

Abstract
Inflammatory myofibroblastoma is a distinctive pseudosarcomatous lesion that occurs primarily in the viscera and soft tissue of children and young adults. It is composed of myofibroblastic spindle cells accompanied by an inflammatory infiltrate of plasma cells, lymphocytes, and eosinophils. We report a 41-year-old man who presented with progressive dyspnea and pain in the right hypochondrium for 6 months. USG abdomen revealed a well-defined hypoechoic rounded mass in the subdiaphragmatic region. CT thorax and abdomen revealed a large hypodense mass in posterosuperior aspect of the right lobe of liver, right pleural effusion and basal consolidation of the right lung. Right thoracotomy was performed. The tumor mass was arising from the right side of diaphragm. Total surgical excision was done. On histomorphology and immunohistochemical analysis, a diagnosis of inflammatory myofibroblastoma was done. Following excision the patient was completely relieved of dyspnea and pain. There was no recurrence on follow-up for 2 years.
Keywords: Inflammatory myofibroblastic tumor, Inflammatory myofibroblastoma, Diaphragm
Introduction
Inflammatory myofibroblastic tumor (IMT) is a distinctive lesion composed of myofibroblastic spindle cells accompanied by an inflammatory infiltrate of chronic inflammatory cells. IMT can occur throughout the body, and the most common sites are the lung, mesentery and omentum. Other sites include soft tissue, mediastinum, gastrointestinal tract, pancreas, genitourinary tract, oral cavity, skin, breast, bone, and nervous system. The site of origin determines the symptom of IMT. Pulmonary IMT is associated with chest pain and dyspnea, but may be asymptomatic. Abdominal tumors may cause gastrointestinal obstruction [1]. Approximately 10–20 % cases are associated with pyrexia, weight loss, elevated ESR, and sometimes anemia. Following excision, 10–25 % of patients develop local recurrence, which may be repeated [2].
Here, we report a rare site of occurrence. In the reviewed literature, single case of inflammatory pseudotumor of diaphragm is reported [3]. Our case may be the second rare case of inflammatory myofibroblastoma of diaphragm.
Case Report
A 41-year-old man presented with history of progressive dyspnea and pain in the right hypochondrium for 6 months. Dyspnea and pain were relatively relieved in the upright or sitting position. There was no history of trauma or pulmonary tuberculosis. General physical examination revealed mild pallor. Respiratory system examination showed decreased air entry on the right lung base. All biochemical and hematological investigations were normal except raised ESR. The chest X-ray showed blunting of right costophrenic angle and compressed right lower lobe of lung. USG abdomen revealed a well-defined hypoechoic rounded mass in the subdiaphragmatic region (Fig. 1). CT thorax and abdomen revealed a large hypodense mass in posterosuperior aspect of the right lobe of liver. There was right pleural effusion and basal consolidation of the right lung (Fig. 2). CT-guided biopsy was done. Histology revealed features of inflammatory myofibroblastoma. Considering the diagnosis of inflammatory myofibroblastoma, surgery for complete excision of the tumor was planned.
Fig. 1.

Photograph of USG abdomen revealed well-defined hypoechoic rounded mass in subdiaphragmatic region
Fig. 2.

Photograph of CT thorax and abdomen revealed a large hypodense mass in posterosuperior aspect of the right lobe of liver and right pleural effusion
Operative Findings
Right thoracotomy was performed with right modified posterolateral thoracotomy incision. A well-defined mass was identified lying on the right side of diaphragm. The tumor mass was separated from lung, pleura, and posterosuperior surface of the right lobe of liver. Around the tumor, the diaphragm tissue showed increased vascularity and areas of hemorrhage. The tumor mass was easily separated from the surrounding tissues and excised along with 2 cm margin of diaphragm. Diaphragm was repaired with edge-to-edge sutures with Prolene number 1 and intermittent polyester. After removal of the tumor, the lower lobe of lung expanded well and no other lesions were found in the diaphragm, lung, or liver.
