

Abstract
Numerous metalloproteins are important therapeutic targets that are gaining increased attention in the medicinal and bioinorganic chemistry communities. This perspectives article describes some emerging trends and recent findings in the area of metalloprotein inhibitor discovery and development. In particular, the increasing recognition of the importance of the metal-ligand interactions in these systems calls for more input and consideration from the bioinorganic community to address questions traditionally confined to the medicinal chemistry community.
Introduction
Metalloproteins are at the heart of numerous biological processes related to disease propagation ranging from gene (mis)regulation, protein matrix degradation, and antibiotic resistance. An ever increasing number of metalloproteins have garnered interest as potential theraputic targets for treating a wide variety of human diseases. Table 1 provides only a partial list of metalloproteins that have been targeted for addressing various pathologies. As can be seen from this list, a host of different metalloenzymes that employ a broad assortment of metal ions are of significant interest as medicinal targets. In this short Perspectives article, some emerging trends in the development of new metalloprotein inhibitors will be described and discussed. This area of research has not attracted the level of attention from the bioinorganic research community that the importance of the subject might command. It is hoped that this short report, which largely focuses on findings from the author’s laboratory, might generate new interest and fresh ideas to tackle some of the most challenging problems faced in the field of metalloprotein-targeted drug design today.
Table 1.
List of metalloprotein targets of interest for pharmaceutical development.
| Metal | Metalloprotein | Indication |
|---|---|---|
| Zn2+ | VanX | Bacterial infection |
| Zn2+ | TNF-α Converting Enzyme | Cancer |
| Zn2+ | Methionyl-tRNA-Synthetase | Bacterial infection |
| Zn2+ | Metallo-β-Lactamases | Bacterial infection |
| Zn2+ | Matrix Metalloprotease | Cancer, arthritis |
| Zn2+ | LpxC | Bacterial infection |
| Zn2+ | Histone Deacetylase | Cancer |
| Zn2+ | Hepatitis C Protease | Hepatitis C |
| Zn2+ | Glyoxalase | Cancer |
| Zn2+ | Farnesyl Transferase | Cancer, malaria |
| Zn2+ | Carbonic Anhydrase | Glaucoma |
| Zn2+ | Botulinum Neurotoxin | Toxin (biodefense) |
| Zn2+ | Anthrax Lethal Factor | Toxin (biodefense) |
| Zn2+ | Angiotensin Converting Enzyme | Hypertension |
| Zn2+ | Aggrecanase | Arthritis |
| Zn2+ | Adenosine Deaminase | Inflammation |
| Ni2+ | Urease | Bacterial infection |
| Ni2+ | Glyoxalase | Bacterial infection |
| Mg2+ | HIV Integrase | HIV infection |
| Fe(heme) | Nitric Oxide Synthase | Inflammation |
| Fe(heme) | Cyclooxygenase | Inflammation |
| Fe2+ | Ribonucleotide Reductase | Cancer, viral infections |
| Fe2+ | Peptide Deformylase | Bacterial infection |
| Fe2+ | Lipoxygenase | Inflammation |
| Fe2+/Mn2+ | Methionine Aminopeptidase | Cancer, bacterial infection |
| Cu2+ | Tyrosinase | Cancer |
Clincially Important Metalloprotein Inhibitors
Inhibitors of metalloproteins have already had a signficant impact on human health. Compounds that inhibit metalloenzymes are used clinically to treat diseases such as fungal infections, hypertension, cancer, HIV, and others. Among the most widely prescribed and successful metalloprotein inhibitors are the angiotensin converting enzyme (ACE) inhibitors.1 ACE is involved in the conversion of angiotensin I into angiotensin II, the latter of which is an octapeptide that is a potent vasoconstrictor. The inhibition of ACE thus prevents the secretion of angiotensin II, allowing the treatment of hypertension as well as congestive heart failure.2 The first FDA approved inhibitor was developed more than thirty years ago by Squibb under the name Captopril (Fig. 1). Importantly, this inhibitor interacts with the active site Zn(II) ion by direct coordination through the thiol metal-binding group (MBG) found in Captopril.3 Second-generation ACE inhibitors ultimately replaced the thiol MBG of Captopril by a carboxylic acid MBG to achieve better pharmacokinetics (Fig. 1).4–6 Many of these ACE inhibitors are formulated as prodrugs, in order to mask the free carboxylic acids and facilitate oral administration.1
Fig. 1.
