

Abstract
Combining long-range magnetic order with polarity in the same structure is a prerequisite for the design of (magnetoelectric) multiferroic materials. There are now several demonstrated strategies to achieve this goal, but retaining magnetic order above room temperature remains a difficult target. Iron oxides in the +3 oxidation state have high magnetic ordering temperatures due to the size of the coupled moments. Here we prepare and characterize ScFeO3 (SFO), which under pressure and in strain-stabilized thin films adopts a polar variant of the corundum structure, one of the archetypal binary oxide structures. Polar corundum ScFeO3 has a weak ferromagnetic ground state below 356 K—this is in contrast to the purely antiferromagnetic ground state adopted by the well-studied ferroelectric BiFeO3.
Introduction
In recent years, multiferroic materials (specifically materials with permanent magnetization and electrical polarization) have attracted great interest, as controlling the magnetic state through the application of an electric field is an enabling feature for novel technologies.1−5 However, materials with multiferroic order above room temperature are rare due to the contraindicated nature of magnetism and polar order.6,7 The most studied systems currently have very small polarizations (structurally undetectable) that are driven by the onset of magnetic order, which lifts the centers of symmetry present in the paramagnetic state.1,4,5,7 In most of the observed cases, the transitions occur well below room temperature, for room temperature magnetoelectric phenomena, currently the best candidates are the hexaferrites and related phases.5,8,9 Many ferroelectrics are based on d0 structures whereas magnetism clearly requires the presence of open shell cations. BiFeO3 can be seen as an archetype for an alternative approach where two sublattices are combined where one is ferroelectrically active and the other magnetically, though in this case, at least near room temperature, the system is purely antiferromagnetically ordered.10 Given the higher ordering temperatures of antiferromagnets, multiferroics with weak ferromagnetism would show promise in this property control.11,12 A two sublattice approach has proved successful in EuTiO313 (where lattice strain in a thin film drives a quantum paraelectric to a ferroelectric phase transition) with the magnetic ordering from Eu2+ occurring at low temperature. High pressure has been used to convert the layered ilmenite FeTiO3 into a polar LiNbO3 polymorph which shows weak ferromagnetism well below room temperature,12 with AF order up to 270 K found in the partially ordered (In1–xMx)MO3 (M = Fe1/2Mn1/2; x = 0.143)14,15 LiNbO3 phases.14−16 Thus, new approaches are needed to increase the temperature of coexistence. We present an alternative approach to generating polarization and magnetization above room temperature by preparing a new polar polymorph of ScFeO3 (SFO) using high pressure synthesis and biaxial strain based on the corundum structure, which exhibits weak ferromagnetism with an ordering temperature of 356 K. The reported SFO structure is in contrast to the perovskite-derived ScCrO317 and ScVO3,18 which are also formed at high pressure for earlier first transition series cations.
Experimental Methods
ScFe1–xCrxO3 (x = 0–1) was prepared from stoichiometric mixtures of Sc2O3 (99.999%, Sigma Aldrich), Fe2O3 (99.999%, Alpha Aesar), and Cr2O3 (99.997%, Alpha Aesar) reacted at 1500 °C (x = 0), 1480 °C (x = 0.03), and 1450 °C (x = 0.10–1) at 6 GPa for 5 min in a Pt-lined Al2O3 crucible within a cylindrical graphite furnace in a Walker-type multianvil press and then quenched to room temperature by turning off the voltage supply to the resistance furnace, which reduces the temperature from 1500 to 25 °C in ∼2 s. The pressure is maintained during the temperature quenching.
Thin films were grown using a Neocera pulsed laser deposition (PLD) chamber equipped with Staib RHEED. A stoichiometric ScFeO3 ceramic target, made by conventional solid state methods, was used in the PLD at a laser energy of 252 mJ over a spot size of approximately 2–3 mm2. The films were grown in an oxygen partial pressure of 2 mTorr. The substrate was held at the temperature 850 °C and the distance from the target 47 mm. A 30 kV electron beam was used in the RHEED at a current of 1.55 A. During cooling, the partial oxygen pressure was held at 150 Torr.
Powder X-ray diffraction (XRD) data were recorded on instrument ID31 (for x = 0 over a range of temperatures, λ = 0.3999 Å) at ESRF, Grenoble. Anomalous scattering data were collected on instrument I11 at Diamond Light Source (DLS). The sample was loaded on the external surface of a quartz capillary (diameter 0.5 mm) to minimize absorption.19 A monochromator scan was used to measure the Fe K edge fluorescence spectrum. High resolution X-ray diffraction patterns were then recorded for 2 h each at six selected energies near the Fe K absorption edge: 7.050, 7.080, 7.112, 7.138, 7.165, and 7.190 keV. The six diffraction patterns were fitted simultaneously using the TOPAS20 software package. The peak shape parameters were constrained to be the same for the six patterns during refinement. Sample absorption was fitted using the Pitschke model21−23 for surface roughness in the TOPAS software package.
Neutron powder diffraction (NPD) data were collected on the POLARIS instrument at the ISIS facility, Rutherford Appleton Laboratories. Diffraction data analysis was performed with the GSAS24 suite of Rietveld analysis programs, using the EXPGUI25 interface. Magnetic structures were modeled using a P1 magnetic only phase:26 atomic positions and lattice tensors were fixed/constrained to maintain the nuclear cell and symmetry. The magnetic models were constructed via the representational analysis of the system using SARAh.27 Structural figures were generated using VESTA.28
Convergent beam electron diffraction (CBED) and energy dispersive spectroscopy (EDS) analyses were performed with a JEOL 2000FX electron microscope. High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were recorded on a Tecnai G2 microscope operated at 200 kV and on a Titan G3 80-300 microscope equipped with the probe aberration corrector and operated at 300 kV. Calculated HAADF-STEM images were obtained using the QSTEM software.29 Imaging of ceramics used in PFM measurements revealed no change in phase composition at the grain boundaries.
Mössbauer data were obtained using a conventional constant acceleration Mössbauer spectrometer incorporating a ∼25 mCi source of Co57 in a Rh matrix. Alternating current and direct current magnetization measurements were carried out with a commercial Quantum Design superconducting quantum interference device (SQUID) magnetometer. We have measured the dielectric and ferroelectric properties of the samples which were sandwiched between Ag paste deposited electrodes, in parallel plate geometry. We used an Agilent E4980A LCR Meter to measure the dielectric properties and a Radiant Precision LC Ferroelectric Tester to measure the P-E loops.
Piezoresponse force microscopy was measured on a 6 × 6 μm2 scan area using a DCP-11 diamond coated tip (NT-MDT, Moscow, Russia) while measuring amplitude and phase (R and θ). It was undertaken on a Park XE-100 (Park Systems, Suwon, South Korea) with a SR830 lock in amplifier (Stanford Research, CA, USA) with a bias voltage of 5 V, a time constant of 1 ms, and a sensitivity of 100 μV).
