

Abstract
Background
Fluticasone furoate (FF; GW685698) is a novel inhaled corticosteroid that is active at 24 h and under development for once-daily administration in combination with the long-acting β2-adrenoceptor agonist vilanterol (GW642444) for chronic obstructive pulmonary disease and asthma. In vitro studies examining the respiratory tissue-binding properties of corticosteroids showed FF to have the largest cellular accumulation and slowest rate of efflux compared with other clinically used inhaled corticosteroids, consistent with greater tissue retention. The enhanced affinity of the glucocorticoid receptor binding of FF, coupled with its extended tissue association, may be expected to lead to greater and more prolonged anti-inflammatory effects and should provide relevant once-daily efficacy.
Objective
The aim of this study was to assess the rate and extent of systemic absorption of FF from the lung following inhaled administration of FF from three exploratory dry powder formulations (via DISKHALER®) compared with inhaled fluticasone propionate (FP) [via DISKHALER®] using deconvolution analysis.
Methods
This open-label, part-randomized, six-way crossover study evaluated three early development dry powder inhaled formulations of FF administered as single doses via DISKHALER®. Healthy male subjects (n = 24) each received FF (2,000 μg; three formulations), inhaled FP (1,000 μg; via DISKHALER®) and 250 μg of each molecule by intravenous infusion. The bioavailability of both inhaled FF and FP represents absorption from the lung as the oral bioavailability from the swallowed portion of the inhaled dose is negligible (<1.5 %). To investigate the absorption kinetics from the lung, the inhaled concentration–time data were subjected to deconvolution analysis using derived pharmacokinetic parameters from fitting of the intravenous concentration–time data.
Results
The terminal elimination half-life (t½β) for inhaled FF was considerably longer (range 17–24 h) than the t½β estimated for intravenous FF (14 h), whereas t½β for FP was similar whether inhaled or given intravenously (11 and 14 h, respectively). This would suggest that FF is exhibiting absorption rate-limited pharmacokinetics following inhaled FF dosing and that the apparent t½β is an estimate of absorption rate. The lung mean absorption time for FF was approximately 7 h irrespective of formulation, which was considerably longer than FP (2.1 h). The time for 90 % absorption from the lung was significantly longer for FF (20–30 h) than for FP (8 h), indicating a significantly longer lung retention time for FF.
Conclusion
In comparison with inhaled FP, inhaled FF (independent of formulation) demonstrated prolonged absorption from the lung into the systemic circulation, indicating a longer lung retention time and suggesting the potential for maintained efficacy with once-daily administration.
Introduction
Fluticasone furoate (FF) is a novel inhaled corticosteroid that is active at 24 h and under development for once-daily administration in combination with the long-acting β2-adrenoceptor agonist vilanterol (GW642444) for chronic obstructive pulmonary disease (COPD) and asthma.
In vitro studies examining the respiratory tissue binding properties of inhaled corticosteroids showed FF to have the largest cellular accumulation and slowest rate of efflux compared with other clinically used inhaled corticosteroids, consistent with greater tissue retention [1]. The enhanced affinity of glucocorticoid receptor binding of FF, coupled with its extended tissue association, may be expected to lead to greater and more prolonged anti-inflammatory effects and could result in once-daily efficacy. For drugs such as inhaled corticosteroids, which exert their pharmacological activity on cells and tissues of the airway walls [2], a longer pulmonary residence time enhances their anti-inflammatory effectiveness. The residence time will be increased for drugs that are absorbed more slowly into the pulmonary circulation after deposition in the airways.
This study was conducted in healthy subjects to investigate the inhaled and intravenous pharmacokinetics of FF compared with those of fluticasone propionate (FP). The pharmacokinetics of three different exploratory dry powder formulations dosed via DISKHALER® were also compared. These formulations utilized different input drug material blended with lactose. In vitro performance data generated using the Anderson Cascade Impactor showed only minimal differences in the respirable fraction [3]. The bioavailability of both inhaled FF and FP predominantly represents absorption from the lung as the oral bioavailability from the swallowed portion of the inhaled dose is negligible (<1.5 %) [4, 5]. This analysis was conducted in healthy subjects to characterize the absorption of inhaled FF and is considered to be representative of subjects with asthma or COPD. Use of analytical deconvolution techniques allowed the rate and duration of the input (absorption) of FF and FP into the systemic circulation after inhalation to be assessed over the entire absorption period.
