

Abstract
Our understanding of major depressive disorder (MDD) has focused on the influence of genetic variation and environmental risk factors. Growing evidence suggests the additional role of epigenetic mechanisms influencing susceptibility for complex traits. DNA sequence within discordant monozygotic twin (MZT) pairs is virtually identical; thus, they represent a powerful design for studying the contribution of epigenetic factors to disease liability. The aim of this study was to investigate whether specific methylation profiles in white blood cells could contribute to the aetiology of MDD. Participants were drawn from the Queensland Twin Registry and comprised 12 MZT pairs discordant for MDD and 12 MZT pairs concordant for no MDD and low neuroticism. Bisulphite treatment and genome-wide interrogation of differentially methylated CpG sites using the Illumina Human Methylation 450 BeadChip were performed in WBC-derived DNA. No overall difference in mean global methylation between cases and their unaffected co-twins was found; however, the differences in females was significant (P=0.005). The difference in variance across all probes between affected and unaffected twins was highly significant (P<2.2 × 10−16), with 52.4% of probes having higher variance in cases (binomial P-value<2.2 × 10−16). No significant differences in methylation were observed between discordant MZT pairs and their matched concordant MZT (permutation minimum P=0.11) at any individual probe. Larger samples are likely to be needed to identify true associations between methylation differences at specific CpG sites.
Keywords: depression, epigenetics, MDD, methylation, twins
Introduction
Major depressive disorder (MDD) is a complex disorder with a considerable impact on the quality of patient's lives1 including an increased rate of mortality.2, 3 Lifetime prevalence is estimated to be 15% and it is twice more common in women than in men.2, 4 Heritability of MDD is estimated to be ∼40%.5 There have been no common genetic variants convincingly associated with MDD,6 and most of the variants identified for complex psychiatric disorders contribute to a small fraction of the genetic variation and explain only a small proportion of the heritability.7 Growing evidence suggests that variations in epigenetic profiles (that is, DNA methylation) influence complex trait variation.8 Epigenetic modifications do not directly interfere with the gene transcript but control tissue and temporal specificity of gene expression.9, 10 DNA methylation is the most common epigenetic variation in the mammalian genome8 and is not entirely stable; stochastic and environmental events can also generate variations over time.11 Non-heritable influences on depression may mediate their biological effects through epigenetic mechanisms such as methylation of CpG dinucleotides.
Several epigenetic studies have attempted to identify differences in DNA methylation associated with complex traits. Associations between variations in DNA methylation profiles and various psychiatric disorders (for example, schizophrenia, MDD, bipolar disorder and so on) have been reported.12, 13, 14, 15, 16, 17 Some limitations should be considered from these studies (for example, small samples sizes, sample heterogeneity, epigenome coverage). Tissues involved in complex psychiatric disorders are not directly accessible from living patients; epigenetic studies often rely on peripheral tissue biomarkers such as buccal, gut and white blood cells (WBC).18 Post-mortem tissue can also be used in epigenetic studies to compare central and peripheral tissue;19 however, samples are not easily accessible. Moreover, post-mortem epigenetic measures may reflect treatment rather than disease. More research is needed to determine the accuracy of peripheral tissue biomarkers.19
Discordant monozygotic twin (MZT) pairs constitute a powerful design in epigenetic studies.20, 21 Single-nucleotide polymorphisms and other DNA sequence variations, which are abundant in singleton-based studies, are not a confounding source of variation in MZT-based epigenetic studies.
The aim of our study was to examine whether diverse epigenetic profiles, specifically DNA methylation profiles, are associated with increased risk of MDD. We analysed genome-wide methylation patterns after bisulphite conversion of CpG sites using the Illumina Human Methylation (HM) 450 BeadChip (HM 450 BeadChip) of WBC-derived DNA from 12 discordant MZT (6 male and 6 female) pairs and 24 concordant MZT pairs.