All DFT calculations have been performed using the CRYSTAL09 program.30 We employed the PBE0 hybrid exchange functional that uses 25% Hartree–Fock exchange; reciprocal space sampling has been performed for all structures using a Monkhorst-Pack grid of 8 × 8 × 8 k-points for all structures. Standard all electron basis sets from the CRYSTAL online database (www.crystal.unito.it) have been used for all elements (indicated by the following labels online: Fe_86–411d41G_towler_1992a, Sc_864–11dG*_harrison_2006, O_8–411d11G_valenzano_2006). Full geometry optimizations have been performed for each structure, followed by a series of constant volume optimizations to evaluate the phase diagrams. Enthalpies have been derived from the energy-volume data, by fitting to the second order Birch–Murnaghan equation of state.
Monte Carlo simulations have been performed using an extended Ising model with coupling parameters fitted to a test set of different disordered structures. Sampling has been performed on cells containing 5184 ion pairs. For each temperature, 10000 sampling points, separated by 4 times the correlation length of the simulation, have been used.
Results and Discussion
Reaction of Sc2O3 with Fe2O3 at 1500 °C for 12 h in air at ambient pressure yields a material adopting the bixbyite structure of Sc2O3 (Figure 1a and Supporting Information Figure S1) which shows weak ferromagnetism with a Tc of 39 K31−33 (Figure S2). The bixbyite-type ScFeO3 is an anion vacancy-ordered derivative of fluorite with a disordered distribution of Sc3+ and Fe3+. This type of structure is a clear candidate for transition to a denser close-packed structure under high pressure by elimination of anion vacancies. Indeed, it has been previously demonstrated that several bixbyite systems convert to corundum type phases at high pressure and temperature.34,35 Reaction of stoichiometric mixtures of Sc2O3 and Fe2O3 at 6 GPa and 1500 °C afforded a material (HP SFO) with a unit cell corresponding to the corundum structure (a = 5.198(1) Å, c = 14.003(1) Å) adopted by α-Fe2O3 (Figure 1b); this hexagonal close-packed structure has 2/3 of the octahedral voids filled by metal cations, which can be considered as dimers of face-sharing MO6 octahedra along the [001] direction of stacking of the close-packed oxide layers, linked by corner-sharing into a three-dimensional framework. Synthesis of the bulk material away from the 1:1 composition in Sc1–xFe1+xO3 (nonstoichiometric Sc1.024Fe0.976O3 (Sc-rich) and Sc0.976Fe1.024O3 (Fe-rich) were prepared under the same conditions as ScFeO3) does change the unit cell dimensions of the corundum–like phase, but it produces Sc2O3 and Fe2O3 impurities even at minor deviations from the fully stoichiometric structure (Figure 2a). The absence of an extensive solid solution in SFO, in contrast to In2O3–Fe2O3 and Ga2O3–Fe2O334 is unexpected given the similar size (0.745 and 0.645 Å, respectively36) and identical charge of Sc3+ and Fe3+, and this indicates the likely presence of short-range ordered structural motifs in SFO (as discussed later).
Figure 1.
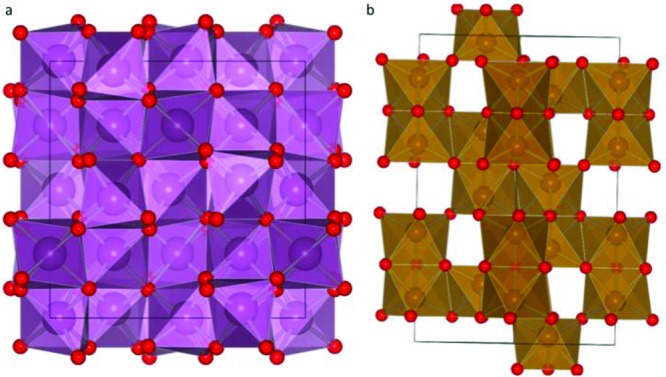
(a) Sc2O3 (bixbyite, Ia3̅) viewed along [100]; (b) α-Fe2O3 (corundum, R3̅c) structure, viewed along [110].
Figure 2.
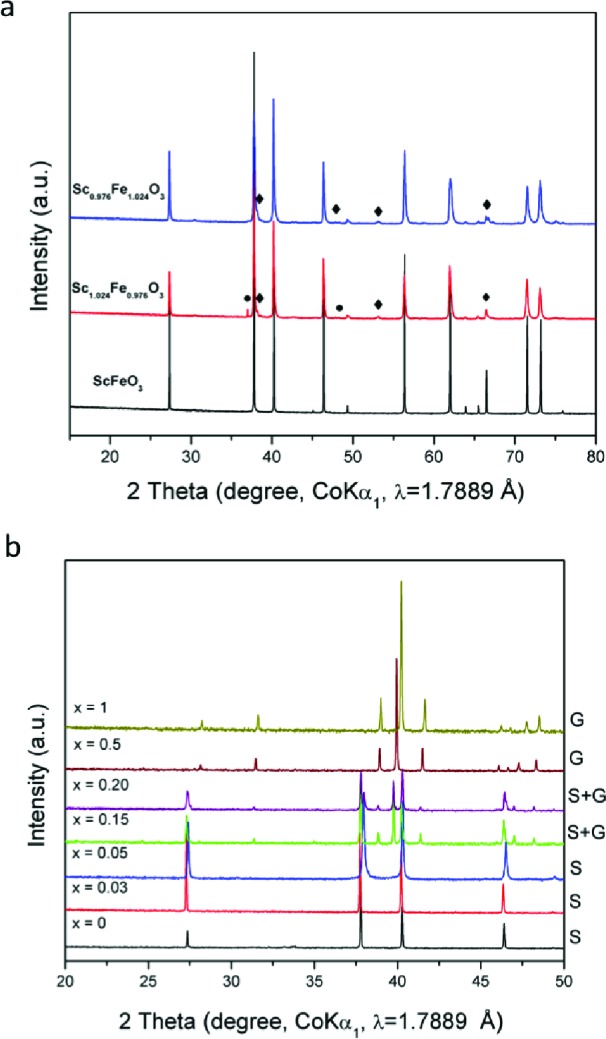
(a) Room temperature XRD patterns for Sc1–xFe1+xO3 (x = 0, ±0.024). Diamonds represent peaks for corundum impurities and dots bixbyite. (b) Room temperature XRD patterns for ScFe1–xCrxO3 (x = 0, 0.03, 0.10, 0.15, 0.20, 0.50, and 1). The resulting phases for each composition are marked on the right side, where S stands for a high pressure ScFeO3-type phase and G for a GdFeO3-type phase.
