

Abstract
To explore the hydrophobic groove subsite within the CB1 cannabinoid receptor we have designed and synthesized a group of tail-substituted anandamide analogs. Our design involves the introduction of aryl or heterocyclic ring as terminal substituents that are connected to the last cis-arachidonyl double bond through aliphatic chains of variable lengths. Our results indicate that there are strict stereochemical requirements for the interaction of such analogs with the CB1 receptor. The optimal pharmacophore includes the phenyl, p-substituted phenyl or 3-furyl substitutents attached to the cis-double bond through a four methylene chain.
Keywords: anandamide, tail modification, CB1 receptor, cannabinoid, endocannabinoid
Anandamide,1 a key endogenous cannabinoid ligand, is a partial CB1 cannabinoid receptor agonist and is known to produce a concentration-dependent inhibition of the electrically evoked twitch response of the mouse vas deferens,2,3 as well as antinociception, hypothermia, hypomotility, and catalepsy in mice.4–8 In brain and liver, anandamide is hydrolyzed enzymatically to yield arachidonic acid and ethanolamine. The reaction is catalyzed by a membrane-bound amidohydrolase9–12 (anandamide amidohydrolase or fatty acid amide hydrolase, FAAH) which has been cloned and fully characterized.13 This enzyme was shown to be co-localized in the same brain regions as the CB1 cannabinoid receptor.12 The hydrolytic breakdown of anandamide can be prevented in vitro by phenylmethanesulfonyl fluoride (PMSF), a general serine protease inhibitor.9,10 PMSF can be included in the competitive binding assays of anandamide analogues where amidase-catalyzed anandamide hydrolysis might be a complicating factor.14 There is also evidence pointing to the existence of carrier-mediated anandamide transport15 which is essential for the termination of the biological effects of anandamide. Thus, anandamide interacts not only with the CB receptors but also possibly with endocannabinoid transporter system and FAAH.
Anandamide exhibits selectivity toward the CB1 cannabinoid receptor. Additionally, structure activity relationship (SAR) studies of anandamide analogs have provided insight into the stereoelectronic requirements for interaction with the CB1 receptor.14,16–27 Although much of the reported work has addressed the head group, SAR of the arachidonoyl side chain has revealed that this hydrophobic chain is also very sensitive to structural modifications.18,22,23 Complete saturation, or replacement of the olefins with alkynes, results in the total loss of receptor affinity. Substitution of the arachidonoyl chain with other fatty acid chains with ω-olefinic bonds, with a trans double bond or analogs with non ω-6 structure leads to a major reduction in affinity for CB1. Earlier studies on the hydrophobic tail of anandamide suggested that the pentyl chain may mimic the five carbon side chain of (−)-Δ9-tetrahydrocannabinol, the principal active ingredient of cannabis.16–18,28 Substitution of the terminal pentyl group of anandamide with a 1,1-dimethyl moiety leads to significant enhancement of its affinity for CB1, as has also been observed with (−)-Δ9-THC.17 The present communication further explores this terrain through the design and synthesis of novel chain modified anandamide analogs.
In earlier work, we have shown that introduction of ω-isothiocyanato and ω-azido groups within the anandamide structure lead to substantial enhancement of CB1 affinity and potency.29 The present work describes the synthesis and evaluation of a series of anandamide analogs of variable chain lengths in which the terminal carbon is functionalized with a phenyl, substituted phenyl or heterocyclic rings. All the compounds carry the R-methylethanolamine moiety, a headgroup which was earlier demonstrated to provide analogs (e.g. methanandamide, AM356) with optimized affinity for the CB1 receptor as well as robust metabolic stability.14 Our results confirm that, as with anandamide, all analogs exhibit CB1 selectivity over CB2. Additionally, our data reveal that the stereochemical features of the anandamide tail may have a major impact on ligand’s affinity for CB1.