Materials and methods
Sample selection/study design
Participants were drawn from the Queensland Twin Registry. Phenotypic information was collected as part of studies undertaken at the Queensland Institute of Medical Research each approved by the Institute's ethics committee. On the basis of the study, depressive symptoms were evaluated with either the Semi-Structured Assessment for the Genetics of Alcoholism,22 (adapted for use in Australia) or the Composite International Diagnostic Interview23 questionnaires. A diagnosis of MDD was constructed from these questionnaires according to the Diagnostic and Statistical Manual of Mental Disorders-IV criteria.24 A total of 17 958 individuals from 6855 families have completed psychiatric interviews, including a total of 1812 MZT pairs, of whom 261 were found to be discordant for MDD.
Six MZT female pairs (including five MZT female pairs previously epityped with the Illumina HM 27 BeadChip; Illumina, San Diego, CA, USA) and six MZT male pairs discordant for MDD were selected as ‘case pairs'. Where possible, we selected case probands with (the more severe) recurrent MDD; all six MZT females and two of the six males met this criterion. Further, we aimed to select twins who were recorded as non-smokers and not alcohol or drug dependent, criteria that imposed severe restrictions on MZT selection, and so selection criteria were extended to include MZT that were matched for these criteria (for example, both smokers with similar daily consumption and age of initiation). Six MZT female pairs and six MZT male pairs concordant for no MDD and low neuroticism were selected as ‘control pairs'. Neuroticism is a domain of personality easily measured by self-report. High neuroticism scores are associated with anxiety and depression; therefore, low scores are expected to represent those with a low liability for depression.25 This gives our study more power to detect variations in methylation status associated with MDD. Environmental factors such as smoking, alcohol or drug abuse are associated with altered methylation patterns.26, 27 Each concordant MZT pair was selected to match as closely as possible to a discordant MZT pair on a number of key variables (Supplementary Tables 1 and 2). Matching criteria were date of blood sample collection, (as methylation differences increase with age20, 25) smoking and alcohol as well as previous history of drug use. A total of 24 MZT pairs (48 individuals) were selected for our study (Table 1). All individuals were of Northern European ancestry.
Table 1. Summary of cases and controls.
| Cases | Controls |
|---|---|
| 6 (5+1) MZT ♀ pairs discordant for MDD | 6 MZT ♀ pairs concordant for no MDD and low neuroticism |
| 6 MZT ♂ pairs discordant for MDD | 6 MZT ♂ pairs concordant for no MDD and low neuroticism |
| Total 24 MZT discordant for MDD | Total 24 MZT concordant for no MDD and low neuroticism |
Abbreviations: MDD, major depressive disorder; MZT, monozygotic twin.
Bisulphite conversions
Bisulphite conversions of previously extracted WBC-derived DNA from the 48 individuals, 6 technical replicate samples and technical control samples (described below) were performed following the protocol and using reagents of the EZ DNA methylation Kit (Zymo Research, Irvine, CA, USA). Each conversion included 10 samples and DNA recovery after conversion was quantified using Nanodrop (Thermo Scientific, Wilmington, DE, USA).
In total, eight bisulphite conversions were undertaken, each comprising three to four MZT pairs, technical replicates and control samples. Later runs repeated eight MZT pairs that had generated <80% DNA recovery in earlier runs (Supplementary Table 3).
Technical controls consisted of commercial DNA samples: Centre Etude Polymorphism Humain (CEPH) male (GM07029), CEPH female (GM06997) and FSK standard (Coriell Institute of Medical Research, Camden, NJ, USA). DNA recovery for the CEPH female sample after bisulphite conversion was ∼50% for this reason the CEPH male and the FSK standard were included in the later bisulphite conversions (Supplementary Table 3). Only FSK standard samples were included in the HM 450 BeadChips as controls because of their high percentage recovery of the bisulphite-converted DNA.