ScFe1–xCrxO3 (x = 0, 0.03, 0.10, 0.15, 0.20, 0.50, 1) solid solutions were also investigated with ScCrO3, which adopts the distorted perovskite GdFeO3 structure at high pressure17 (Figure 2b) The HP SFO corundum-like structure was retained for x ≤ 0.10 (reacted at 1500 °C (x = 0), 1480 °C (x = 0.03), and 1450 °C (x = 0.10)). The HP SFO and GdFeO3-type structures coexist for x = 0.15 and 0.2. At x ≥ 0.50, only the GdFeO3-type phase was formed. For x = 0, the material starts to decompose on heating; at about 200 °C, the peaks begin to broaden; and by 500 °C, the material has decomposed into Sc2O3 and Fe2O3 (Figure S3). Both x = 0.03 (Supporting Information Figure S4) and x = 0.10 (Figure S5) are significantly more robust, not decomposing below 800 °C.
SFO thin films (∼25 nm thick) were grown on SrTiO3 (001) substrates using pulsed laser deposition (PLD, 850 °C deposition temperature, oxygen partial pressure of 2 mTorr) with an SrRuO3 buffer layer (typically 3–5 unit cell monolayers) through substrate-imposed strain engineering37 (Figure S6). The lattice mismatch of 2.7% between the pseudocubic perovskite cell of corundum ScFeO3 (ap = 3.802 Å, α = 86.2°; Figure 3d and e) and (001) SrTiO3 is less than that for the ambient pressure bixbyite structure (14.7% in the 45° cube on cube configuration). The observed 3.806 Å out-of-plane lattice parameter (Figure 3a) is consistent with the high-pressure synthesized bulk material.
Figure 3.

Characterization data for thin films of ScFeO3 grown on SrTiO3 (100) substrates. The epitaxial relationship of the film to substrate was [2–21]SFO (cor)||[001]STO and (012)SFO (cor)|| (010)STO. (a) out-of-plane X-ray diffraction scan indexed to the perovskite pseudocubic cell (gray italic indices indicate a reflection from the substrate) with lattice vector relations of [0 0 1]per ∥ [2 −2 1]corand (0 1 0)per ∥(0 1 2)cor. (b) Reciprocal space map about the (0–33) reflection of the substrate showing coherent alignment of Fe2O3 (FO) with ScFeO3 (0 3 −12)SFO (cor)∥ (1 3 10)FO The ScFeO3 peak is indexed to the perovskite pseudocubic cell. The breadth of the film peak is assigned to strain gradients moving away from the substrate and the domain structure within the film. (c) High resolution HAADF-STEM image of the substrate film interface, the white arrow indicates a boundary between two twin domains. This twinning is due to the rhombohedral distortion having four possible orientations when growing on the cubic SrTiO3 surface. (d) Schematic indicating the registry of the ScFeO3 thin films (top) to the SrTiO3 substrate (bottom). The perovskite pseudocubic cell is indicated and its relationship to the corundum cell. For clarity, the Sc and Fe site are shown disordered with the Sc octahedra removed. Perovskite cell highlighted with respect to the substrate (e) and orientation of the rhombohedral cell with respect to the perovskite cell. In the Figure the out-of-plane lattice parameter is that determined experimentally and the in-plane is set equal to SrTiO3 with γ = 90° rather than the bulk rhombohedral angle. The experimentally determined γ = 89.2°.
The cell is distorted from the bulk cell due to substrate strain with α determined (Supporting Information p S9) as 89.2° at the substrate film interface. Off-axis reciprocal space maps (RSM) (Figure 3b) reveal Fe2O3 coherently aligned with ScFeO3, consistent with EDX measurement of the film composition as Sc0.87Fe1.13O3, attributed to incomplete stoichiometry transfer from the target and consistent with the restricted compositional variation found in bulk high pressure synthesis. Vertical twin planes separate the film into columnar domains, originating at misfit dislocations that relax the epitaxial strain and are present every 4–10 nm (Figure 3c).
The 1:1 stoichiometric ratio of Sc and Fe observed in HP SFO suggests ordering of these cations within the dimers of face-shared octahedra of the corundum lattice. Density functional theory (DFT) calculations, employing the PBE0 hybrid-exchange functional under periodic boundary conditions, show that structures with only heteronuclear (Sc,Fe) dimers are at least 1.0 eV per dimer more stable than structures with homonuclear (2Fe or 2Sc) pairs. Homonuclear dimers are thus expected only in very low concentration, which explains the lack of solid solutions away from the 1:1 ratio. However, inverting the position of Sc and Fe ions within a heteronuclear dimer has much smaller energetic cost (Supporting Information Table S2) and is likely to occur extensively; we will henceforth refer to this defect as the antisite dimer.
Ordered decoration of the corundum lattice with the energetically favored heteronuclear dimers affords two candidate structures. The ilmenite structure (Figure 4a, left) arises when the two cations form alternate layers perpendicular to c (space group R3̅). If instead the dimers are arranged to avoid octahedral edge-sharing by like cations (Figure 4a, right), we obtain the LiNbO3 structure with three-dimensional self-connectivity of each cation (space group R3c). DFT calculations (Supporting Information pp S11–12) show an energetic preference of 0.13 eV/formula unit (f.u.) for the R3c variant.
Figure 4.

(a) Polyhedral representation of SFO slabs demonstrating the cation order in ilmenite (left) and LiNbO3 (right). The black circle indicates an antisite dimer defect in polar corundum SFO. (b) [11̅0] zone axis noncentrosymmetric CBED pattern. A mirror plane and two GjonnesMoodie (GM) lines parallel to it are highlighted which correspond to the c glide plane. (c) The whole [11̅0] zone axis CBED pattern showing a mirror plane. The five CBED patterns shown here and in Supporting Information Figure S5 are all collected from different crystallites. (d) TEM SAED along the [11̅0] zone axis—the spots due to double diffraction are marked by arrows. (e) [241] = ⟨100⟩p zone axis HAADF-STEM image of ScFeO3. (f) [121̅] = ⟨110⟩p zone axis HAADF-STEM image of ScFeO3. The calculated images for the ordered and disordered structure models are superimposed.
Convergent beam electron diffraction (CBED) allows unique determination of point groups and was thus applied to determine the presence or absence of an inversion center.38 CBED on bulk ScFeO3 revealed the noncentrosymmetric 3m point group and the presence of c-glide planes in the structure (Figure 4 and Supporting Information Figure S7)—the (006) and (006̅) disks in the [11̅0] zone axis CBED pattern (Figure 4b and c) have different structures, reflected in other differences between (hkl) and (hkl) disks, demonstrating the absence of an inversion center. Together with the selected area electron diffraction data (Figure 4d), the noncentrosymmetric space group R3c was determined. The space group and unit cell correspond to those of ferroelectric LiNbO3 and multiferroic BiFeO3. These can be described either as cation ordered corundums (see above) or alternatively as a derivative of perovskite, where a combination of ferroelectric Γ4– (x,x,x shift) cation displacement from the AO3 close-packed layer and R4 (a–a–a–) octahedral tilting leads to the creation of an octahedral site for the initially 12 coordinate perovskite A cation.10,39 The polar nature of ScFeO3 thus suggests some Sc/Fe order in the corundum-like structure. Assuming a completely cation-ordered LiNbO3-type structure, one would expect the projected Sc and Fe columns to be imaged as dots of different brightness on the high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images due to a difference in the atomic number of Sc (Z = 21) and Fe (Z = 26). However, no noticeable difference in the brightness of the dots associated with the Sc and Fe columns was observed on the experimental images (Figure 4e and f), indicating that the order of Sc and Fe in ScFeO3 is incomplete.
