

Summary
Domchek et al provide a case report of a 28 year-old woman with congenital abnormalities, inherited ovarian cancer, and carboplatin hypersensitivity. Interestingly, the woman has validated germline mutations in both BRCA1 alleles. These findings further implicate BRCA1 in the Fanconi Anemia/BRCA pathway and have important implications for BRCA1 genetic testing.
Significant advances in our understanding of breast and ovarian cancer resulted from the genetic linkage mapping and cloning of two major tumor suppressor genes BRCA1 and BRCA2. Mutations in BRCA1 and BRCA2 account for approximately 16% of the familial risk of breast cancer (1). Moreover, heterozygous carriers of mutations in BRCA1 and BRCA2 have an 82% lifetime risk of breast cancer and a 54% and 23% risk of ovarian cancer, respectively (2). Biallelic germline mutations in BRCA2 account for a rare and highly cancer-prone subtype of Fanconi Anemia (3); however, humans with validated biallelic germline mutations in BRCA1 have not been identified, suggesting that at least one functional allele of BRCA1 is required for human embryogenesis and development.
Fanconi Anemia (FA) is an autosomal recessive or X-linked recessive genetic disease characterized by multiple congenital anomalies, bone marrow failure, cancer predisposition, and cellular hypersensitivity to DNA crosslinking agents, such as Mitomycin C and Cisplatin. There are fifteen known FA genes, and the encoded fifteen FA proteins (Figure 1, green) cooperate in a common pathway, the so-called Fanconi Anemia/BRCA Pathway (4), required for the repair of DNA crosslinks (5) (Figure 1). Additional proteins (red) such as BRCA1, while not bonafide FA proteins (i.e., not encoded by genes known to have germline mutations in FA patients) are also required for the activity of the pathway and are therefore candidate FA proteins. Interestingly, at least four of the FA genes FANCD1/BRCA2, FANCN/PALB2, FANCJ/BRIP1, and FANCO/RAD51C are also breast/ovarian cancer susceptibility genes. Heterozygote carriers of germline mutations in these genes carry an increased cancer risk, albeit at highly variable penetrance. A new study by Domchek et al (6) demonstrates that the BRCA1 protein is also a critical component of this pathway and that BRCA1 may itself be an FA gene.
Figure 1. BRCA1 cooperates in the Fanconi Anemia/BRCA1 pathway.
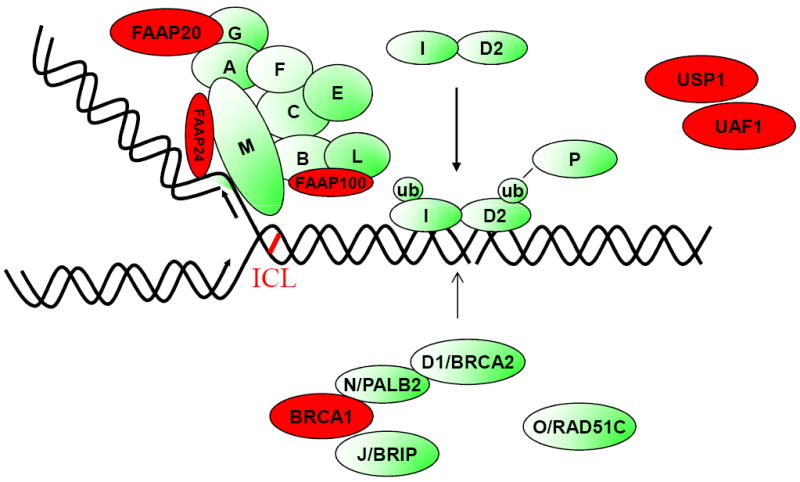
The fifteen known FA proteins (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P) cooperate in a common DNA repair pathway (in green). The pathway is activated when a replication fork stalls at a cisplatin interstrand crosslink (ICL). The FA core complex binds to the stalled fork, leading to monoubiquitination and recruitment of the I and D2 proteins. The downstream FA proteins (D2, N, J, and O) are recruited to DNA repair complexes. Additional proteins (in red), while not bonafide FA proteins, are also required for function of the FA pathway and for repair of DNA crosslinks. The BRCA1 protein is an FA-like protein and binds in a complex with at least three bonafide FA proteins. Biallelic mutations in BRCA1 (see text) result in an inherited ovarian cancer (FA-like) syndrome.
The interaction of BRCA1 with other proteins in the FA pathway has been suspected for several years. BRCA1, like bonafide FA proteins, has a well known function in the maintenance of genomic stability, through homologous recombination repair, and in the promotion of DNA repair of interstrand crosslinks (7). Moreover, knockdown of BRCA1 in tumor cells results in a reduction of FANCD2 monoubiquitination and nuclear FANCD2 DNA repair foci (8), suggesting that BRCA1 is an amplifier of the FA/BRCA pathway. Perhaps most interestingly, BRCA1 is a component of a large nuclear protein complex (Figure 1), consisting of at least three other bonafide FA proteins, BRCA2 (FANCD1), PALB2 (FANCN), and BRIP1 (FANCJ). PALB2 was originally identified in a screen of BRCA2 binding proteins (9), and it binds to the extreme N terminus of BRCA2. PALB2 also binds to the C-terminal BCRT repeats of BRCA1, and BRCA1, in turn, binds directly to BRIP1 (J) (Figure 1). This intimate interaction of BRCA1 with other bonafide FA proteins, which are themselves breast/ovarian cancer susceptibility proteins, further suggested that BRCA1 directly participates in the FA/BRCA pathway and that biallelic BRCA1 mutations might result in FA or an FA-like syndrome.