Infinium methylation assay
Bisulphite-converted DNA from 24 MZT pairs, 4 technical replicate samples and 4 FSK standard controls were hybridised to the Illumina HM 450 BeadChip, following the Illumina Infinium HD methylation protocol and using reagents and conditions supplied by Illumina. The HM 450 BeadChip-assessed methylation status was interrogated at 485 577 CpG sites across the genome. It provides coverage of 99% of RefSeq genes. Methylation scores for each CpG site are obtained as a ratio of the intensities of fluorescent signals and are represented as β-values.
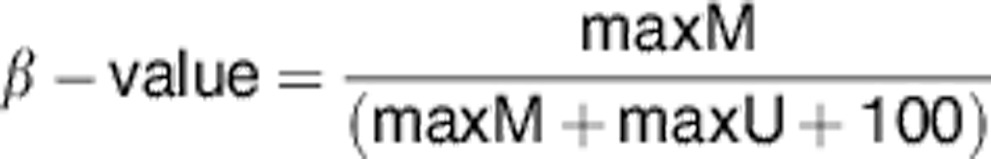 |
where max=maximum intensity of fluorescence, M=methylated allele and U=unmethylated allele.28 The 100 in the denominator prevents division by 0 when both max M and max U are very small, but has little impact otherwise as max M+max U>1000 in >95% of sites.29 β-values range between 0 and 1, where 0 means that no DNA molecules with a methylated CpG at the site of interest were detected in the sample, while 1 means that all DNA molecules had a methylated CpG site in the sample.
Each MZT pair was placed on the same array and in neighbouring wells to avoid batch effects contributing to differences between pairs. A total of five HM 450 BeadChips were used for this study. Two of the replicate samples were placed on the same array in non-adjacent wells from the corresponding MZT whereas the other two replicate samples were placed on different arrays (Supplementary Table 4). The FSK male and female samples were also duplicated and placed on different chips. A total of 56 samples were included in the arrays. Arrays were scanned with the Illumina iScan (Software Version 3.3.28) and the intensities of the images were extracted using the GenomeStudio (2010.3) methylation module (1.8.5) software.
Quality control
Background was subtracted from our data using GenomeStudio (2010.3). Illumina's specification for the HM 450 BeadChip is 98% detection of CpG with P<0.05. All of our samples included in the five BeadChips exceeded specification, with >99% of CpG loci detected; therefore, no samples were excluded from the study. Three hundred seventy-three probes for which 75% of the samples had a detection P-value>0.05 were removed. A total of 485 204 probes remained.
The β-values were transformed using a logit transformation in the IMA30 package in the R environment for statistical computing (version 2.13.2); http://www.R-project.org). Transforming via a logit transformation increases the validity of statistical tests of differential methylation.29 Supplementary Figure 1 shows a histogram of the transformed β-values across all samples. The peak correction method of Deuderwaerder et al.31 was applied to correct for differences in results between the two chemistries utilised on the HM 450 chip. QC plots produced by the IMA package are shown in Supplementary Figures 2–4. Unsupervised clustering of all samples showed that male and female samples clustered together. Furthermore, replicate samples and twin samples clustered together providing an indication of the accuracy of the methylation detection procedure.
To improve the accuracy of our results, we excluded potential sources of technical and biological biases before analysing the methylation data. There is currently no standard procedure for filtering the samples and probes before methylation analysis.32 Our experimental design, including replicate samples enabled stringent quality control (QC) steps. These QC steps are described in detail in the Supplementary Methods. The number of sites annotated by probe type that were removed by the QC steps are shown in Table 2. After removing probes with large amounts of missing data and high levels of discordance between replicate pairs, a total of 462 002 (95.1%) probes remained for analysis.
Table 2. HM 450 BeadChip distribution of probes.
| Annotation | Before QC | QC1 | QC2 |
|---|---|---|---|
| Island | 150 254 | 143 391 | 138 738 |
| Shelf | 47 161 | 46 447 | 45 691 |
| Shore | 112 072 | 109 736 | 107 394 |
| NCpG | 176 277 | 172 695 | 170 178 |
| Total | 485 557 | 472 269 | 462 001 |
Abbreviations: HM, human methylation; QC, quality control.