The very similar X-ray and neutron scattering factors of Sc3+ and Fe3+ prevent unambiguous assignment of the ordering extent, based purely on either neutron or nonresonant X-ray diffraction (qualities of Rietveld fits were insensitive to occupancy). Therefore, anomalous scattering diffraction data at the Fe K edge were collected on the I11 instrument at the Diamond Light Source at six energies near the Fe K absorption edge (Figure 5a). Anomalous scattering experiments exploit the change in the X-ray scattering power of an element as the X-ray wavelength moves through the absorption edge,40 and they are used here to increase the contrast between Sc and Fe by working at the Fe K edge. The data were refined to a composition [Sc0.54(2)Fe0.46(2)] [Sc0.46(2)Fe0.54(2)]O3(Figure 5b), confirming that only a small degree of order is observed (8(2)% defined as the difference in occupancy), consistent with the polar symmetry observed in CBED, but lack of observable cation order on the HAADF-STEM images due to the negligibly small difference of the scattering power at these positions. These cation site occupancies were then fixed in the refinement of the neutron and nonresonant synchrotron powder diffraction data. The refined structure and coordination environments are shown in Figure 5c–e with refined coordinates, bond lengths, and angles given in Tables 1 and 2 and the cif file provided in the Supporting Information (where the magnetic moments can be found in the gsas .lst file). The Sc3+ site is larger (2.104 Å) than the Fe3+ site (2.067 Å), as might be expected from simple ionic radii arguments. This is consistent with the DFT-optimized bond lengths (0 K) of 3 × 2.074 Å, 3 × 2.167 Å (Sc–O) and 3 × 1.953 Å, 3 × 2.152 Å (Fe–O) for a fully ordered structure (Supporting Information Tables S3 and S4). The Sc3+ site is also more distorted (3.49% vs 3.39%)—the refined bond lengths are thus consistent with the identified polar order. The magnetic structure is that described for α-Fe2O3 above the transition Morin transition.41 The observed reduced moment is consistent with the mean field estimate for T/TN = 0.84 of 2.8 μB/Fe3+. The term “polar corundum” is therefore suggested to reflect the combination of partial cation ordering resulting in a noncentrosymmetric corundum-type structure. In polar corundum ScFeO3, the distinction between oxygen coordination of the two cation sites is less pronounced than in LiNbO3 and BiFeO3, favoring incomplete cation order rather than a perovskite-based description.
Figure 5.

(a) Fluorescence intensity versus energy at the Fe K edge for ScFeO3 on instrument I11 at DLS. The six selected energy levels (En = 7.050, 7.080, 7.112, 7.138, 7.165, and 7.190 keV for n = 1–6, respectively) used for diffraction studies near the Fe K absorption edge were marked by arrows (↓) and stars (☆). (b) Simultaneous Rietveld refinement of X-ray diffraction data for ScFeO3 collected at six selected energy levels near the Fe K absorption edge, giving a partially ordered composition of [Sc0.54(2)Fe0.46(2)] [Sc0.46(2)Fe0.54(2)]O3. The inset (38.6–39.8°) shows the peak evolution and changes in diffraction intensity due to the energy variation of resonant scattering. (c) Rietveld refinement of neutron powder diffraction data (POLARIS 145° bank) for ScFeO3; black crosses represent observed data, the red line the calculated fit, the blue line the difference, upper black tick marks the allowed nuclear Bragg peaks, and the lower ones the allowed magnetic peaks; the inset shows longer d-spacing data from the 35° bank of POLARIS. (d) Polyhedral representation of the final structure viewed along [110]. Brown polyhedral are 54% Fe, 46% Sc, and purple polyhedra are 54% Sc, 46% Fe. Arrows represent the refined magnetic moment orientations. Local coordination environment of (e) the scandium rich site Sc2 and the iron rich site Fe2 viewed perpendicular to the c axis.
Table 1. Refined Structural Parameters for Polar Corundum ScFeO3 (R3c, a = 5.202475(4) Å, c = 14.01449(10) Å, V = 328.4944(25) Å3, χ2 = 1.047, Rwp = 6.1%)a.
| x | Y | z | 100Ui/Ue | M (μB) | |
|---|---|---|---|---|---|
| Sc1 | 0 | 0 | 0.04769(5) | 0.378(5) | |
| Fe1 | 0 | 0 | 0.257903b | 0.510(5) | 2.72(4) |
| O1 | 0.3424(2) | –0.0260 (1) | 0.8176(2) | 0.482(5) | |
| Fe2 | 0 | 0 | 0.04769(5) | 0.378(5) | 2.72(4) |
| Sc2 | 0 | 0 | 0.257903b | 0.510(5) |
Further details of the refinement can be found in the cif file. Sc1 and Fe2 are 46% occupied whilst Sc2 and Fe1 are 54% occupied as determined from the anomalous scattering experiment.
Fixed to fix the origin.
Table 2. Refined Bond Lengths for Polar Corundum ScFeO3.
| Fe2|Sc1 | O1 | 3× | 1.9948(20) Å |
| O1 | 3× | 2.1391(27) Å | |
| Sc2|Fe1 | O1 | 3× | 2.0324(17) Å |
| O1 | 3× | 2.1752(27) Å |
The isolation of polar corundum SFO under high P/T reaction conditions can be understood by DFT calculations of the enthalpies of competing structure types (perovskite, ilmenite, polar corundum, postperovskite (CaIrO3), and YMnO3) in comparison with a mixture of the binary oxides at up to 40 GPa (Figure 6). Ferro- (FM) and antiferromagnetic (AFM) phases have been considered for all structures. Taking only 0 K effects into account (upper diagram), the polar corundum structure is predicted not to form a stable phase. Instead, the mixture of AFM binary oxides is stable up to 10.5 GPa, when it transforms into an AFM perovskite, which finally converts into the CaIrO3 postperovskite structure at pressures above 41 GPa. Temperature effects on the phase stability arise from configurational and magnetic terms. The enthalpy of the paramagnetic phase is approximated as the mean of the AFM and FM enthalpies of each polymorph. Configurational effects are not important for polymorphs where Fe and Sc are in substantially different environments (perovskite, postperovskite, and YMnO3).
Figure 6.
