

Abstract
Papillary renal cell carcinoma (PRCC) is traditionally classified into type 1 and type 2. Recently, an oncocytic variant of PRCC has been described. We report a series of 6 oncocytic renal papillary tumors (OPRCC) which tended to occur in older patients (mean, 56.8 years) with a male preference (male-to-female ratio is 5:1). All 6 patients are alive with no evidence of disease after initial resection, showing an indolent clinical behavior. Histologically, tumors exhibited predominant papillary structure with delicate fibrovascular cores. Papillae were lined by single layers of cells with large, deeply eosinophilic and finely granular cytoplasms and round regular nucleus. The phagocytosis of tumor cells was frequently and evidently seen in our cases that hemosiderin-laden tumor cells and foamy tumor cells were noticed in five and four cases respectively. All tumors were immunoreactive for racemase, vimentin, CD10, and MET and negative for CD117. While E-cadherin, EMA, and cytokeratin 7 exhibited variable immunopositivity. FISH analysis was performed in five of six cases and found heterogeneous results. Trisomy of chromosomes 7 was found in three cases and trisomy of chromosomes 17 in two cases. Loss of chromosome Y was noted in one of four tumors in male patients. MET gene status was also investigated by direct sequencing in all 6 cases and found no distinct mutation in any case. These results suggest that OPRCC shows distinct morphology, indolent clinical behavior, and similar immunohistochemical and cytogenetic features with PRCC, seems to be a variant in the PRCC group. Whether the strong expression of MET indicates a potential therapeutic target is still unknown and requires further investigation in clinical trials.
Keywords: Kidney, papillary renal cell carcinoma, oncocytic tumors, MET
Introduction
Papillary renal cell carcinomas (PRCC) is a well-established subtype of RCC with characteristic gross and histological features and is further subdivided into 2 subtypes, type 1 and 2, for its distinct morphological feature and prognostic implications. Type 1 PRCC consist of small cells with low nuclear grade and a scant amount of cytoplasm arranged in a single layer, whereas type 2 PRCC tumor cells are larger, with abundant eosinophilic cytoplasm, higher nuclear grade, and nuclear pseudostratification. The two types of PRCC also demonstrate different clinical behavior. Patients with type 2 have a poorer prognosis than those with type 1 [1]. Therefore, accurate subtyping of PRCC is important for prognosis and proper patient management.
Recently, a new histopathologic variant of PRCC named oncocytic PRCC (OPRCC) has been described. It was first reported by Lefevre et al. in 2005 that 10 cases of RCC with the features of prominent papillary architecture, abundant granular eosinophilic cytoplasm and low-grade nonoverlapping nuclei [2]. These tumors exhibited histological features overlapping those of type 1 (low nuclear grade and a single layer) and type 2 (abundant eosinophilic cytoplasm) PRCC, and characterized by strong expression of CD10, vimentin, and AMACR. While none showed the genetic changes of trisomy 7 or 17, which were reported in more than 90% of type 1 and 70% of type 2 PRCC. Lefevre et al. regarded these tumors as an independent subtype of PRCC. After then, a few similar tumors have been reported as OPRCC, but showed heterogeneous clinicopathologic features. Their immunoreactivity seemed conflict and variable. And their cytogenetic data remained controversial that most cases showed trisomy of chromosome 7 and 17, while some cases did not [3-7]. In this article, we reported 6 such oncocytic papillary renal tumors. For these cases, we documented distinct histopathology, immunophenotype, molecular genetic features, and clinical behavior.
Materials and methods
Patients
We retrieved approximate 1500 RCCs between 1997 and 2011 from the files of Departments of Pathology at Nanjing Jingling Hospital (China) and selected 6 cases with the presence of both prominent papillary architecture and abundant oncocytic cytoplasm with low-grade nonoverlapping nuclei. The clinicopathologic features such as age, sex, disease histology, treatment, and the final follow-up dated from the time of initial diagnosis were recorded.