QC1 represents remaining probes after QC1. QC2 represents remaining probes after QC2.
Statistical analysis
A two-sample t-test was used to test for differences in overall mean methylation between monozygotic cases and their unaffected co-twin. In order to test for differences in variance between cases and their unaffected co-twin, the variance for each probe was computed in cases and their unaffected co-twin separately, and the distributions were compared using the Wilcoxon signed-rank test. All statistical tests were implemented in R.
Testing for differences between MZTs owing to MDD at each individual probe
In order to test for differences in methylation between monozygotic co-twins owing to MDD, a linear model was fitted. For each probe, the absolute difference between each twin pair was taken and regressed on matched twin set and case–control status.
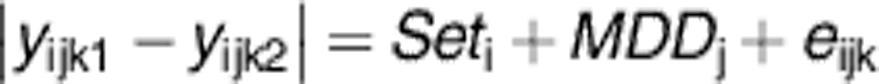 |
where, yijkl=the residual transformed β-value of a probe after adjusting for batch and chip effects of the lth (l=1,2) twin of the jth twin pair (j=1,2) from the ith set (i=1,..,12). MDDj is an indicator such that MDDj=1 for the discordant pairs and MDDj=0 for the concordant pair eijk is the random error term. By taking the absolute difference in the residuals of the transformed β-value between MZT, the test can reflect both probes that are associated with more or less methylation or probes that are more variably methylated probes associated with MDD. Under the MZT design we assume that any confounding variables are independent of the differences between MZT.
To establish the significance of the top results, a set of 100 permutations were performed. In each permutation, the concordant pair and discordant label were randomly assigned within each twin pair set, the analysis was performed for each probe, and the P-value for case–control status was extracted. The results of each permutation were extracted and ranked. The mean of the P-values with the same rank across all 100 permutations was calculated and the distribution of the means was taken as the null distribution. An empirical P-value was calculated by counting the proportion of permutations that had at least one probe more significant than the top probe identified in the real data set.
Pathway analysis
All genes for which at least one probe had a P-value<0.001 for association with MDD status were uploaded to the commercial pathway analysis software Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA) and the freely available software DAVID,33 to test for enrichment of associations in functional pathways and gene ontology terms.
Results
Global differences in mean and variance
There was no significant difference in overall methylation between discordant pairs (P=0.56). As there were highly significant differences in overall methylation of autosomal probes between males and females (P<2.2 × 10−16; Supplementary Material) as previously reported by others,34, 35 we tested for differences in overall methylation levels by sex within discordant pairs. The difference in mean methylation was not significant (P=0.05), but was significant in females (P=0.005), with cases being less methylated when compared with controls (whereas the trend of differences in males was the opposite).
The difference in variance between cases and their control co-twins was highly significant (P<2.2 × 10−16), with 52.4% of probes having higher variance in cases (binomial P-value 2.2 × 10−16). This increased variance in cases was consistent across all annotated probe types (Supplementary Table 6). The result was significant in males and females (both P<2.2 × 10−16). A jackknifing procedure where one twin pair was removed and the data reanalysed also gave significant results across all 12 replicates, indicating that the significant results are not attributable to one specific pair.
The mean of the probe variances was 0.09 with s.d. 0.27 with range 0.001–10.54. The distribution was highly skewed (Supplementary Figures 6a and b), with the majority of probes having a small variance compared with a much smaller number of probes that are highly variable. In order to test whether there were differences in the distribution of variances between cases and controls in probes with both high and low overall variance, we subdivided the probes into deciles based on their variance. We then performed the Wilcoxon signed-rank test for differences in variance in each of the probe sets. Across all of the deciles, more than half of the probes had a higher variance in cases than in controls. The differences in variance were significant in each of the deciles except for the ninth (Table 3).