Methods
This was an open-label, part-randomized, single-dose, six-way crossover study in healthy male subjects. The study was approved by the local ethics committee. All subjects gave their written informed consent before participating in the trial. Study FFA10003 was conducted at a single site between 28 May and 13 October 2002.
Healthy male subjects (n = 24) were randomized to receive the four FF treatments in the first four periods followed by the FP treatments in the last two periods. The three exploratory inhaled formulations of FF investigated FF drug material of different input particle size blended with lactose in ROTADISK®. For the FF treatments subjects received the following in a randomized fashion:
single inhaled dose of formulation 1 FF (2,000 μg) via DISKHALER®;
single inhaled dose of formulation 2 FF (2,000 μg) via DISKHALER®;
single inhaled dose of formulation 3 FF (2,000 μg) via DISKHALER®; or
single intravenous dose of FF (250 μg). 1 mL of 250 μg/mL in 100 % propylene glycol was administered at a constant rate of infusion over 20 min using a syringe and pump.
Subsequently, subjects then received the following in a randomized fashion:
single inhaled dose of FP (1,000 μg) via DISKHALER®; or
single intravenous dose of FP (250 μg). 0.5 mL of 500 μg/mL in 100 % propylene glycol was administered at a constant rate of infusion over 20 min using a syringe and pump.
There was a washout period of at least 1 week between treatment periods. The duration of the study was approximately 8 weeks for each subject. The clinical phase of the study was conducted over a 20-week period.
Venous blood samples (2.7 mL) for analysis of plasma drug concentrations were collected in potassium EDTA (KEDTA) tubes pre-dose and at 5, 10, 20, 30 and 45 min and 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 32, 48 and 72 h after the start of dosing. Additionally samples were collected at 22 and 25 min following the start of the intravenous infusion. The blood samples were put on ice until centrifugation at 1,500 g for approximately 10 min at 4 °C. The plasma was transferred into polypropylene containers, frozen at approximately −20 °C.
Bioanalytical Methods
Plasma samples were analysed for FF or FP, as appropriate, using [13C3]CCI18781 as internal standard, by solid-phase extraction followed by high-performance liquid chromatography with tandem mass spectrometry using a Perkin Elmer Sciex API 3000 (York Bioanalytical Solutions, York, UK). A gradient system using ammonium formate pH 5.0 buffer (26:74, v/v with methanol) and methanol was run with column Phenomex Prodigy, ODS3, 5 μm, 2.0 mm (internal diameter) × 150 mm running at 40 °C. The ion transition for FF was m/z 539–313 and for FP was m/z 501–313. The validation range of the assays was 10–1,000 pg/mL for both FF and FP. Where reported, concentrations were above the higher limit of quantification the plasma samples were diluted, as appropriate, to provide concentrations within the validated range. Interbatch precision was ≤7.9 % coefficient of variation (CV) over the assay range; the lower limit of quantification for FF was 10 pg/mL. Interbatch precision was ≤12.1 % CV over the assay range; the lower limit of quantification for FP was 10 pg/mL. Quality controls prepared at three different concentrations were analysed with each batch of samples against separately prepared calibration standards to assess the day-to-day performance of the assay. Quality control results from this study met the acceptance criteria of no more than one third of the quality control results deviating from the nominal concentration by more than 15 %, with at least one quality control result acceptable at each concentration.