Indeed, in their case report, Domchek et al determine that the proband with ovarian cancer has biallelic mutations in BRCA1. While one mutant allele is clearly deleterious (the known C 2457 del C allele), the other allele is a variant of unknown significance (VUS), encoding a BRCA1 protein with a V1736A amino acid substitution. The authors provide several lines of reasoning to determine that this VUS allele is also deleterious and encodes a dysfunctional BRCA1 protein. First, they examined other kindreds, which carry this BRCA1 variant allele. These kindreds have family members with breast and ovarian cancer, and the combined odds ratio, in favor of the variant allele being pathogenic, was elevated (ratio of 234:1). Second, the V1736A mutation falls in one of the C-terminal BRCT domains of BRCA1. Although the mutation does not directly disrupt the BRCT binding surface of BRCA1, it indirectly alters the binding affinity of the BRCT domain for RAP80, another BRCA1 interacting protein. Furthermore, the presence of the V1736A mutation reduces BRCA1 localization to genomic sites of DNA double-strand breaks. Interestingly, recent studies indicate that mice with homozygous mutations corresponding to this BRCT region of BRCA1 yield viable animals, albeit with increased cancer risk (10). Third, and most convincingly, the ovarian tumor derived from the proband does not have loss of heterozygosity (LOH) at the BRCA1 locus. This finding is in contradistinction to the ovarian tumors derived from other family members, which do exhibit loss of the wildtype BRCA1 allele and persistence of the V1736A mutant allele.
Importantly, this case report does not indicate that BRCA1 is an FA gene, per se. The proband has a FA-like syndrome (some congenital abnormalities, ovarian CA, carboplatin hypersensitivity) but some clear differences from FA. The patient did not exhibit spontaneous bone marrow failure, though she did exhibit significant mucositis and enhanced bone marrow toxicity (prolonged neutropenia) following carboplatin treatment. Interestingly, carboplatin hypersensitivity is not a feature of BRCA1 heterozygote carriers (11), further supporting the argument that this patient had biallelic deleterious BRCA1 mutations. The patient died before the definitive diagnostic test for FA- namely, the DEB chromosome breakage test (12) - could be performed. If the patient had survived, the DEB test could have been performed on the primary peripheral blood lymphocytes and on a patient-derived ovarian tumor cell line. There is a high probability that these patient-derived cells would have been hypersensitive to DEB or cisplatin in vitro, and that transfection with the wild-type BRCA1 cDNA would have rescued this cellular phenotype. Still, the patient’s cells also carried a variant allele of BRCA2 which may have contributed, at least in part, to the hypersensitivity to cisplatin.
Although FA patients are prone to myeloid leukemia and squamous cell carcinomas, they generally do not develop breast or ovarian cancer. Several explanations may resolve this paradox. First, FA females are hypogonadal, and their low serum estrogen level may protect these individuals from these cancers. Second, FA females have decreased breast and ovarian tissue mass, perhaps further reducing their cancer risk. Third, FA patients often die young from complications of bone marrow failure or other cancers. As FA females live longer, due to improved supportive care, careful screening for breast and ovarian cancer in these patients may be warranted. Recently, there have been anectodal reports of women with subclinical FA (ie, FANCA -/- patients) who have presented with breast cancer, despite the absence of more obvious FA phenotypic features. Taken together, any women with ovarian or breast cancer who have a suspicious phenotype (ie, very early onset of cancer, characteristic developmental abnormalities, or profound carboplatin sensitivity) should be screened for mutations in BRCA1 or BRCA2 and perhaps other genes in the FA/BRCA pathway. Similarly, presentation with an FA phenotype (i.e. bone marrow failure and developmental defects) should warrant investigation for mutations in these same FA/BRCA genes.
In summary, this important new work by Domchek et al identifies a new autosomal recessive genetic syndrome (developmental defects, inherited ovarian cancer susceptibility, and crosslinker hypersensitivity caused by biallelic mutations in BRCA1). The work also describes a systematic approach for testing variant BRCA1 proteins for functional activity in vitro. As more variant BRCA1 alleles are identified through expanding genome sequencing efforts, there will be a growing need for better functional assays to distinguish deleterious from benign variants.
Footnotes
Disclosure of Potential Conflicts of Interest:
A.D. D’Andrea has no potential conflicts of interest.
References
- 1.Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nature genetics. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 2.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–6. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 3.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–9. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 4.D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nature reviews Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 5.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. The Journal of pathology. 2012;226:326–37. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 6.Domchek S. this issue. Cancer Discovery. 2012 [Google Scholar]
- 7.Silver DP, Livingston DM. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012;2:679–84. doi: 10.1158/2159-8290.CD-12-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Molecular cell. 2012;46:125–35. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular cell. 2006;22:719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 2011;334:525–8. doi: 10.1126/science.1209909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes & development. 2005;19:2925–40. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 12.Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Experimental hematology. 1993;21:731–3. [PubMed] [Google Scholar]