The synthesis of the anandamide analogs is illustrated in Scheme 1. Methyl 5-hexynoate 1 was coupled with 4-chloro-butyn-1-ol in the presence of copper (I) iodide30 and the alcohol intermediate was converted to the corresponding bromide using CBr4/PPh3. Bromide 2 was coupled in an analogous procedure with 3-butyn-1-ol to afford alcohol 3 which underwent hydrogenation with Lindlar’s catalyst in the presence of quinoline to afford triene 4 in high yield.31,32 Dess-Martin periodinane33 oxidation of alcohol 4 resulted in the formation of the unstable aldehyde 5 which was immediately subjected to Wittig olefination with various phosphonium salts to furnish cis-alkenes 6. The methanandamide analogs were obtained by direct amidation of the corresponding esters with R-(−)-alaninol catalyzed by sodium cyanide.34 The iodo-substituted compound 7j was prepared from the corresponding bromo compound 9 via the tin intermediate.
Scheme 1.

Reagents and conditions: (i). 4-chloro-butyn-1-ol, CuI, NaI, K2CO3, DMF, rt, overnight, 84%; (ii). PPh3, CBr4, rt, 95%; (iii). 3-butyn-1-ol, CuI, NaI, K2CO3, DMF, rt, overnight, 80%; (iv). H2, Lindlar catalyst, quinoline, ether, 0–10 °C, 86%; (v). Dess-Martin reagent, CH2Cl2, rt, 30 min, 86%; (vi). Wittig reagent from the corresponding phosphonium salt (see scheme 2) with n-BuLi in THF at −78° C, 60–80%;(vii). R-(−)-2-amino-propanol, NaCN (cat), Methanol, 55°C, sealed vial, 74%; (viii). bis-tributyltin, Pd(PPh3)4, toluene, reflux, 40%; (ix). I2, CH2Cl2, rt, 95%.
Scheme 2 illustrates the preparation of the phosphonium salts utilized in Scheme 1. Starting with the coupling of properly functionalized iodobenzene 8 and hex-5-yn-1-ol 9 catalyzed by tetrakistriphenylphosphinepalladium(0),35 the resulting alcohols were hydrogenated to give ω- phenyl alcohols 11. Iodination of alcohol 11 by I2/PPh3 followed by heating with PPh3 gave the desired phosphonium salts 13 in high yields (85–95%). All the other phosphonium salts were obtained from commercially available iodide and triphenylphosphine, and were used for the Wittig reaction without further purification. The furyl derivatives 7o–7s were synthesized by utilizing an approach published earlier by our research group.29 This involves the coupling of furyl substituted terminal alkynes (14 or 15) with 13-bromo-trideca-5,8,11-triynoic acid methyl ester 16 followed by partial hydrogenation with Lindlar’s catalyst to give esters 17 (Scheme 3) which were converted to the corresponding amides using the same method described in the scheme 1.
Scheme 2.

Reagents and conditions: (i). Pd(PPh3)4, CuI, piperidine, DMF, 0 °C-rt, 50–82%; (ii). H2, 5% Pd-C, methanol, rt, 85–90%; (iii). I2, PPh3, imidazole, 0 °C, CH2Cl2, rt, 80–92%; (iv). PPh3, neat, 110 °C, 80–92%.
Scheme 3.

Reagents and conditions: (i) CuI, NaI, K2CO3, DMF, rt, overnight, 60%; (ii). H2, Lindlar catalyst, quinoline, ether, 0–10 °C, 94%.