Pharmacokinetic Analysis
FF and FP concentration–time data were subjected to non-compartmental analysis using WinNonlin® Pro v2.1. For each inhaled treatment in each individual subject, the absolute bioavailability (F) was estimated using the following formula (Eq. 1):
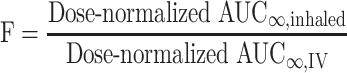 |
1 |
where AUC∞,inhaled is the area under the concentration–time curve from time zero to infinity (AUC∞) for the inhaled drug and AUC∞,IV is the AUC∞ for the intravenous drug.
Deconvolution analysis was used to characterize the input rate and cumulative absorption of FF and FP from the lung into the systemic circulation. The inhaled concentration–time data were subjected to deconvolution analysis using derived pharmacokinetic parameters from fitting of the intravenous concentration–time data using the computer program PCModfit [6]. This deconvolution procedure utilizes equations developed by Loo and Riegelman [7] and requires concentration–time data following extra-vascular dosing and assumes that the intravenous pharmacokinetics have been studied. The micro-constants describing distribution and elimination were obtained by fitting a two-compartment infusion model to FP concentration–time data and a three-compartment infusion model to FF concentration–time data following intravenous dosing by iterative weighted non-linear least-squares regression analysis with PCModfit for Windows® version 1.70 [6].
The inhaled concentration–time data were subjected to deconvolution analysis utilizing the Loo-Riegelman model with the same number of compartments as the infusion model and percent remaining to be absorbed versus time data was generated. The absorption rate constant (ka) was obtained by fitting a mono-exponential function to the percent remaining to be absorbed versus time data visually assessed to lie on the linear portion of the semi-logarithmic plots. The absorption half-life (t½abs) was calculated as the ratio of ln2/ka and this was used to calculate the time for 90 % of the drug to be absorbed (T90).
The input profiles were presented graphically after normalizing for the estimated absolute bioavailability (F) and the administered dose, i.e. (Eq. 2):
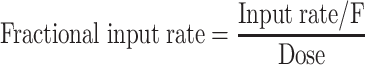 |
2 |
Mean absorption time (MAT) was derived from the observed mean residence time for FF and FP after inhalation (MRTinhaled) and intravenous administration (MRTIV), according to Eq. 3:
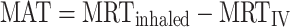 |
3 |
For each of the derived parameters, summary statistics have been calculated for each treatment group: median, maximum, minimum, arithmetic mean, standard deviation, CV, geometric mean, 95 % confidence interval for the geometric mean and standard deviation of logarithmically transformed data.
Results
All subjects were White Caucasian males: mean age 32 years (range 20–53), mean bodyweight 78.1 kg (range 65.1–92.7) and mean body mass index 24.9 kg/m2 (range 21.8–29.7).
The median plasma concentration–time profiles (Fig. 1) and pharmacokinetic parameters were very similar for intravenous FF and FP (Table 1). Both FF and FP showed high plasma clearance, a large volume of distribution and a longer terminal elimination half-life (t½β) of on average 14 h following intravenous administration.
Fig. 1.
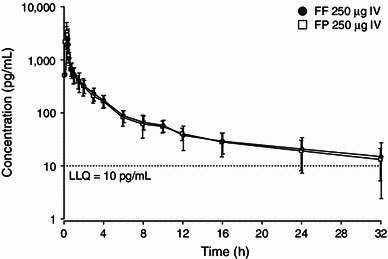
Mean (±SD) plasma concentration–time profile following 250 μg single intravenous fluticasone furoate and fluticasone propionate administration. FF fluticasone furoate, FP fluticasone propionate, IV intravenous, LLQ lower limit of quantitation
Table 1.