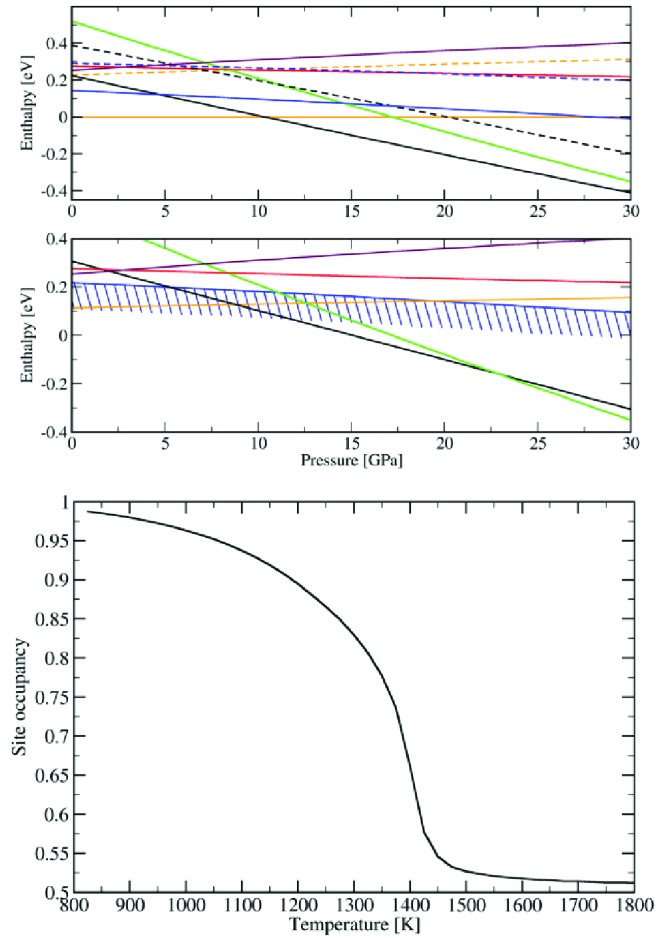
Calculated enthalpies as a function of pressure for ScFeO3 polymorphs: polar corundum (blue), perovskite (black), YMnO3 (purple), ilmenite (red), and postperovskite (green) relative to the binary oxides (yellow). Upper plot: 0 K results, for FM (solid lines) and AFM (dashed lined) phases. Mid plot: 1773 K results, for the paramagnetic phase, where the blue area represents the contribution of the configurational entropy to the polar corundum, with the lower bound shown representing the maximum possible stabilizing contribtion. Both magnetic and configurational terms are essential for the polar corundum to become the stable phase between 0 and 12 GPa. Bottom diagram: temperature dependence of cation order in polar corundum SFO from MC simulations, showing a transition from a mainly ordered to a mainly disordered structure between 1300 and 1450 K.
For ilmenite and polar corundum, the results above show that antisite disorder is important. A first order approximation of the entropy can be obtained by describing the material as a solid solution AxB(1–x)O3 of the Fe–Sc dimers, where A and B correspond to the two possible orientations of each dimer. This entropy leads to a stabilization of 0.105 eV/f.u. at 1773 K for ilmenite and polar corundum. Taking configurational and magnetic terms into account stabilizes the polar corundum structure at modest pressures with respect to the binary oxides (Figure 6).
Antisite disorder in the polar corundum structure has been quantified by a series of Monte Carlo (MC) simulations. Interaction parameters are based on an extended Ising model and have been chosen to reproduce the relative energy of different magnetic phases and antisite defects from the DFT calculations (reported in Supporting Information Table S2). The temperature dependence of antisite disorder with respect to the R3c structure (Figure 6) shows a sharp drop in cation order from 0.82 to 0.54 between 1300 and 1450 K. At the synthesis temperature, the MC model predicts the material to be paramagnetic (neither short nor long-range magnetic order remains appreciable in the magnetic RDF) and gives a composition of [Sc0.515Fe0.485][Sc0.485Fe0.515]O3, in good agreement with experiment. This indicates that increased cation order can be obtained by annealing the material below 1300 K, but the reduced configurational entropy under these conditions provides insufficient stabilization of the polar corundum structure with respect to the binary oxides.
Having ascertained the structural details of SFO as a polar partially disordered phase compatible with ferroic behavior, we measured its functional response to applied electric and magnetic fields. The polarization of the ideal crystal structure was computed using a Wannier function approach. The same inversion mechanism was found as in LiNbO342 via a migration of an octahedral ion through the face of the octahedron perpendicular to the c-axis (barriers are discussed in Supporting Information Figures S11 and S12). For the fully ordered R3c ScFeO3 structure, the polarization was computed as 3.3 μC cm–2. This small value originates from the relative polarity of Fe–O and Sc–O bonds and the difference in the off-center displacements of Sc3+ and Fe3+, where Fe3+ is more able than most magnetic transition metal cations to tolerate irregular environments due to its spherically symmetric d5 high spin (HS) configuration. The ionic contribution to the polarization was calculated as 4.0 μC cm–2 with a formal point charge model at the geometry found in the DFT calculations using the program PSEUDO.43 Experimental measurements made use of piezoresponse force microscopy (PFM).44,45 The combination of a high level of structural disorder and small polarization even within a fully ordered ScFeO3 makes PFM observation of polar domains difficult, and in general, no response was observed. In some regions of the ceramic, however, clear 180° domains were seen in the PFM phase image (Figure 7, Figure S11), showing equal amplitude domains separated by a zero amplitude domain wall. The small effective d33 of ∼0.1 pm V–1 in the ceramic (estimated from a comparison with periodically poled LiNbO3) reinforces the difficulty of observing the polar domains. PFM spectroscopy was performed on a thin film sample where the PFM amplitude was measured as a function of the ac voltage applied at a single point. The response is ∼5 pm V–1 and is linear, as would be expected from a piezoelectric material, as opposed to a quadratic response characteristic of electrostriction that occurs in all materials.
Figure 7.

Piezoresponse force microscopy of ScFeO3. (a) Topography. (b) Vertical PFM phase. (c) Point spectroscopy of the vertical piezoresponse amplitude as a function of ac drive amplitude. (d) Vertical PFM amplitude. PFM images a,b, and d were measured on a ceramic sample; PFM spectroscopy c was measured on a thin film sample. White circles highlight two domains of equal amplitude but opposite phase and are separated by a domain wall of zero amplitude.