Binding affinities for the CB1 and CB2 receptors were determined according to previously reported procedures.36,21,23 For the CB1 receptor, binding data were obtained using a rat brain membrane in the presence of phenylmethanesulfonyl fluoride (PMSF) a general serine protease inhibitor to protect the analogues from the hydrolytic activity of fatty acid amide hydrolase. CB2 data were obtained using a membrane preparation from mouse spleen known to be rich in CB2 receptor. [3H]CP-55,940, a most widely used radioligand, was chosen as a competing ligand for the assays as it has high affinity for both CB1 and CB2 receptors and is nonselective.37
The binding affinities of novel anandamide analogues are summarized in Table 1, where (R)-methanandamide is included for comparison. It is apparent from the CB2 affinities reported here that these novel anandamide analogues show selectivity for the CB1 receptor. Analogs 7a–7e were synthesized to assess the optimal length for the phenyl group at the hydrophobic tail for receptor affinity. The binding results show that 7d with four methylene carbons separating C15 from the phenyl group exhibited comparable activity (Ki = 13.4 nM) for the CB1 receptor as (R)-methanandamide (Ki = 17.9 nM in the presence of PMSF). Decreasing the number of methylene carbons between C15 cis-double bond and the phenyl group gave analogs 7a–7c and showed progressively weaker affinities for the CB1 receptor with decreasing length of the alkyl chain. Conversely, compound 7e which differs from 7d by one extra methylene group displayed approximately over 5-fold lower affinity compared to 7d. These results clearly indicate that a four methylene linker is optimal between the distal arachidonyl double bond and the pendant phenyl moiety. In an effort to enhance the polar features of ananadamide analogs, we have substituted the phenyl ring with the more polar furan moiety. A similar trend was observed with the 3-furyl derivative (7o). However, surprisingly the corresponding 2-furyl derivative (7q) showed a substantially weaker affinity. Introduction of terminal α-naphthyl group (7f) resulted in the loss of CB1 receptor affinity.
Table 1.
Affinity (Ki)a of new anandamide analogues for CB1 and CB2 receptors
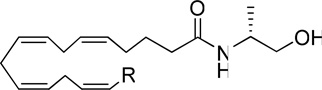 | |||
|---|---|---|---|
| Analogues | R | CB1 Ki (nM) (PMSF) |
CB2 Ki (nM) |
| (R)-methanandamide | (CH2)4CH3 | 17.9 | 868 |
| 7a | CH2Ph | 348.5 | 826.3 |
| 7b | (CH2)2Ph | 241.2 | 335 |
| 7c | (CH2)3Ph | 69.2 | 533.7 |
| 7d | (CH2)4Ph | 13.3 | 1117 |
| 7e | (CH2)5Ph | 76.6 | 240.6 |
| 7f | (CH2)4-α-naphthyl | 678.3 | 3996 |
| 7g | (CH2)4Ph-m-F | 30.4 | 987.3 |
| 7h | (CH2)4Ph-m-Cl | 89.4 | 605.1 |
| 7i | (CH2)4Ph-m-Br | 95.3 | 1054 |
| 7j | (CH2)4Ph-m-I | 533.3 | 1564 |
| 7l | (CH2)4Ph-m-Me | 344.3 | 839.7 |
| 7m | (CH2)4Ph-o-Br | 170.0 | N.R. |
| 7n | (CH2)4Ph-p-Br | 8.9 | 250.4 |
| 7o | (CH2)4(3-furyl) | 12.0 | 1027 |
| 7p | (CH2)5(3-furyl) | 170 | 869 |
| 7q | (CH2)4(2-furyl) | 325 | 749.8 |
| 7s | (CH2)5(2-furyl) | 561.2 | 3255 |
CB1 affinities were determined using rat brain membranes and 0.8 nM [3H] CP-55,940 as the radioligand. Mouse spleen was used as source of CB2 receptor. Data were analyzed using nonlinear regression analysis. Ki values were obtained from a minimum of two independent experiments run in duplicate.
Earlier data has suggested the presence of a hydrophobic groove subsite capable of accommodating the side chain of classical cannabinoids and possibly the terminal five carbon alkyl group of anandamide.38 Our present results are congruent with the existence of the hydrophobic subsite for the anandamide tail within the CB1 receptor capable of accommodating a terminal phenyl group. It can be argued that a short chain linking the phenyl ring with the last double bond as seen in 7a (n=1) and 7b (n=2), may not occupy a sufficient portion of this groove, resulting in low binding affinity. Conversely, a linker that is too long, as seen in 7e (n=5), may be too large to be fully encompassed by the subsite. The ideal length, given by 7d, completely fills the groove and yet does not incur any steric penalties. The groove also accommodates the 3-furyl moiety, but not the 2-furyl isomer suggesting that the relative position of the furyl oxygen within the receptor subsite may affect the overall ability of the ligand for an optimal interaction with the receptor. Substitution of the phenyl ring with an α-naphthyl moiety results in severe loss in affinity in CB1, an effect which is probably due to its large size.