Pharmacokinetic parameters of fluticasone furoate and fluticasone propionate following intravenous administration
| Parameter (units) | Fluticasone furoate 250 μg | Fluticasone propionate 250 μg | ||
|---|---|---|---|---|
| Geometric mean (95 % CI) | CV (%) | Geometric mean (95 % CI) | CV (%) | |
| AUC∞ (pg·h/mL) | 3,484 (3,136, 3,870) | 21 | 3,910 (3,464, 4,413) | 21 |
| t½β (h) | 13.6 (10.6, 17.6) | 42 | 14.0 (10.4, 18.9) | 58 |
| CL (L/h) | 71.8 (64.6, 79.7) | 22 | 63.9 (56.7, 72.2) | 22 |
| Vss (L) | 704 (543, 914) | 47 | 577 (453, 736) | 48 |
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, CL clearance, CV coefficient of variation, t ½β terminal elimination half-life, V ss volume of distribution at steady state
The plasma concentration–time profile for inhaled FF appeared to be multi-exponential for all formulations, consisting of an initial rapid phase followed by a second slower phase. Peak FF plasma concentrations were observed at median times of 10–45 min, dependent upon formulation (Table 2). There were notable differences in FF plasma concentration–time profiles for the three formulations of FF (Fig. 2). The extent of systemic exposure for the exploratory FF formulations administered via DISKHALER® were different (Table 2) with values for absolute bioavailability being, on average, 6.3, 13.3 and 18.4 % for formulations 1, 2 and 3, respectively (Table 2). The absolute bioavailability for FP was, on average, 9.0 %.
Table 2.
Pharmacokinetic parameters of fluticasone furoate and fluticasone propionate following inhaled administration
| Parameter (units) | FF 2,000 μg | FP 1,000 μg | ||
|---|---|---|---|---|
| Formulation 1 | Formulation 2 | Formulation 3 | ||
| Cmax (pg/mL) | 140 (116, 170) | 478 (402, 569) | 707 (579, 864) | 334 (277, 401)a |
| tmax (h)b | 0.75 (0.08–3.00) | 0.17 (0.07–1.50) | 0.17 (0.08–2.03) | 0.75 (0.17–2.02) |
| AUC∞ (pg·h/mL) | 2,168 (1,704, 2,759) | 3,677 (3,274, 4,131) | 5,144 (4,538, 5,831) | 2,432 (1,918, 3,083)a |
| t½β (h) | 23.9 (19.8, 28.9) | 20.3 (17.8, 23.2) | 17.3 (15.0, 20.0) | 10.8 (8.5, 13.7) |
| MAT (h)c | 7.2 (4.4, 10.7) | 6.5 (4.1, 8.7) | 7.1 (4.8, 7.9) | 2.1 (0.2, 3.4) |
| F (%) | 6.3 (5.2, 7.6) | 13.3 (11.0, 16.0) | 18.4 (15.2, 22.1 | 9.0 (6.9, 11.7) |
Values are expressed as geometric means (90 % CI) unless specified otherwise
aFP AUC∞ and Cmax dose normalized to 2,000 μg
bValues are expressed as median (range)
cValues are expressed as median (95 % CI)
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, C max maximum observed plasma concentration, F absolute bioavailability, FF fluticasone furoate, FP fluticasone propionate, MAT mean absorption time, t ½β terminal elimination half-life, t max time to Cmax
Fig. 2.
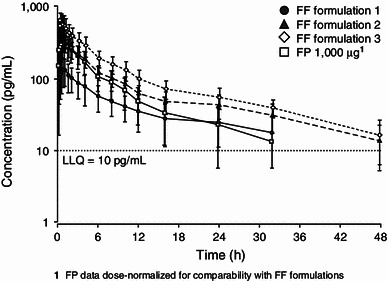
Mean (± SD) concentration–time profile following inhaled fluticasone furoate (2,000 μg) and fluticasone propionate (1,000 μg) administration. FF fluticasone furoate, FP fluticasone propionate, LLQ lower limit of quantitation
The apparent t½β of inhaled FF was longer than the terminal elimination t½β estimated after intravenous dosing. This would suggest that FF is exhibiting absorption rate-limited pharmacokinetics following inhaled FF dosing and that the apparent t½β is an estimate of absorption rate. Estimates of this apparent t½abs for FF following inhaled administration was independent of formulation. In contrast, the FP estimates of apparent t½β were similar for the two administration routes.