Polar corundum structured ScFeO3 itself displays poor dielectric characteristics with large and frequency-dependent relative permittivity and loss (Figure S8(a)), together with “dead short” P(E) behavior.46 Annealing to reduce the oxygen vacancy content proposed as the origin of the high loss is not possible due to the decomposition of SFO. Substitution of Fe3+ by Cr3+ in ScFe1–xCrxO3 is possible up to x = 0.1 (Figure 2b) and increases the ambient pressure thermal stability, permitting annealing at 750 °C in oxygen for 6 h (Figure S4). This decreases the dielectric loss significantly (tan δ < 0.1 at 1 kHz), and the (relative) dielectric permittivity attains a frequency-independent high frequency limit of 60 above 1 kHz (Figure 8b), within the range reported for BiFeO310,47,48—this value is suggested to be intrinsic. The divergence of the dielectric constant and dielectric loss at low frequency is ascribed to hopping conductivity,49 which is also common for BiFeO3.50,51 Cole–Cole plots for ScFe1–xCrxO3 (x = 0 and 0.1) are shown and discussed in the Supporting Information on page S15. P(E) loops close at high frequencies, suggesting removal of the influence of extrinsic charge carriers, but they do not saturate, reaching 1.4 μC/cm2 at 50 kV/cm and 100 Hz (Figure S9 is consistent with alignment of the intrinsic polarization without sufficient field being applied to switch it. Nonsaturated P(E) loops are common in polycrystalline BiFeO3 and explained in terms of high conductivity or large coercive fields due to pinned domain walls.52−55 Ferroelectricity is not demonstrated by these measurements.)
Figure 8.

Frequency dependent dielectric constant and dielectric loss of (a) ScFeO3 and (b) ScFe0.9Cr0.1O3.
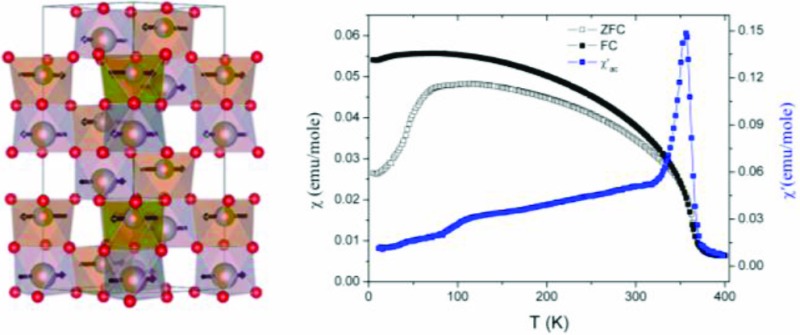
Neutron powder diffraction (NPD) shows that polar corundum ScFeO3 is magnetically ordered at 300 K, adopting a G-type antiferromagnetic (AFM) structure with antiparallel spins for all nearest neighbors: the observed moment orientation within the close-packed layers is the same as that found in the high temperature phase of α-Fe2O3: this orientation permits a weak ferromagnetic (WFM) canting.56 Alternating current susceptibility measurements on ScFeO3 in a 5 Oe magnetic field reveal a cusp at 356 K consistent with the onset of antiferromagnetic order (Figure 9; for Cr doped samples, see Figure 10 and Table 3). Below this temperature, the FC and ZFC DC magnetization data measured at 1000 Oe diverge, and hysteresis is observed in M(H) loops (Ms reaching 0.0106 μB/formula unit at 5 K—this small moment canting would not be directly detectable in the NPD measurement), consistent with weak ferromagnetism occurring simultaneously with the antiferromagnetic order. Similar ordering temperatures and magnetization values were obtained in three distinct samples. The bulk nature of the antiferromagnetic order and coupled WFM is proved by Mössbauer spectroscopy (Figure 9c), which reveals TN = 360 ± 5 K, consistent with the magnetization data. Mössbauer spectroscopy is a local probe of the magnetic field sensed by each iron nucleus and is thus the technique of choice to avoid confusion by small quantities of highly magnetic impurities, which is a potential problem with bulk magnetization measurements used in isolation. The isomer shift indicates Fe3+, and the observation of a quadrupole splitting is consistent with the observed distorted octahedral environment. The absence of a center of symmetry on the Fe–O–Fe exchange path allows weak ferromagnetism via the Dzialoshinskii–Moriya interaction. The antisite defects create local FM moments associated with the ilmenite-like environment.
Figure 9.

Magnetic behavior of high pressure ScFeO3. (a) Temperature dependence of the ZFC and FC dc susceptibility measured at 1000 Oe, and the real part of the ac susceptibility measured at 5 Oe. (b) Temperature dependence of the saturation magnetization, extracted from the M–H curves, by extrapolating the high-field magnetization. The inset shows a magnified version of 5 and 300 K M(H) data; M(H) measurements at other temperatures, used to construct the Msat vs T plot, are given in Figure S8. (c) Mössbauer spectra recorded at four different temperatures. The green and red subspectra at 350 K represent fits to the ordered and nonordered components, respectively, while the solid lines are guides to the eye.
Figure 10.
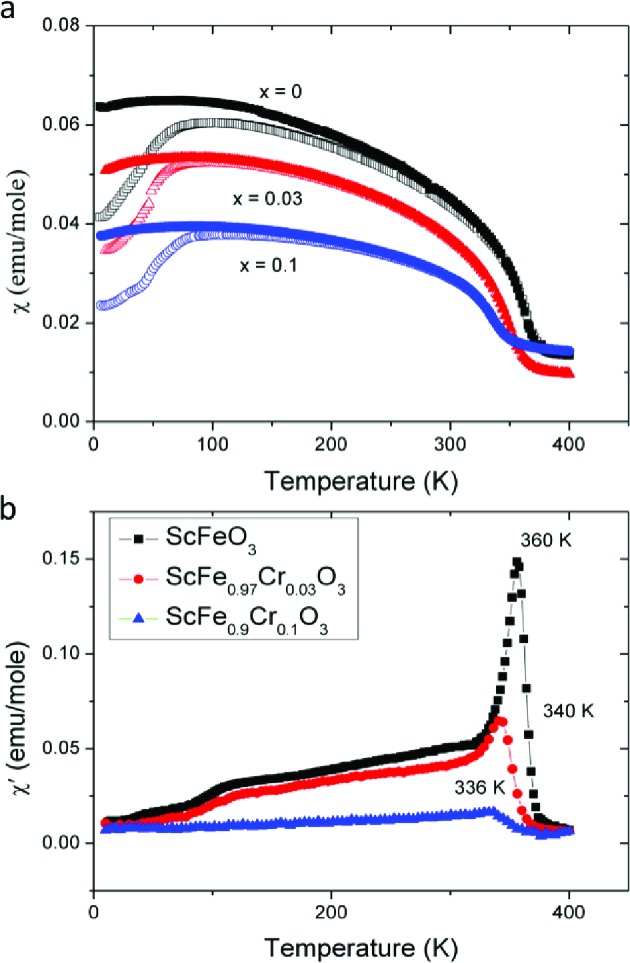
(a) Temperature dependence of the ZFC and FC dc magnetic susceptibility of ScFe1–xCrxO3 (x = 0, 0.03, and 0.10), measured at 1000 Oe. Open and closed symbols represent ZFC and FC data, respectively. (b) Temperature dependence of the real part of the ac susceptibility of ScFe1–xCrxO3 (x = 0, 0.03, and 0.10), measured at 5 Oe.