To further probe this CB1 receptor subsite, different substituents were introduced on the pendent phenyl ring. Our results indicate that, while p-phenyl substituents are tolerated in the receptor subsite, meta or ortho substitution leads to a substantial reduction in affinity. The most potent analog is that with the p-bromophenyl group (7n). Substitution at the meta and ortho postion is not tolerated. However, the introduction of bromine at the para position results in an increase in CB1 receptor affinity. The above stereochemical postulate for the anandamide distal tail pharmacophore is pictorially summarized in figure 1.
Figure 1.

Pictorial representation of the CB1 binding groove for the tail of (R)-methanandamide analogs. 7a (green) 7b (cyan), 7c (magenta), 7d (yellow), 7e (pink), 7i (grey), 7m (blue), 7n (orange). Structures were minimized with the OPLS_2005 force field in Macromodel.39
In conclusion, new anandamide analogues with structural modifications at the hydrophobic tail were synthesized and evaluated for their affinities for the cannabinoid receptors. Introduction of a pendant phenyl, aryl or furyl group separated from the last cis double bond of arachidonic acid by methylene chains of variable lengths were used to probe the cannabinoid side-chain subsite with CB1 and CB2 receptors. Our results suggest that, as with the anandamide head group, the stereochemical requirements for the anandamide hydrophobic tail are stringent.
The series of analogs described here provides further insight into the SAR of the hydrophobic tail in anandamide. The results also reveal the presence of a hydrophobic subsite at the distal end of the groove capable of accommodating a phenyl or furyl substituent as well as a p-substituted phenyl group. Such information will further the understanding of the binding motif of anandamide and aid in the design of novel molecular probes for the CB1 receptor.
Acknowledgements
The authors are grateful to Pusheng Fan for the biochemical assays of these compounds and to Richard Duclos for helpful discussions. This work was supported by grants from the National Institute on Drug Abuse DA03801 and DA07215.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 2.Pertwee RG, Fernando SR, Griffin G, Abadji V, Makriyannis A. Eur. J. Pharmacol. 1995;272:73. doi: 10.1016/0014-2999(94)00618-h. [DOI] [PubMed] [Google Scholar]
- 3.Pertwee RG, Stevenson LA, Elrick DB, Mechoulam R, Corbett AD. Br. J. Pharmacol. 1992;105:980. doi: 10.1111/j.1476-5381.1992.tb09088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawley JN, Corwin RL, Robinson JK, Felder CC, Devane WA, Axelrod J. Pharmacol. Biochem. Behav. 1993;46:967. doi: 10.1016/0091-3057(93)90230-q. [DOI] [PubMed] [Google Scholar]
- 5.Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R, Martin BR. J. Pharmacol. Exp. Ther. 1994;270:219. [PubMed] [Google Scholar]
- 6.Pertwee RG, Stevenson LA. In: Temperature Regulation: Recent Physiological and Pharmacological Advances. Milton AS, editor. Basel: Birkhäuser; 1994. pp. 177–182. [Google Scholar]
- 7.Welch SP, Dunlow LD, Patrick GS, Razdan RK. J. Pharmacol. Exp. Ther. 1995;273:1235. [PubMed] [Google Scholar]
- 8.Romero J, Garcia-Palomero E, Lin SY, Ramos JA, Makriyannis A, Fernandez-Ruiz JJ. Life Sci. 1996;58:1249. doi: 10.1016/0024-3205(96)00086-0. [DOI] [PubMed] [Google Scholar]
- 9.Deutsch DG, Chin SA. Biochem. Pharmacol. 1993;46:791. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- 10.Childers SR, Sexton T, Roy MB. Biochem. Pharmacol. 1994;47:711. doi: 10.1016/0006-2952(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 11.Lang W, Qin C, Hill WA, Lin S, Khanolkar AD, Makriyannis A. Anal. Biochem. 1996;238:40. doi: 10.1006/abio.1996.0247. [DOI] [PubMed] [Google Scholar]