From visual inspection of the absorption profiles obtained from deconvolution analysis, the absorption of FF appeared to be multi-exponential for all formulations, consisting of an initial rapid phase followed by a second slower phase. Absorption of the bioavailable dose for FF was virtually complete by 32 h, compared with 16 h for FP (Fig. 3). The lung MAT for FF was approximately 7 h irrespective of formulation, which was considerably greater than FP (2.1 h) (Table 2). The T90 from the lung was significantly greater for FF (19–32 h) compared with FP (11 h), indicating a significantly longer lung retention time for FF (Table 3).
Fig. 3.
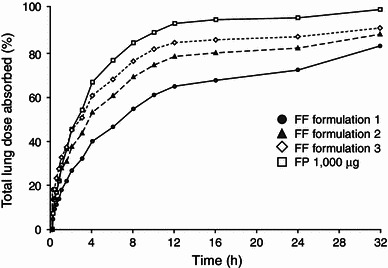
Fraction (%) of inhaled fluticasone furoate (2,000 μg) and fluticasone propionate (1,000 μg) absorbed from the lung. FF fluticasone furoate, FP fluticasone propionate
Table 3.
Summary of parameters devised from deconvolution analysis
| Parameter | FF 2,000 μg: formulation 1 | FF 2,000 μg: formulation 2 | FF 2,000 μg: formulation 3 | FP 1,000 μg |
|---|---|---|---|---|
| t½abs (h) | 12.1 (10.2, 14.3) | 10.1 (8.57, 11.8) | 10.7 (8.92, 12.8) | 4.81 (3.20, 7.22) |
| T90 (h) | 31.7 (25.4, 39.6) | 23.4 (19.1, 28.6) | 19.1 (15.2, 23.9) | 10.5 (1.67, 14.4) |
Values are expressed as geometric means (90 % CI)
FF fluticasone furoate, FP fluticasone propionate, T90 time for 90 % of the drug to be absorbed, t ½abs absorption half-life
Single-dose FF and FP following inhaled and intravenous dosing was well tolerated. Twenty-three of the 24 enrolled subjects successfully completed all doses as planned. One subject withdrew after one treatment period for personal reasons unrelated to study medications (this was his first study and he could not tolerate the repeated cannulation and venesection procedures). There were no serious adverse events reported and no clinically significant changes in biochemical, haematological or urinary parameters.
Discussion
This study was conducted in healthy male subjects to investigate the inhaled and intravenous pharmacokinetics of FF, compared with those of FP. Use of analytical deconvolution techniques allowed the rate and duration of the input (absorption) of FF and FP into the systemic circulation after inhalation to be assessed over the entire absorption period. The results showed that the estimate of apparent t½β following single-dose, inhaled administration of FF was notably longer than that seen following intravenous dosing, whereas for FP the t½β values were similar following the two routes. This suggests that FF is exhibiting absorption rate-limited pharmacokinetics and that the apparent t½β is an estimate of absorption rate following inhaled administration. MAT for FF was, on average, 6–7 h and independent of formulation. This was considerably longer than that for FP which was, on average, 2.1 h. As oral absorption for both FF and FP is in the region of <1.5 % [4, 5], the majority of exposure from inhaled dosing, as with FP, would appear to be from the lung. The t½abs and T90 from the lung were similar for all FF treatments and significantly longer than those for FP. These parameters would suggest that, independent of formulation, a significant amount of FF remains in the lung for up to 24 h post-dose (Fig. 3) and would also translate to the final formulation and device. This greater residence of FF in the lung may result in a longer duration of action than with FP (t½abs of 10.1–12.1 h compared with 4.8 h). Deconvolution analyses have previously been reported for FP and the results for FP in this study are consistent with those previous results. [8, 9]
Systemic exposure results for each of the different FF treatments were notably different, with values for absolute bioavailability being, on average, 6.3, 13.3 and 18.4 % for formulations 1, 2 and 3, respectively. In vitro performance data generated showed only minimal differences in the respirable fraction and, hence, differences in lung dose provided only partial explanation for the differences in bioavailability. It is evident that there are other factors such as lung deposition and the distribution of particle mass material that may also contribute to these differences.