Immunohistochemistry
Tissues were fixed in 10% formalin and embedded in paraffin. Sections of 3-mm thickness were stained for hematoxylin and eosin and Prussian blue. Immunohistochemical analysis used the following antibodies: Vimentin (V9, Zymed, 1:200), racemase (P-504S, Zeta, Sierra Madre, 1:50), EMA (E29, DAKO, Glostrup, 1:1000), CK7 (OV-TL12/30, Zymed, 1:300), CD10 (56C6, Novocastra, 1:100), E-Cadherin (18-0223, Zymed, 1:100), CD117 (Polyclonal, Dako, 1:100), MET (24H2, Cell Signaling Technology, 1:100). Immunoreaction was performed using the labeled streptavidin-biotin method. Diaminobenzidine (3,3’-diaminobenzidine) was used for visualization. The interpretation of immunoreactivity was performed in a semiquantitative manner by analyzing the extent of the staining positivity of the tumor cells. The interpretation score was as follows: 0 or negative ≤ 5% tumor cell positivity; 1+ or focal = 5% to 10% tumor cell positivity; 2+ or moderate = 11% to 50% tumor cell positivity; and 3+ or diffuse > 50% tumor cell positivity.
MET mutation analysis
Genomic DNA were extracted from the formalin-fixed, paraffin-embedded tissue samples of the tumor and nonneoplastic tissue by the DNeasy Blood &; Tissue Kit (QIAgen, Hilden, Germany), according to the manufacturer’s protocol. In the composite tumors, genomic DNA of different components was extracted respectively from corresponding blocks or areas. The 14-21 exons of MET were amplified by PCR using the previously described primer pairs [8]. For sequence analysis, the PCR products were purified using the Wizard PCR Preps Purification System (Promega Corp.). Sequencing was performed using Big Dye Terminator and an ABI Basecaller (Applied Biosystems).
Fluorescence in situ hybridization
Deparaffinized slides containing tumor tissue were subjected to heat pretreatment (pressure cooking for 10 min at full pressure) in distilled water and then digested using 0.25% pepsin (Sigma, Taufkirchen, Germany) and 0.01 M HCl for 15 min at 37°C. After rinsing twice in 2 × SSC for 5 min, slides were dehydrated by immersing in 70%, 85%, and 100% ethanol for 1 min each at room temperature and then air-dried. Slides containing tissue DNA probe (10 μl/slide) were codenatured in an in situ thermocycler (Perkin Elmer, System 1000, Germany) at 88°C for 9 min, annealed at 37°C and hybridized in a humid chamber at 37°C overnight. After posthybridization washing in 0.4 × SSC (°C 2 min), and 2 × SSC (room temperature 2 min), slides were coverslipped by 10 ml of 4,6 diamino-2-phenylindole counterstain.
Spectrum Green centromeric probes (CEP) for chromosomes 7, 17, and Y (Vysis, Downers Grove, IL) were used. Each probe was diluted 1:30 in tDenHyb1 buffer (Insitus, Albuquerque, NM). Ten microliters of diluted probe was applied to each slide and cover slips were placed over the slides. Denaturation was achieved by incubating the slides at 83°C for 12 minutes in a humidified box, then hybridization was done at 37°C over night. The cover slips were then removed and the slides were immersed at room temperature in 0.5 × SSC for 2 minutes, in 50% formamide/1 × SSC for 5 minutes, and in 2 × SSC for 2 minutes. The slides were air-dried and counterstained with 10 lL DAPI/Antifade (DAPI in Fluorguard, 0.5 μg/mL, Insitus).
FISH evaluation
For each case, a minimum of 100 tumor nuclei were examined with probes by fl uorescence microscopy at × 1000 magnification. Nonneoplastic kidney parenchyma was used as control tissue. Only nonoverlapping tumor nuclei were evaluated. Definitions of chromosomal gains and losses were based on the Gaussian model and related to the nonneoplastic controls. Any tumor with a signal score beyond the cutoff value was considered to have had a gain or loss of the specific chromosome. The cutoff value was set at the mean ± 3 standard deviations (SDs) of the control values [9-13].
Results
Clinical features
The clinical features are summarized in Table 1. The 6 cases of OPRCC were diagnosed from 1990 to 2011 at Nanjing Jinling Hospital, accounting for 0.5% of all renal tumors diagnosed during this period. The patients’ ages ranged from 40 to 68 years (mean, 56.8; median, 59.5 years). The male-to-female ratio was 5:1, exhibiting a male preference. None of the patients had bilateral or multifocal neoplasms. Nephrectomy was performed at the time of diagnosis for all patients. None of these patients was treated with chemotherapy or radiation therapy after surgery. Follow-up was available for 6 patients (range, 33 to 121; mean, 82; median, 84 months). All 6 patients were alive with no evidence of disease after initial resection.