Table 3. Results from comparing distribution of variances in cases and controls by decile.
| Decile | Wilcoxon signed-rank test P-value | Proportion of probes with increased variance in cases |
|---|---|---|
| 1 | <2.2 × 10E−16 | 0.545 |
| 2 | <2.2 × 10E−16 | 0.539 |
| 3 | <2.2 × 10E−16 | 0.539 |
| 4 | <2.2 × 10E−16 | 0.530 |
| 5 | <2.2 × 10E−16 | 0.531 |
| 6 | <2.2 × 10E−16 | 0.522 |
| 7 | <2.2 × 10E−16 | 0.519 |
| 8 | 6.50E−04 | 0.508 |
| 9 | 8.79E−01 | 0.501 |
| 10 | 1.25E−04 | 0.504 |
In order to test whether the differences in variance could be attributed to use of antidepressants, we tested for differences in variances between cases who had been prescribed antidepressants (n=6) and their co-twin control, and separately for cases who had not been prescribed antidepressants (n=6) and their co-twin. Cases who had been prescribed antidepressants had increased variance at 51.5% of probes and the difference in variances was highly significant (P<2.2 × 10−16). Cases that had not been prescribed antidepressants had increased variance relative to their unaffected co-twins at 51.4% of probes and the difference in variances was highly significant (P<2.2 × 10−16).
MDD discordance at individual probes
The QQ plot for the genome-wide methylation analysis of concordant and discordant twins is shown in Supplementary Figure 5a. The observed distribution of P-values for the probes did not deviate from null distribution. The most significant probe was cg15083522 (P=1.38 × 10−6, Table 4), found on chromosome 10 in a CpG island upstream of the MAST4 gene. This probe was not significant when compared against the null distribution of all probes (P=0.11) or when compared with all probes in shores (P=0.06) (Supplementary Figures 5b–d). The MAST4 gene is expressed in a distinct pattern in white matter-containing regions of the brain in rodents.36 Other members of the MAST family have been implicated in dopaminergic signalling in dendrites.37 However, this gene has not previously been linked to MDD or other mood disorders.
Table 4. Probes that are most significantly differentiated between discordant and concordant pairs (P<10−4).
| ILMNID | CHR | MAPINFO | UCSC_REFGENE_NAME | UCSC_REFGENE_ACCESSION | RELATION_TO_UCSC_CPG_ISLAND | P-value |
|---|---|---|---|---|---|---|
| cg10665379 | 5 | 65891878 | MAST4 | TSS1500 | Island | 1.38E−06 |
| cg17222196 | 1 | 155057324 | EFNA3 | Body | Island | 1.89E−05 |
| cg08985282 | 7 | 2089600 | MAD1L1 | Body | Island | 2.16E−05 |
| cg25372357 | 4 | 56659770 | NA | NA | Island | 2.59E−05 |
| cg22864266 | 12 | 49319673 | FKBP11 | TSS1500 | S_Shore | 2.68E−05 |
| cg10946423 | 20 | 44565633 | PCIF1 | 5'UTR | S_Shore | 2.98E−05 |
| cg11843516 | 1 | 17023283 | ESPNP | Body | Island | 3.19E−05 |
| cg22374165 | 2 | 39199173 | LOC100271715 | 3'UTR | NA | 3.65E−05 |
| cg22231758 | 17 | 69199815 | NA | NA | NA | 3.93E−05 |
| cg14852276 | 3 | 197684729 | IQCG | 5'UTR | N_Shelf | 4.37E−05 |
| cg00270520 | 2 | 241579262 | NA | NA | N_Shore | 4.54E−05 |
| cg06339265 | 16 | 11369615 | PRM2 | 3'UTR | N_Shore | 5.76E−05 |
| cg04145601 | 20 | 44641469 | MMP9 | Body | N_Shore | 5.98E−05 |
| cg09858767 | 1 | 14742375 | NA | NA | NA | 8.21E−05 |
| cg22618878 | 6 | 27156656 | NA | NA | NA | 8.28E−05 |
| cg07048516 | 22 | 24105087 | C22orf15 | TSS200 | NA | 9.08E−05 |
| cg03666973 | 2 | 27008764 | CENPA | TSS200 | Island | 9.48E−05 |
Among the genes that harbour probes that are most significantly differentiated between discordant pairs, MAD1L1 is the only gene that has been previously implicated in the aetiology of mood disorders. The 3rd most significant probe was located in the MAD1L1 gene (cg08985282, P=2.16 × 10−5, Table 4). Recently, two independent genome-wide significant hits for schizophrenia in this gene were found,38 and evidence of association with bipolar disorder was found in a recent meta-analysis (rs4332037, P=6.3 × 10−6).39
A total of 211 genes were uploaded to Ingenuity and DAVID pathway analysis packages. Neither pathway analysis package identified any canonical pathways or biological functions to be significantly enriched after setting a false discovery rate of 5%.
