fukushima Y, Abe M, Shojima N, Kikuchi M, Oka Y, Hirai H, Asano T. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J Biol Chem. 2003;278:25802–25807. doi: 10.1074/jbc.M301127200. [DOI] [PubMed] [Google Scholar]
- 33.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 34.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–5882. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 36.Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lubbert H, Stichel CC. Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci. 2005;30:249–264. doi: 10.1016/j.mcn.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 37.Siraskar B, Volkl J, Ahmed MS, Hierlmeier M, Gu S, Schmid E, Leibrock C, Foller M, Lang UE, Lang F. Enhanced catecholamine release in mice expressing PKB/SGK-resistant GSK3. Pflugers Arch. 2011;462:811–819. doi: 10.1007/s00424-011-1006-6. [DOI] [PubMed] [Google Scholar]
- 38.Foller M, Kempe DS, Boini KM, Pathare G, Siraskar B, Capuano P, Alesutan I, Sopjani M, Stange G, Mohebbi N, Bhandaru M, Ackermann TF, Judenhofer MS, Pichler BJ, Biber J, Wagner CA, Lang F. PKB/SGK-resistant GSK3 enhances phosphaturia and calciuria. J Am Soc Nephrol. 2011;22:873–880. doi: 10.1681/ASN.2010070757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho JH, Johnson GV. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem. 2004;279:54716–54723. doi: 10.1074/jbc.M403364200. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Mao H, Zou H, Jin W, Ni L, Ke K, Cao M, Shi W. Up-regulation of ski-interacting protein in rat brain cortex after traumatic brain injury. J Mol Histol. 2013;44:1–10. doi: 10.1007/s10735-012-9444-9. [DOI] [PubMed] [Google Scholar]