Table 1.
Clinicopathological data
| Case No. | Age/Sex | Size (mm) | TNM Stage | Fuhrman | Hemorrhage | Necrosis | Foamy Macrophages | Foamy Tumor Cells | Hemosiderin-laden tumor cells | Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 67/M | 32 | pT1N0M0 | 2 | Present | Absent | Present | Present | Present | RN | NED 121 months |
| 2 | 67/M | 28 | pT1N0M0 | 2 | Absent | Absent | Absent | Present | Absent | RN | NED 82 months |
| 3 | 52/F | 50 | pT1N0M0 | 3 | Present | Absent | Absent | Absent | Present | RN | NED 86 months |
| 4 | 40/M | 25 | pT1N0M0 | 2 | Present | Absent | Present | Present | Present | RN | NED 74 months |
| 5 | 68/M | 65 | pT1N0M0 | 2 | Present | Absent | Present | Absent | Present | RN | NED 33 months |
| 6 | 47/M | 40 | pT1N0M0 | 2 | present | Absent | Present | Present | present | RN | NED 96 months |
RN, radical nephrectomy; NED, no evidence of disease.
Morphology
The histological features are summarized in Table 1. These tumors ranged from 25 to 65 mm in size (mean, 40; median, 36 mm). All tumors showed a predominant papillary structure with delicate fibrovascular cores, while diffuse growth pattern was also seen in 4 cases. Papillae were lined by single layers of cells with large, deeply eosinophilic and finely granular cytoplasm, a low nucleocytoplasmic ratio, and round to oval nuclei. All tumors were Fuhrman grade 2, except for one tumor that was focally grade 3 due to huge nucleolus. Most of our cases (4/6) showed foamy macrophages within papilla cores. Likewise, hemorrhages were observed in five of six cases. No case had either necrosis or psammoma bodies (Figure 1A and 1B).
Figure 1.
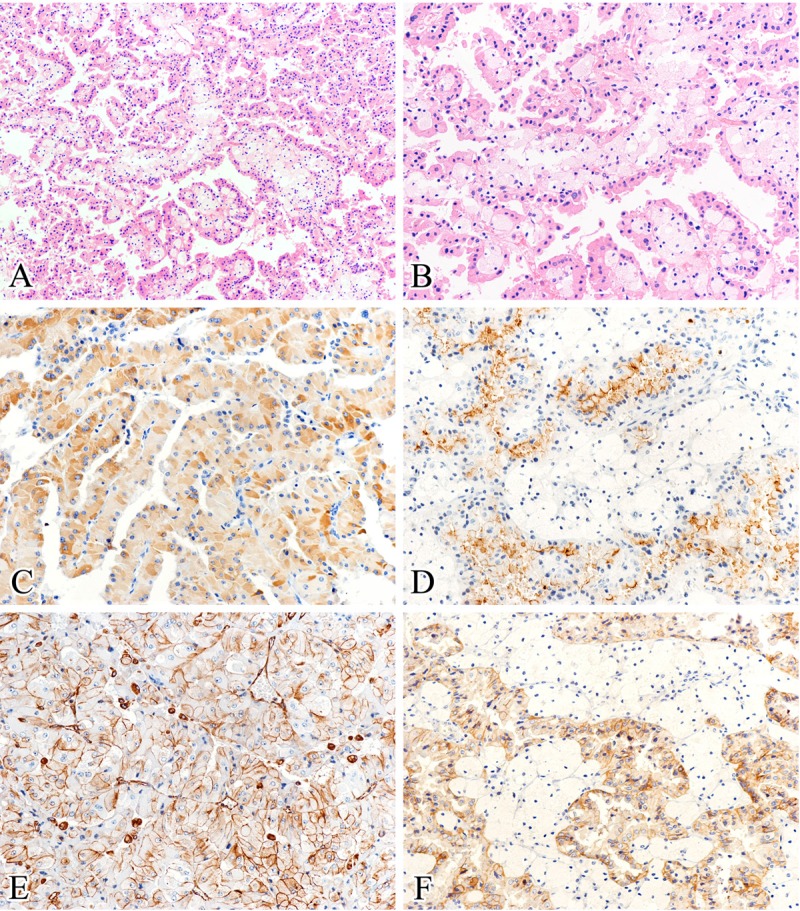
Histological and immunohistochemical features. A: All tumors showed a predominant papillary structure and delicate fibrovascular cores, with foamy macrophages usually seen in the papilla cores (hematoxylin and eosin). B: Papillae were lined by single layers of tumor cells with large, deeply eosinophilic and finely granular cytoplasm, and low grade nuclei (hematoxylin and eosin). C-F: All cases exhibited strong, diffuse positive for racemase (C), CD10 (D), vimentin (E), and MET (F) staining (immunohistochemical staining). (Original magnifications: A × 100; B-F × 200).