- 12.Desarnaud F, Cadas H, Piomelli D. J. Biol. Chem. 1995;270:6030. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 13.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 14.Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG, Makriyannis A. J. Med. Chem. 1994;37:1889. doi: 10.1021/jm00038a020. [DOI] [PubMed] [Google Scholar]
- 15.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Science. 1997;277:1094. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 16.Ryan WJ, Banner WK, Wiley JL, Martin BR, Razdan RK. J. Med. Chem. 1997;40:3617. doi: 10.1021/jm970212f. [DOI] [PubMed] [Google Scholar]
- 17.Seltzman HH, Fleming DN, Thomas BF, Gilliam AF, McCallion DS, Pertwee RG, Compton DR, Martin BR. J. Med. Chem. 1997;40:3626. doi: 10.1021/jm9702950. [DOI] [PubMed] [Google Scholar]
- 18.Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK, Martin BR. J. Pharmacol. Exp. Ther. 1995;273:1172. [PubMed] [Google Scholar]
- 19.Pinto JC, Potie F, Rice KC, Boring D, Johnson MR, Evans DM, Wilken GH, Cantrell CH, Howlett AC. Mol. Pharmacol. 1994;46:516. [PubMed] [Google Scholar]
- 20.Adams IB, Ryan W, Singer M, Razdan RK, Compton DR, Martin BR. Life Sci. 1995;56:2041. doi: 10.1016/0024-3205(95)00187-b. [DOI] [PubMed] [Google Scholar]
- 21.Khanolkar AD, Abadji V, Lin S, Hill WA, Taha G, Abouzid K, Meng Z, Fan P, Makriyannis A. J. Med. Chem. 1996;39:4515. doi: 10.1021/jm960152y. [DOI] [PubMed] [Google Scholar]
- 22.Wise ML, Soderstrom K, Murray TF, Gerwick WH. Experientia. 1996;52:88. doi: 10.1007/BF01922423. [DOI] [PubMed] [Google Scholar]
- 23.Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, Makriyannis A. J. Med. Chem. 1998;41:5353. doi: 10.1021/jm970257g. [DOI] [PubMed] [Google Scholar]
- 24.Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. J. Pharmacol. Exp. Ther. 1999;289:1427. [PubMed] [Google Scholar]
- 25.Hampson AJ, Hill WA, Zan-Phillips M, Makriyannis A, Leung E, Eglen RM, Bornheim LM. Biochim. Biophys. Acta. 1995;1259:173. doi: 10.1016/0005-2760(95)00157-8. [DOI] [PubMed] [Google Scholar]
- 26.Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. J. Med. Chem. 1997;40:659. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- 27.Goutopoulos A, Fan P, Khanolkar AD, Xie XQ, Lin S, Makriyannis A. Bioorg. Med. Chem. 2001;9:1673. doi: 10.1016/s0968-0896(01)00088-8. [DOI] [PubMed] [Google Scholar]
- 28.Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7656. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Xu W, Vadivel SK, Fan P, Makriyannis A. J. Med. Chem. 2005;48:6423. doi: 10.1021/jm050272i. [DOI] [PubMed] [Google Scholar]
- 30.Lapitskaya MA, Vasiljeva LL, Pivnitsky KK. Synthesis. 1993;1:65. [Google Scholar]
- 31.Lindlar H. Helv. Chim. Acta. 1952;35:446. [Google Scholar]
- 32.Vasiljeva LL, Manukina TA, Demin PM, Lapitskaya MA, Pivnitsky KK. Tetrahedron. 1993;49:4099. [Google Scholar]
- 33.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155. [Google Scholar]
- 34.Hoegberg T, Stroem P, Ebner M, Raemsby S. J. Org. Chem. 1987;52:2033. [Google Scholar]
- 35.Sonogashira K. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. New York: Wiley-VCH; 1998. Chapter 5. [Google Scholar]
- 36.Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Mol. Pharmacol. 1988;34:605. [PubMed] [Google Scholar]
- 37.Palmer SL, Thakur GA, Makriyannis A. Chem. Phys. Lipids. 2002;121:3. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- 38.Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. Mol. Pharmacol. 2005;68:1623. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- 39.Macromodel, version 9.5. New York, NY: Schrodinger, LLC; 2007. [Google Scholar]