Discussion
Previous studies involving MZT pairs discordant for psychiatric disorders have been limited to a small number of twin pairs (for example, two MZT pairs for schizophrenia40), or have assessed only a fraction of the genome-wide CpG sites.15, 16 This study represents the largest twin study to date of genome-wide associated methylation differences in MZT pairs discordant for MDD. Furthermore, this is one of the first methylation studies in psychiatry that utilises the HM 450 Beadchip, which provides coverage of a much larger number of CpG sites throughout the genome.
Our analyses included sets of discordant MZT pairs and its matched concordant pairs. Although our sample size was not sufficient to identify methylation differences that could be attributed to MDD, it revealed that cases show a highly significantly increased variation in methylation throughout the genome (P<2.2 × 10−16) when compared with their control co-twins. This finding suggests that the methylome in patients with MDD may be undergoing more changes as a result of environmental influences than that in controls as a result of a loss of epigenetic stability. Increased variability of methylation in cases is seen across many cancers,41 but has not been previously investigated in studies of psychiatric disorders. One potential reason for this increased variance in cases may be due to various forms of treatment including the use of medications such as antidepressants by affected twins that are not used by the unaffected twins. However, the result remained significant when comparing those reporting antidepressant use with their co-twins and also in those reporting no antidepressant use. No information was collected on the length of time that the drugs were used for or the dosage that was prescribed. Antidepressant drugs are known to induce epigenetic changes in the brain42 and may also do so in blood samples.
Our results also showed that female cases had significantly increased methylation throughout the genome when compared with their unaffected co-twin (P=0.005). This significant difference was not detected when comparing male cases with their unaffected co-twins or when analysing males and females together. We detected distinct patterns of methylation in male and female twins across a large number of autosomal probes, which suggests that analysing methylation differences across the genome by sex is appropriate.
There are a number of important limitations of the design and analysis of this study. First, the tissue available for methylation analysis in this study imposes limitations. Methylation levels are known to vary among tissues.13 Brain tissue would be the tissue of choice for the study of psychiatric disorders. However, biopsy is not an option and there is no available collection of post-mortem MZT brain tissue discordant for psychiatric disorders.43 For epigenetic studies of disorders related to neurodevelopment, it has been argued that cells that are derived from the same embryonic origin as the cells involved in the disease should be studied;8 for example, buccal cells have the same embryonic origin as brain cells. This argument is relevant for psychiatric disorders like schizophrenia where interest may focus on neurodevelopment. However, if environmental stresses influencing the liability of MDD occur after the early stages of development (for example, childhood trauma), then cells from the same embryonic origin as brain tissue (that is, buccal cells) seem no more relevant than WBC. Reliance on DNA from WBC means that the inferences that can be made regarding pathways involving the brain are limited. However, disease-related pathways can still be identified from peripheral tissues.43 DNA methylation differences identified using peripheral tissue may not be explanatory or causal of changes in gene expression taking place in the brain, but they could be used as a diagnostic tool.
A second important limitation of our study is the small sample size. The discordant MZT model is considered to be powerful for detecting epigenetic differences21 because the cases and controls are matched on their genetic complement eliminating this as a confounding variable. In order to eliminate confounding environmental risk factors, we imposed stringent criteria on the selection of MZT. Only a small number of MZT pairs fulfilled these criteria despite selection from one of the largest twin collections worldwide. Larger samples can only be achieved through international consortia or through relaxation of the selection criteria.