Interestingly, the phagocytosis of tumor cells was frequently and evidently seen in our cases. Hemosiderin-laden tumor cells were exhibited in five cases (Figure 2A, 2B and 2D), while foamy tumor cells were noticed in four cases (Figure 2C).
Figure 2.

The phagocytosis of tumor cells. A, B: Hemosiderin-laden tumor cells were frequently and evidently seen in our cases (hematoxylin and eosin). C: Foamy tumor cells were noticed in four cases (hematoxylin and eosin). D: Prussian blue staining was used to reveal intracellular hemosiderin (Prussian blue staining). (Original magnifications: A, D × 100; B × 200; C × 400).
Immunohistochemistry
The results of immunohistochemistry are summarized in Table 2. The neoplastic oncocytic cells in all cases exhibited strong, diffuse positivity for vimentin, CD10, racemase and MET. Tumorous cells demonstrated variable positivity for E-cad (5/6, 83%), CK7 (1/6, 17%), and EMA (2/6, 33%). CD117 was negative in all cases (Figure 1C-F).
Table 2.
Immunohistochemical Profiles and Fluorescence In Situ Hybridization Analysis
| Case No. | Immunohistochemistry | Fluorescence In Situ Hybridization | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| racemase | CD10 | vimentin | CK7 | MET | CD117 | E-cadherin | EMA | CEP 7 | CEP 17 | CEP Y | |
| 1 | ++ | + | +++ | 0 | +++ | 0 | + | 0 | - | - | - |
| 2 | ++ | ++ | ++ | 0 | +++ | 0 | ++ | 0 | Disomic | Disomic | Lost |
| 3 | ++ | +++ | +++ | 0 | +++ | 0 | + | 0 | Trisomic | Trisomic | Female |
| 4 | ++ | + | +++ | 0 | +++ | 0 | +++ | 0 | Trisomic | Disomic | Not Lost |
| 5 | ++ | ++ | ++ | ++ | +++ | 0 | + | + | Disomic | Trisomic | Not Lost |
| 6 | +++ | + | ++ | 0 | +++ | 0 | 0 | + | Trisomic | Disomic | Not Lost |
0, null; +, mild; ++, moderate; +++, intense staining.
MET mutation analysis
We analyzed the 14-21 exons of MET gene by direct sequencing in all 6 cases and found no distinct mutation in any case.
FISH analysis
The summary of FISH data is presented in Table 2. FISH analysis was performed in five of six cases, demonstrating trisomy of chromosomes 7 in three cases and trisomy of chromosomes 17 in two cases. Loss of chromosome Y was noted in one of four tumors in male patients.
Discussion
OPRCC was first reported by Lefevre et al. in 2005 with the histological features overlapping those of type 1 (low nuclear grade and a single layer) and type 2 (abundant eosinophilic cytoplasm) PRCC, and regarded as an independent subtype of PRCC not only for its distinct pathological features but also for its indolent clinical behavior [2]. After then, a few correlational research were published and 41 similar tumors have been reported as OPRCC [3-5,7].
Herein, we describe 6 patients with OPRCC, accounting for 0.5% of RCCs in our renal tumor series. The clinical and histological features are consistent with previous studies. Patients in our study tend to be older in age (mean, 56.8; median, 59.5 years) with a male preference (male-to-female ratio is 5:1). All 6 patients are alive with no evidence of disease after initial resection, showing an indolent clinical behavior. Histologically, tumors exhibit predominant papillary structure with delicate fibrovascular cores. Papillae are lined by single layers of cells with large, deeply eosinophilic and finely granular cytoplasm. In some studies, it was described that the nuclei of OPRCC tend to be at the periphery rather than at the center of the cells, suggesting an inverted nuclear pattern [7]. But in our series, such phenomenon is not frequently seen. It is of interest that the phagocytosis of tumor cells is frequently and evidently seen in our cases, which has not been described previously. Hemosiderin-laden tumor cells were exhibited in five cases, while foamy tumor cells were noticed in four cases. In our experience, it is not usually found in the majority of RCCs, such as conventional RCC or typical PRCC. These clues may be of some help in pointing to the correct diagnosis.