Table 3. Change of Ordering Temperature in ScFe1–xCrxO3 (x = 0, 0.03, and 0.10).
| composition | TN (K) |
|---|---|
| ScFeO3 | 356 |
| ScFe0.97Cr0.03O3 | 340 |
| ScFe0.9Cr0.1O3 | 336 |
The simultaneous appearance of WFM with the main AFM order (Figure 9) confirms that WFM is an intrinsic property of SFO. The ordering temperature is an order of magnitude higher than that found in the bixbyite polymorph of ScFeO3 because of the more favorable Fe–O–Fe angles that allow the extremely strong d5–d5 AFM superexchange to operate. The dilution of Fe3+ with nonmagnetic Sc3+ reduces the magnetic ordering temperature from 948 K in the nonpolar R3̅c corundum α-Fe2O3 to ∼360 K in ScFeO3, but the strength of the exchange interaction and retention of the 3D network of Fe3+ connectivity in the polar corundum still places the ordering above room temperature. Unlike the case of α-Fe2O3 (where canting disappears below the 260 K Morin spin reorientation transition due to changes in the anisotropy aligning the moments along the c-axis), WFM in SFO persists to 5 K. The Cr3+-doped compounds on which the permittivity measurements were performed display WFM order above room temperature with coupled TN and Tc demonstrated by ac and dc magnetization measurements, with both Ms (judged by the low T temperature invariant FC magnetization) and TN decreasing as the Cr3+ content increases (Figure 10, Table 3).
Conclusions
Corundum is an archetypal oxide structure, and the observation of a polar variant in SFO is unexpected. CBED unambiguously demonstrates the polar nature of high pressure ScFeO3, accounted for by partial Sc/Fe order over the two distinct octahedral cation sites. The extent of the cation site order observed is quantitatively consistent with DFT calculations of antisite dimer defect creation enthalpies. The absence of extensive solid solution in both bulk and thin film is consistent with each dimer containing one Sc and one Fe—the small degree of ordering can then be assigned to disorder in the orientation of these dimers—which agrees with the high energy of homoatomic occupation of the face-sharing octahedral dimers in DFT calculations. The resulting heteroatomic dimers have two possible polar orientations, so the observed antisite disorder entropically stabilizes the polar corundum structure and is intrinsic to its formation. The combination of high pressure (to eliminate vacancies in the bixbyite structure where face-sharing octahedra are absent) and high temperature (to generate sufficient though not complete antisite disorder) is thus required to stabilize the polar corundum structure. In contrast to the high pressure LiNbO3 polymorph of FeTiO312 and both ordered and corrundum type (In1–xMx)MO3 (M = Fe1/2Mn1/2; x = 0.143),14,15 weak ferromagnetism persists above room temperature and polar distortions are maintained at high levels of disorder. The calculated theoretical electrical polarization value is smaller than that of BiFeO3 (60 μC/cm2)52 while it is 3 orders of magnitude larger than those of multiferroics where a spin spiral breaks the inversion symmetry.1,4,5 The observed Ms at 5 K (0.01 μB/f.u.) is of the same magnitude (0.008 μB/f.u) as the Ms measured for high pressure FeTiO3 at 100 K.12
The difference in polarity of the Sc–O and Fe–O bonds, and between Sc3+ and Fe3+ off-center displacements, thus creates the observed polarization, assigned to the different bonding preferences of d0 Sc3+ and d5 Fe3+. The Fe3+ 3d5 electrons are connected three-dimensionally in the polar corundum, resulting in magnetic order above room temperature. Calculations show antisite defects to have FM nature, and as in α-Fe2O3 the absence of an inversion center in the Fe–O–Fe superexchange pathway allows weak ferromagnetism to occur simultaneously with the antiferromagnetic long-range order. Unlike corundum-structured α-Fe2O3, weak ferromagnetism in polar corundum ScFeO3 persists to 5 K, consistent with different local environments of the Fe3+ moments in the two materials. Polar corundum is thus a platform for the wide-temperature coexistence of polarity and ferromagnetism, as, in contrast to other mechanisms for combining polarity and permanent magnetization above room temperature,8,9 it does not rely on symmetry breaking associated with magnetic order, resulting in a polar distortion detectable by both structural and functional (piezoresponse force microscopy) measurements. Further modification of the polar corundum structure based on SFO is required to permit switching of the polarization.
Acknowledgments
We acknowledge the UK EPSRC for support under EP/H000925 and the ERC for support under ERC Grant agreement 227987. We wish to thank the STFC for access to DLS (where Dr. Stephen Thompson and Prof. Chiu Tang are thanked for assistance on beamline I11), ISIS (where Dr. Ron Smith is thanked for assistance on the POLARIS), and ESRF (where Dr. Adrian Hill and Prof. Andy Fitch are thanked for assistance on ID31).
Supporting Information Available
Eleven figures and 4 tables plus gsas list files and powder cif for the final neutron refinement, together with the full author list for ref 13. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Cheong S. W.; Mostovoy M. Nat. Mater. 2007, 6, 13. [DOI] [PubMed] [Google Scholar]
- Choi Y. J.; Yi H. T.; Lee S.; Huang Q.; Kiryukhin V.; Cheong S. W. Phys. Rev. Lett. 2008, 100, 047601. [DOI] [PubMed] [Google Scholar]
- Hur N.; Park S.; Sharma P. A.; Ahn J. S.; Guha S.; Cheong S. W. Nature 2004, 429, 392. [DOI] [PubMed] [Google Scholar]
- Kimura T. Annu. Rev. Mater. Res. 2007, 37, 387. [Google Scholar]
- Kimura T.; Goto T.; Shintani H.; Ishizaka K.; Arima T.; Tokura Y. Nature 2003, 426, 55. [DOI] [PubMed] [Google Scholar]
- Hill N. A.; Filippetti A. J. Magn. Magn. Mater. 2002, 242, 976. [Google Scholar]
- Khomskii D. I. J. J. Magn. Magn. Mater. 2006, 306, 1. [Google Scholar]
- Kitagawa Y.; Hiraoka Y.; Honda T.; Ishikura T.; Nakamura H.; Kimura T. Nat. Mater. 2010, 9, 797. [DOI] [PubMed] [Google Scholar]
- Ishiwata S.; Taguchi Y.; Murakawa H.; Onose Y.; Tokura Y. Science 2008, 319, 1643. [DOI] [PubMed] [Google Scholar]
- Catalan G.; Scott J. F. Adv. Mater. 2009, 21, 2463. [Google Scholar]
- Fennie C. J. Phys. Rev. Lett. 2008, 100, 167203. [DOI] [PubMed] [Google Scholar]
- Varga T.; Kumar A.; Vlahos E.; Denev S.; Park M.; Hong S.; Sanehira T.; Wang Y.; Fennie C. J.; Streiffer S. K.; Ke X.; Schiffer P.; Gopalan V.; Mitchell J. F. Phys. Rev. Lett. 2009, 103, 047601. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; et al. Nature 2010, 466, 954. [DOI] [PubMed] [Google Scholar]
- Belik A. A.; Furubayashi T.; Yusa H.; Takayama-Muromachi E. J. Am. Chem. Soc. 2011, 133, 9405. [DOI] [PubMed] [Google Scholar]
- Belik A. A.; Furubayashi T.; Matsushita Y.; Tanaka M.; Hishita S.; Takayama-Muromachi E. Angew. Chem. 2009, 48, 6117. [DOI] [PubMed] [Google Scholar]
- Belik A. A.; Matsushita Y.; Tanaka M.; Takayama-Muromachi E. Angew. Chem. 2010, 49, 7723. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Parise J. B. Mater. Res. Bull. 1997, 32, 1617. [Google Scholar]
- Castillo-Martinez E.; Bieringer M.; Shafi S. P.; Cranswick L. M.; Alario-Franco M. A. J. Am. Chem. Soc. 2011, 133, 8552. [DOI] [PubMed] [Google Scholar]
- Richard J. G.; et al. J. Phys.: Condens. Matter 2005, 17, 7633. [Google Scholar]
- Coelho A. TOPAS—Academic, General Profile and Structure Analysis Software for Powder Diffraction Data, version 4.1; 2007.