A third limitation of our study was the variability between samples in factors that may affect methylation. For example, first, the age of the subjects ranged from 31 to 63 (but was not significantly different between the sexes). Second, although it is estimated that 96% of the participants in the Queensland Institute of Medical Research studies have North European ancestry,44 ancestry differences between twin pairs cannot be excluded. Third, environmental factors (that is, alcohol abuse and smoking) are associated with altered DNA methylation patterns. We matched pairs as closely as possible; however, some of the individuals in our sample were smokers or alcohol dependent. These examples show confounding factors, which may influence our results especially because our sample size is small. Future studies would ideally control for these and other confounding factors.
Another limitation of our design of this study is that blood samples were collected after the onset of depression. Therefore, any differences in methylation associated between MZTs associated with MDD may reflect a range of factors including treatment. A prospective study design, in which samples are taken before first onset of MDD and then after disease onset, but before treatment would aid in the identification of epigenetic biomarkers associated with MDD. Further collection of samples collected during and after treatment may also help implicate sites that show the greatest changes in methylation after successful treatment of the disorder.
The genetic architecture of complex traits is very heterogeneous. Genome-wide association studies have been successful in identifying susceptibility variants for complex traits.45 However, most of the variants identified have very small effect sizes. It is likely that DNA methylation differences involved in complex psychiatric disorders will also have small effects, and that sample sizes commensurate with those utilised in genome-wide association studies may be required to detect methylation differences at individual CpG islands.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Translational Psychiatry website (http://www.nature.com/tp)
Supplementary Material
References
- Tamsen L, Eneroth P. Serum levels of pregnancy-specific beta 1-glycoprotein (SP1) and human chorionic gonadotropin (beta-hCG) in women using an intrauterine device. Contraception. 1986;33:497–501. doi: 10.1016/s0010-7824(86)80008-7. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Craddock N, Forty L. Genetics of affective (mood) disorders. Eur J Hum Genet. 2006;14:660–668. doi: 10.1038/sj.ejhg.5201549. [DOI] [PubMed] [Google Scholar]
- Weissman MM, Bland R, Joyce PR, Newman S, Wells JE, Wittchen HU. Sex differences in rates of depression: cross-national perspectives. J Affect Disord. 1993;29:77–84. doi: 10.1016/0165-0327(93)90025-f. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- Psychiatric_GWAS_Consortium_Major_Depressive_Disorder A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2012;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Daly MJ, O'Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–551. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl C, Guldberg P. DNA methylation analysis techniques. Biogerontology. 2003;4:233–250. doi: 10.1023/a:1025103319328. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353–2364. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]
- Holliday R. Epigenetics: an overview. Dev Genet. 1994;15:453–457. doi: 10.1002/dvg.1020150602. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351. doi: 10.1016/s0168-9525(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Petronis A, Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GHT, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- Uddin M, Koenen KC, Aiello AE, Wildman DE, de los Santos R, Galea S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol Med. 2011;41:997–1007. doi: 10.1017/S0033291710001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratomi G, Iwamoto K, Bundo M, Kusumi I, Kato N, Iwata N, et al. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol Psychiatry. 2008;13:429–441. doi: 10.1038/sj.mp.4002001. [DOI] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabunciyan S, Aryee MJ, Irizarry RA, Rongione M, Webster MJ, Kaufman WE, et al. Genome-wide DNA methylation scan in major depressive disorder. PloS one. 2012;7:e34451. doi: 10.1371/journal.pone.0034451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham KE. Stage-specific and cell type-specific aspects of genomic imprinting effects in mammals. Differentiation. 1995;59:269–282. doi: 10.1046/j.1432-0436.1996.5950269.x. [DOI] [PubMed] [Google Scholar]