The immunoprofiles of OPRCC are generally similar to typical PRCC. Of all the reported cases of OPRCC, including the six described here, 94% (45/48), 85% (35/41) and 79% (38/48) of the cases showed diffuse positivity for AMACR, vimentin and CD10, respectively. CD117 is always negative in such tumors although one study found that seven of seven tumors exhibited CD117 expression. But CK7 is variable expressed among reports, ranging from 17% to 100% [2-5,7]. The present study find CK7 inmmunopositivity in only one of the six tumors (17%), exhibiting a slight difference with typical PRCC, which is always diffusely positive for CK7 [1]. These confl icting results can be explained because OPRCC represents a heterogeneous group.
Otherwise, we find uniform positive immunostaining for MET in all cases, indicating MET overexpression contrast to adjacent normal tissue. MET is the receptor for the hepatocyte growth factor (HGF), belongs to the family of tyrosine kinase (TK). The HGF/MET signaling pathway is involved in proliferation, survival, cell growth, differentiation, and cell migration. MET was reported to be overexpression in 80-90% of sporadic RCC by the immunohistochemistry. A germline mutation of MET gene of 7q31 has been detected in patients with hereditary PRCC as well as a small portion (5-13%) of sporadic PRCC. Most sporadic PRCCs show trisomy of chromosome 7 without MET mutation, and the increased MET gene copy number may also lead to MET overexpression in protein level. Most MET mutations that correlate to PRCC are missense, located within the TK domain [8,14-16]. We performed the DNA mutation analysis by direct sequencing of exon 14-21 of MET gene which included the TK domain, and found no mutation in any case. We also performed FISH analysis of chromosome 7, 17 and Y in five of six cases. The results demonstrate trisomy of chromosomes 7 in three cases and trisomy of chromosomes 17 in two cases. Loss of chromosome Y was noted in one of four tumors in male patients. The cytogenetic features of OPRCC are consistent with that of typical PRCC, supporting the diagnosis of OPRCC as a subtype of PRCC. In present study, gains of chromosomes 7 can partly explain the overexpression of MET. Whereas, there are two cases showed high level of MET protein but were absent for chromosomes 7 gain or MET gene mutation. Their mechanisms of MET overexpression remain unexplained. It may be because of other events such as upstream activation and translational control deregulation. Recently, considerable attention has been focused on the role of MET overexpression and amplification in tumor genesis. MET-inhibitors have been in trial and demonstrated antitumor activity for papillary RCC [17,18]. For MET is high expressed in OPRCC, it may be a potential therapeutic target for this tumor.
In conclusion, we reported a distinct subset of PRCC with oncocytic features, strong diffuse expression of racemase, vimentin, CD10, and MET, but negative for CK7 in most cases. Trisomy for chromosome 7 and 17 and loss of chromosome Y exhibited variable results. All cases have an indolent clinical behavior. Whether the strong expression of MET indicates a potential therapeutic target is still unknown and requires further investigation in clinical trials.
Disclosure of conflict of interest
The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article. Supported by National Natural Science Foundation of China (81171391; XJ Zhou), National Natural Science Foundation of China (81101933; Q Rao), Natural Science Foundation of Jiangsu Province, China (BK2010463; Q Rao), and Maixin fund (m1203; SS Shi).