- Suortti P. J. Appl. Crystallogr. 1972, 5, 325. [Google Scholar]
- Pitschke W. M. N.; Hermann H. Powder Diffr. 1993, 8, 6. [Google Scholar]
- Pitschke W.; Collazo J. A. L.; Hermann H.; Hildenbrand V. D. J. Appl. Crystallogr. 1996, 29, 561. [Google Scholar]
- Larson A. C.; Von Dreele R. B. General Structure Analysis System (GSAS). Los Alamos Natl. Lab., [Rep.] LA (U. S.) 2000, 86–748. [Google Scholar]
- Toby B. H. J. Appl. Crystallogr. 2001, 34, 210. [Google Scholar]
- Cui J.; Huang Q.; Toby B. H. Powder Diffr. 2006, 21, 71. [Google Scholar]
- Wills A. S. Phys. B (Amsterdam) 2000, 276–278, 680. [Google Scholar]
- Momma K.; Izumi F. J. Appl. Crystallogr. 2008, 41, 653. [Google Scholar]
- Koch C. Ph.D. thesis, Arizona State University, 2002. [Google Scholar]
- Dovesi R.; Saunders V. R.; Roetti C.; Orlando R.; Zicovich-Wilson C. M.; Pascale F.; Civalleri B.; Doll K.; Harrison N. M.; Bush I. J.; D’Arco Ph.; Llunell M.. CRYSTAL09; University of Torino: Torino, 2009.
- Geller S.; Sherwood R. C.; Williams H. J. J. Chem. Phys. 1961, 35, 1908. [Google Scholar]
- Grant R. IEEE Trans. Magn. 1972, 8, 637. [Google Scholar]
- Breard Y.; Fjellvag H.; Hauback B. Solid State Commun. 2011, 151, 223. [Google Scholar]
- Prewitt C. T.; Shannon R. D.; Rogers D. B.; Sleight A. W. Inorg. Chem. 1969, 8, 1985. [Google Scholar]
- Shannon R. D.; Prewitt C. T. Mater. Res. Bull. 1969, 4, 57. [Google Scholar]
- Shannon R. D.; Prewitt C. T. Acta Crystallogr., B 1969, B 25, 925. [Google Scholar]
- Salvador P. A.; Doan T.-D.; Mercey B.; Raveau B. Chem. Mater. 1998, 10, 2592. [Google Scholar]
- Spence J. C. H.; Zuo J. M.. Electron Microdiffraction; Plenum Press: New York, 1992. [Google Scholar]
- Stokes H. T.; Kisi E. H.; Hatch D. M.; Howard C. J. Acta Crystallogr., B 2002, 58, 934. [DOI] [PubMed] [Google Scholar]
- Waseda Y.Anomalous X-Ray Scattering for Materials Characterization; Springer: 2002 [Google Scholar]
- Shull C. G.; Strauser W. A.; Wollan E. O. Phys. Rev. 1951, 83, 333. [Google Scholar]
- Ohkubo Y.; Murakami Y.; Saito T.; Yokoyama A.; Uehara S.; Kawase Y. Phys. Rev. B 2002, 65, 052107. [Google Scholar]
- Capillas C.; Tasci E. S.; de la Flor G.; Orobengoa D.; Perez-Mato J. M.; Aroyo M. I. Z. Kristallogr. 2011, 226, 186. [Google Scholar]
- Kholkin A.; Kalinin S. V.; Roelofs A.; Gruverman A.. Scanning Probe Microscopy: Electrical and Electromechanical Phenomena at the Nanoscale; Springer: New York, 2007. [Google Scholar]
- Burnett T. L.; Weaver P. M.; Blackburn J. F.; Stewart M.; Cain M. G. J. Appl. Phys. 2010, 108, 042001. [Google Scholar]
- Dawber M.; Rabe K. M.; Scott J. F. Rev. Mod. Phys. 2005, 77, 1083. [Google Scholar]
- Kamba S.; Nuzhnyy D.; Savinov M.; Sebek J.; Petzelt J.; Prokleska J.; Haumont R.; Kreisel J. Phys. Rev. B 2007, 75, 024403. [Google Scholar]
- Lu J.; Gunther A.; Schrettle F.; Mayr F.; Krohns S.; Lunkenheimer P.; Pimenov A.; Travkin V. D.; Mukhin A. A.; Loidl A. Eur. Phys. J. B 2010, 75, 451. [Google Scholar]
- Lunkenheimer P.; Krohns S.; Riegg S.; Ebbinghaus S. G.; Reller A.; Loidl A. Eur. Phys. J.—Spec. Top. 2010, 180, 61. [Google Scholar]
- Chung C. F.; Lin J. P.; Wu J. M. Appl. Phys. Lett. 2006, 88, 242909. [Google Scholar]
- Wang Y. P.; Zhou L.; Zhang M. F.; Chen X. Y.; Liu J. M.; Liu Z. G. Appl. Phys. Lett. 2004, 84, 1731. [Google Scholar]
- Lebeugle D.; Colson D.; Forget A.; Viret M. Appl. Phys. Lett. 2007, 91, 022907. [Google Scholar]
- Pradhan A. K.; Zhang K.; Hunter D.; Dadson J. B.; Loiutts G. B.; Bhattacharya P.; Katiyar R.; Zhang J.; Sellmyer D. J.; Roy U. N.; Cui Y.; Burger A. J. Appl. Phys. 2005, 97, 093903. [Google Scholar]
- Rojac T.; Kosec M.; Budic B.; Setter N.; Damjanovic D. J. Appl. Phys. 2010, 108, 074107. [Google Scholar]
- Scott J. F. J. Phys.: Condens. Matter 2008, 20, 021001. [Google Scholar]
- Lovesey S. W.; Rodriguez-Fernandez A.; Blanco J. A. Phys. Rev. B 2011, 83, 054427. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.