- Olsson CA, Foley DL, Parkinson-Bates M, Byrnes G, McKenzie M, Patton GC, et al. Prospects for epigenetic research within cohort studies of psychological disorder: a pilot investigation of a peripheral cell marker of epigenetic risk for depression. Biol Psychol. 2010;83:159–165. doi: 10.1016/j.biopsycho.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Gordon L, Joo JE, Andronikos R, Ollikainen M, Wallace EM, Umstad MP, et al. Expression discordance of monozygotic twins at birth: effects of intrauterine environment and a possible mechanism for fetal programming. Epigenetics. 2011;6:1–13. doi: 10.4161/epi.6.5.15072. [DOI] [PubMed] [Google Scholar]
- Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends Genet. 2011;27:116–125. doi: 10.1016/j.tig.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, Jr., et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Ustun TB. The World Mental Health (WMH) Survey Initiative Version of the World Health Organization (WHO) Composite International Diagnostic Interview (CIDI) Int J Methods Psychiatr Res. 2004;13:93–121. doi: 10.1002/mpr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagnostic and statistical manual of mental disorders4th edn,American Psychiatric Association: Washington DC; 1994 [Google Scholar]
- Duggan C, Sham P, Lee A, Minne C, Murray R. Neuroticism: a vulnerability marker for depression evidence from a family study. J Affect Disord. 1995;35:139–143. doi: 10.1016/0165-0327(95)00062-3. [DOI] [PubMed] [Google Scholar]
- Tao MH, Marian C, Shields PG, Nie J, McCann SE, Millen A, et al. Alcohol consumption in relation to aberrant DNA methylation in breast tumors. Alcohol. 2010;45:689–699. doi: 10.1016/j.alcohol.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L, Boukouvala S, Sim E. Characterisation of CpG methylation in the upstream control region of mouse Nat2: Evidence for a gene-environment interaction in a polymorphic gene implicated in folate metabolism. Gene. 2010;452:16–21. doi: 10.1016/j.gene.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Kuan PF, Wang S, Zhou X, Chu H. A statistical framework for Illumina DNA methylation arrays. Bioinformatics. 2010;26:2849–2855. doi: 10.1093/bioinformatics/btq553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yan L, Hu Q, Sucheston LE, Higgins MJ, Ambrosone CB, et al. IMA: an R package for high-throughput analysis of Illumina's 450 K Infinium methylation data. Bioinformatics. 2012;28:729–730. doi: 10.1093/bioinformatics/bts013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450 K technology. Epigenomics. 2011;3:771–784. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2011;22:407–419. doi: 10.1101/gr.119867.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists Nucleic Acids Res 200735(Web Server issue):W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PloS one. 2010;5:e10028. doi: 10.1371/journal.pone.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PloS one. 2009;4:e6767. doi: 10.1371/journal.pone.0006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland P, Quraishe S, French P, O′Connor V. Expression of the MAST family of serine/threonine kinases. Brain Res. 2008;1195:12–19. doi: 10.1016/j.brainres.2007.12.027. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Hemmings HC, Jr, Greengard P, Nairn AC. Beyond the dopamine receptor: regulation and roles of serine/threonine protein phosphatases. Front Neuroanat. 2011;5:50. doi: 10.3389/fnana.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dushlaine C, Ripke S, Moran J, Chambert K, Sklar P, Purcell S, et al. Proceedings of the World Congress of Psychiatric Genetics. Hamburg, Germany; 2012. Genome-wide Association Study of 32,143 Individuals Reveals Several Novel Associations in Schizophrenia. [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronis A, Gottesman II, Kan P, Kennedy JL, Basile VS, Paterson AD, et al. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance. Schizophr Bull. 2003;29:169–178. doi: 10.1093/oxfordjournals.schbul.a006988. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci USA. 2008;105:13614–13619. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Birley AJ, Sullivan PF, Visscher PRM, Martin NG. Genetic and phenotypic stability of measures of neuroticism over 22 years. Twin Res Hum Genet. 2007;10:695–702. doi: 10.1375/twin.10.5.695. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.