The intravenous pharmacokinetics of FF were very similar to those for FP. Volume of distribution at steady state for FF and FP (geometric mean 704 and 577 L, respectively) was greater than that quoted for total body water (42 L) for a 70 kg man [10], indicating extensive distribution into tissues and total plasma clearance for FF and FP (average 71.8 and 63.9 L/h, respectively) approached liver blood flow (87 L/h) [10]. The absolute bioavailability for FP was, on average, 9.0 % and the pharmacokinetic characteristics of inhaled and intravenous FP in this study are consistent with previous data [11, 12].
Conclusion
Inhaled FF (independent of formulation), in comparison with inhaled FP, demonstrated prolonged absorption from the lung into the systemic circulation, indicating a longer lung retention time and suggesting the potential for maintained efficacy with once-daily administration.
Acknowledgments
Bioanalytical support for the fluticasone furoate and fluticasone propionate analyses was provided by York Bioanalytical Solutions, UK. Editorial support in the form of copyediting and graphic services was provided by David Cutler at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline.
Conflict of interest
All authors are employed by and hold stocks/shares in GlaxoSmithKline.
References
- 1.Salter M, Biggadike K, Matthews JL, et al. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L660–L667. doi: 10.1152/ajplung.00108.2007. [DOI] [PubMed] [Google Scholar]
- 2.Schleimer RP. Effects of glucocorticoids on inflammatory cells relevant to their therapeutic applications in asthma. Am Rev Respir Dis. 1990;141:S59–S69. [PubMed] [Google Scholar]
- 3.Data on file, GlaxoSmithKline, 2002 May.
- 4.Hughes SC, Shardlow PC, Hollis FJ, et al. Metabolism and disposition of fluticasone furoate, an enhanced-affinity glucocorticoid, in humans. Drug Metab Dispos. 2008;36:2337–2344. doi: 10.1124/dmd.108.022137. [DOI] [PubMed] [Google Scholar]
- 5.Falcoz C, Oliver R, McDowall J, et al. Bioavailability of orally administered micronised fluticasone propionate. Clin Pharmacokinet. 2000;39(Suppl 1):9–15. doi: 10.2165/00003088-200039001-00002. [DOI] [PubMed] [Google Scholar]
- 6.Allen GD. Modfit: a pharmacokinetics computer program. Biopharm Drug Dispos. 1990;11:477–498. doi: 10.1002/bdd.2510110603. [DOI] [PubMed] [Google Scholar]
- 7.Loo JCK, Riegelman S. New method for calculating the intrinsic absorption rate of drugs. J Pharm Sci. 1968;57:918–928. doi: 10.1002/jps.2600570602. [DOI] [PubMed] [Google Scholar]
- 8.Allen DB, Bielory L, Derendorf H, et al. Inhaled corticosteroids: past lessons and future issues. J Allergy Clin Immunol. 2003;112:S1–S40. doi: 10.1016/S0091-6749(03)01859-1. [DOI] [PubMed] [Google Scholar]
- 9.Krishnaswami S, Hochhaus G, Möllman H, et al. Interpretation of absorption rate data for inhaled fluticasone propionate obtained in compartmental pharmacokinetic modelling. Int J Clin Pharmacol Ther. 2005;43:117–122. doi: 10.5414/cpp43117. [DOI] [PubMed] [Google Scholar]
- 10.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 11.Holliday SM, Faulds D, Sorkin EM. Inhaled fluticasone propionate: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in asthma. Drugs. 1994;47:318–331. doi: 10.2165/00003495-199447020-00007. [DOI] [PubMed] [Google Scholar]
- 12.Mackie AE, Ventresca GP, Fuller RW, et al. Pharmacokinetics of intravenous fluticasone propionate in healthy subjects. Br J Clin Pharmacol. 1996;41:539–542. doi: 10.1046/j.1365-2125.1996.36110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]