Gross
The tumor mass was well-circumscribed, encapsulated, grayish white in color, measuring 11.5 cm × 10.5 cm × 7.5 cm. Cut surface showed a grayish white, solid fleshy mass with whorled appearance (Fig. 3). Surrounding compressed muscle fibers showed areas of hemorrhage. There were no areas of hemorrhage and necrosis in the tumor.
Fig. 3.
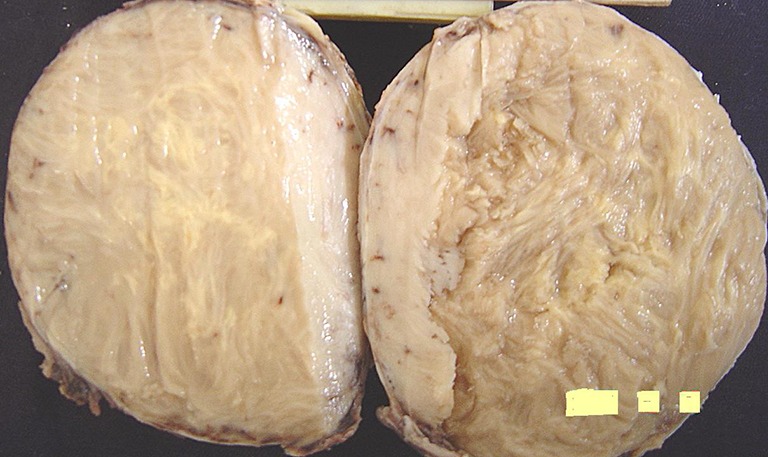
Gross photograph showing well-circumscribed, grayish white tumor mass with fleshy and whorling appearance on cut surface
Microscopy
Sections from different areas of the tumor mass revealed a well-circumscribed mass showing peripheral rim of focal microcalcification. It was composed of admixture of spindle cells, ovoid cells, and prominent lymphoplasmacytic inflammatory component with a few blood vessels in an edematous or sclerotic background. The spindle cells having vesicular nuclei were arranged in interlacing fascicles (Fig. 4). There was mild to moderate atypia in a few spindle cells and mitoses were observed in a few cell nuclei. A number of plasma cells and lymphocytes were seen interspersed with spindle cells (Fig. 5). Large foamy cells and focal giant cells were also observed in some areas. The tumor mass was surrounded by skeletal muscle fibers of diaphragm showing congestion and hemorrhagic necrosis. This histological finding was suggestive of the origin of tumor in the diaphragm. Immunohistochemistry showed strong and diffuse cytoplasmic reactivity for vimentin, which is typical for all IMTs. Spindle cells focally expressed desmin. Smooth muscle actin was positive. Alk1, S-100 protein, C-Kit, myogenin, and myoglobin were negative. Histological features and immunohistochemical findings clinched the diagnosis of inflammatory myofibroblastoma.
Fig. 4.
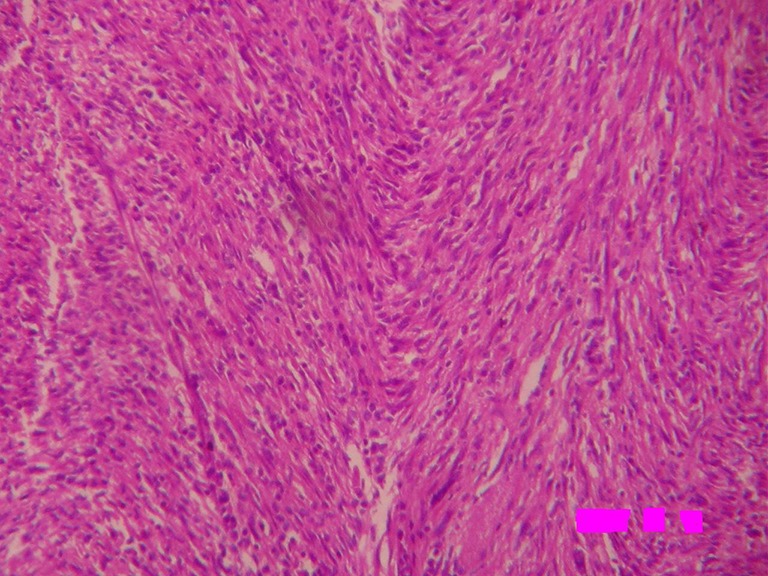
Photomicrograph showing fascicular architecture with numerous admixed plasma cells and lymphocytes (H&E × 100)
Fig. 5.