References
- 1.Lopez-Beltran A, Scarpelli M, Montironi R, Kirkali Z. 2004 WHO classification of the renal tumors of the adults. Eur Urol. 2006;49:798–805. doi: 10.1016/j.eururo.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 2.Lefevre M, Couturier J, Sibony M, Bazille C, Boyer K, Callard P, Vieillefond A, Allory Y. Adult papillary renal tumor with oncocytic cells: clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol. 2005;29:1576–1581. doi: 10.1097/01.pas.0000184821.09871.ec. [DOI] [PubMed] [Google Scholar]
- 3.Hes O, Brunelli M, Michal M, Cossu Rocca P, Hora M, Chilosi M, Mina M, Boudova L, Menestrina F, Martignoni G. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol. 2006;10:133–139. doi: 10.1016/j.anndiagpath.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Kunju LP, Wojno K, Wolf JS Jr, Cheng L, Shah RB. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol. 2008;39:96–101. doi: 10.1016/j.humpath.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 5.Mai KT, Kohler DM, Robertson SJ, Belanger EC, Marginean EC. Oncocytic papillary renal cell carcinoma with solid architecture: mimic of renal oncocytoma. Pathol Int. 2008;58:164–168. doi: 10.1111/j.1440-1827.2007.02205.x. [DOI] [PubMed] [Google Scholar]
- 6.Okada A, Sasaki S, Fujiyoshi Y, Niimi K, Kurokawa S, Umemoto Y, Kohri K. A case of oncocytic papillary renal cell carcinoma. Int J Urol. 2009;16:765–767. doi: 10.1111/j.1442-2042.2009.02336.x. [DOI] [PubMed] [Google Scholar]
- 7.Park BH, Ro JY, Park WS, Jee KJ, Kim K, Gong G, Cho YM. Oncocytic papillary renal cell carcinoma with inverted nuclear pattern: distinct subtype with an indolent clinical course. Pathol Int. 2009;59:137–146. doi: 10.1111/j.1440-1827.2009.02341.x. [DOI] [PubMed] [Google Scholar]
- 8.Duh FM, Scherer SW, Tsui LC, Lerman MI, Zbar B, Schmidt L. Gene structure of the human MET proto-oncogene. Oncogene. 1997;15:1583–1586. doi: 10.1038/sj.onc.1201338. [DOI] [PubMed] [Google Scholar]
- 9.Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. Mod Pathol. 2003;16:1060–1063. doi: 10.1097/01.MP.0000090923.50509.55. [DOI] [PubMed] [Google Scholar]
- 10.Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fl uorescent in situ hybridization study. Mod Pathol. 2003;16:1053–1059. doi: 10.1097/01.MP.0000090924.90762.94. [DOI] [PubMed] [Google Scholar]
- 11.Brunelli M, Eble JN, Zhang S, Martignoni G, Delahunt B, Cheng L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol. 2005;18:161–169. doi: 10.1038/modpathol.3800286. [DOI] [PubMed] [Google Scholar]
- 12.Jones TD, Eble JN, Wang M, MacLennan GT, Delahunt B, Brunelli M, Martignoni G, Lopez-Beltran A, Bonsib SM, Ulbright TM, Zhang S, Nigro K, Cheng L. Molecular genetic evidence for the independent origin of multifocal papillary tumors in patients with papillary renal cell carcinomas. Clin Cancer Res. 2005;11:7226–7233. doi: 10.1158/1078-0432.CCR-04-2597. [DOI] [PubMed] [Google Scholar]
- 13.Gobbo S, Eble JN, Grignon DJ, Martignoni G, MacLennan GT, Shah RB, Zhang S, Brunelli M, Cheng L. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol. 2008;32:1239–1245. doi: 10.1097/PAS.0b013e318164bcbb. [DOI] [PubMed] [Google Scholar]
- 14.Lubensky IA, Schmidt L, Zhuang Z, Weirich G, Pack S, Zambrano N, Walther MM, Choyke P, Linehan WM, Zbar B. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–526. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JS, Kim MK, Seo JW, Choi YL, Kim DH, Chun YK, Ko YH. MET expression in sporadic renal cell carcinomas. J Korean Med Sci. 2006;21:672–677. doi: 10.3346/jkms.2006.21.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvi A, Marchina E, Benetti A, Grigolato P, De Petro G, Barlati S. Germline and somatic c-met mutations in multifocal/bilateral and sporadic papillary renal carcinomas of selected patients. Int J Oncol. 2008;33:271–276. [PubMed] [Google Scholar]
- 17.Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:553–572. doi: 10.1517/14728222.2012.680957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Posadas EM, Figlin RA. Understanding the role of MET kinase in cancer therapy. J. Clin. Oncol. 2013;31:169–170. doi: 10.1200/JCO.2012.46.7738. [DOI] [PubMed] [Google Scholar]