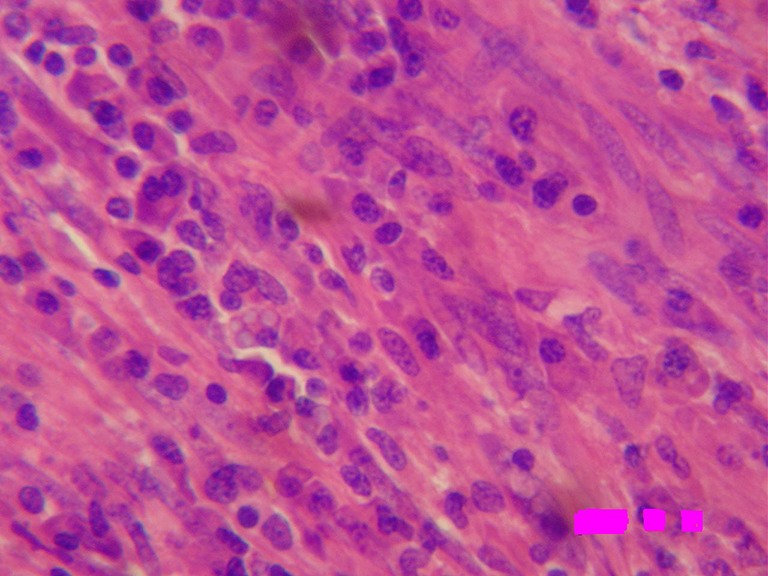
Photomicrograph showing spindle-shaped cells in interlacing fascicles, intimately admixed with the inflammatory infiltrate predominantly of plasma cells (H&E × 400)
Discussion
An inflammatory myofibroblastic tumor is a distinctive pseudosarcomatous lesion that occurs primarily in the viscera and soft tissue of children and young adults. The lesion has a distinctive histological appearance and usually pursues a benign clinical course. The lesion has many names including plasma cell granuloma, plasma cell pseudotumor, inflammatory myofibrohistiocytic proliferation, omental-mesenteric myxoid hamartoma, and most commonly inflammatory pseudotumor [4]. The inflammatory myofibroblastic tumor is a subgroup of the broad category of inflammatory pseudotumors. The inflammatory pseudotumor has been applied to diverse entities including reparative pseudosarcomatous lesions, infectious lesions including those secondary to mycobacterium avium intracellulare infections, and Ebstein–Barr virus associated follicular dendritic cell tumors usually found in liver or spleen and inflammatory myofibroblastoma [4]. Grossly the lesions are circumscribed or multinodular, firm white or tan mass with a whorled fleshy/or myxoid cut surface [1]. The tumor ranges in size from 2 to 20 cm, but most are 5–10 cm [4]. It is composed of a variable mixture of collagen, inflammatory cells, and usually cytologically bland spindle cells showing myofibroblastic differentiation [5].
IMTs have been studied cytogenetically and found to have clonal abnormalities, especially at 2p22–24, supportive of a neoplastic process. The anaplastic lymphoma kinase (ALK) gene, located on 2p23, has been implicated in the pathogenesis of this lesion, and a significant percentages of intra-abdominal IMTs which have been studied show immunohistochemical expression of ALK [4].
IMT contains a mixture of spindle cells showing fibroblastic and myofibroblastic differentiation arrayed in fascicles or with storiform architecture. The spindle cells have oval nuclei, fine chromatin, inconspicuous nucleoli, and abundant bipolar lightly eosinophilic cytoplasm. Mitoses are infrequent. Cytological atypia is not obvious. Admixed with the spindle proliferation and often obscuring it, is an inflammatory infiltrate containing lymphocytes, plasma cells, and histiocytes including Touton type giant cells. Plasma cells are often prominent [5]. All the above-mentioned histological features were present in our case.
Pulmonary and extrapulmonary IMTs show similar immunoprofiles. Spindle cells express strong and diffuse staining with vimentin and rarely desmin as seen in our case. They fail to express myogenin, myoglobin, CD117(c-Kit), and S-100 proteins. Focal cytokeratin reactivity is noted in about one third of cases, and CD68 is detected in up to 25 % of cases [4, 5]. Immunohistochemistry was also favoring the diagnosis of inflammatory myofibroblastoma in our case. P53 immunoreactivity is rare and reported in association with recurrence and malignant transformation [4, 5], but these features were also seen in over half of the clinically benign IMTs, limiting their prognostic utility. Furthermore, in a recent study of atypical and/or clinically aggressive IMTs, half of the metastasizing cases showed no atypical features. There is no clear-cut relationship between ALK expression and prognosis in IMT [6].
Differential diagnosis of IMT includes various benign and malignant mesenchymal tumors. Inflammatory malignant fibrous histiocytoma is characterized by bizarre pleomorphic cells. In leiomyosarcoma, nuclei are cigar shaped and arranged in a more regular fascicular growth pattern. In addition, leiomyosarcoma is not typified by a conspicuous inflammatory infiltrate. In inflammatory fibrosarcoma, multinucleate giant cells are rare and chronic inflammatory cells are particularly lymphocytes [4].
In most instances, complete surgical excision of IMT leads to excellent survival. Extrapulmonary IMT has a recurrence rate of approximately 25 %. Kubal et al reported a locally aggressive and recurrent pleural inflammatory myofibroblastoma [7].
Recurrence usually occurs in cases which are incompletely excised. Rare cases (<5 %) also metastasize. Evidence suggests that a combination of atypia, ganglion-like cells, TP53 expression, and aneuploidy may help to identify IMT with a more aggressive potential. Unfortunately, it is difficult to predict on the basis of histopathological findings alone in an individual case whether recurrence or malignant transformation occurs. Although surgery is the principal treatment, regression and response to corticosteroids and nonsteroidal inflammatory agents have been noted in rare cases [1, 5].
We report here a rare site of occurrence. The first case of the inflammatory pseudotumor of diaphragm has been reported in a 5-year-old boy by Hoer et al. In our case following excision of tumor, the patient was completely relieved of dyspnea and pain. There was no recurrence on follow-up for 2 years.
References
- 1.Coffin CM, Fletcher JA. Inflammatory myofibroblastic tumor. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumors of soft tissue and bone. Lyon: IARC Press; 2002. pp. 91–93. [Google Scholar]
- 2.Fletcher CDM. Soft tissue tumors. In: Fletcher CDM, editor. Diagnostic Histopathology of Tumors. 3. Elsevier: Churchill Livingstone; 2007. pp. 1527–1592. [Google Scholar]
- 3.Hoer J, Steinau G, Fuzesi L, Gunawan B, Schumpelick V. Inflammatory pseudotumor of diaphragm. Pediatr Surg Int. 1999;15:387–390. doi: 10.1007/s003830050607. [DOI] [PubMed] [Google Scholar]
- 4.Abdul-Karim KW, Chang AE, Fletcher JA, Folpe AL, Geisinger KR, Gown AM, et al. Fibrous tumors of infancy and childhood. In: Weiss SW, Goldblum JR, et al., editors. Enzinger and Weiss’s soft tissue tumors. 4. St Louis: Mosby; 2001. pp. 347–408. [Google Scholar]
- 5.Yousen SA, Tazelaar HD, Manabe T, Dahner LP. Inflammatory myofibroblastic tumor. In: Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC (eds) World Health Organization classification of tumors. Pathology and genetics of tumors of the lung, pleura, thymus and heart. IARC Press, Lyon, pp. 105–106
- 6.Gleason BC, Hornick JL. Inflammatory myofibroblastic tumors: where are we now? J Clin Pathol. 2008;61:428–437. doi: 10.1136/jcp.2007.049387. [DOI] [PubMed] [Google Scholar]
- 7.Kubal C, Ghotkar S, Gosney J, Carr M. Pleural Inflammatory myofibroblastoma: a locally aggressive intrathoracic tumor. J Cardiothorac Surg. 2007;2:29. doi: 10.1186/1749-8090-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]