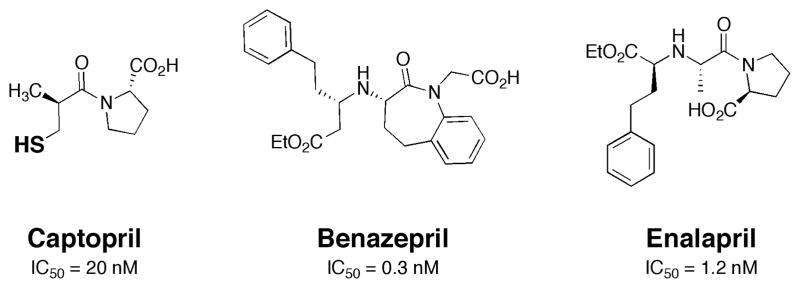
Structure of Captopril and second-generation (prodrug) ACE inhibitors. The donor atom of the thiol MBG in Captopril is shown in bold. IC50 values are shown for each inhibitor (IC50 = concentration of inhibitor required to achieve 50% enzyme inhibition).5, 6
More recent developments in metallprotein inhibitors have made headway against oncology and viral targets. In the realm of oncology, the first inhibitor of a histone deacetylase (HDAC) was approved for clinical use in 2006. HDACs are a class of proteins involved in the deacetylation of histones (lysine residues). The acetylation of histones alters chromatin structure, thus influencing transcriptional regulation.7 A subclass of HDACs are Zn(II)-dependent hydrolytic enzymes that have been targeted by a variety of compounds. The clinically approved compound suberoylanilide hydroxamic acid (SAHA, commercial name Vorinostat, Fig. 2) was developed by Aton Pharmaceuticals (later acquired by Merck), and is presently used for treating cutaneous T cell lymphoma. The structure of Vorinostat follows that of the canonical HDAC inhibitor and includes a capping group, linker, and metal-binding group (Fig. 2).8 Vorinostat uses a hydroxamic acid moiety as the MBG.9 Hydroxamic acids were first popularized as MBGs for use in metalloenzyme inhibitors due to their widespread use in the development of matrix metalloproteinase (MMP) inhibitors,10 and have since been used in inhibitors of many other zinc metalloenzymes.11 Despite the pervasive use of hydroxamic acids as MBGs, they frequently display poor biooavailibility and pharmacokinetics. Indeed, Vorinostat is the only FDA approved drug that contains a primary hydroxamic acid moiety as a MBG.
Fig. 2.
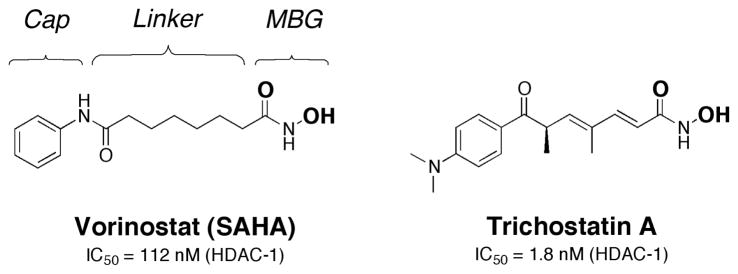
Structure of Vorinostat and a related HDAC inhibitor, Trichostatin A. The donor atoms of the hydroxamic acid MBG are shown in bold. The IC50 values shown are against partially purified HDAC-1 from mouse liver.8, 12
One of the more exciting recent discoveries in metalloenzyme inhibition is the approval of a first-in-class HIV integrase inhibitor. HIV integrase (HIV-IN) is an important viral enzyme required for integrating the viral genome with the host genome.13 The enzyme utilizes a dinuclear Mg(II) site to achieve integrase activity. In 2007, Merck received approval for the clinical use of Raltegravir,14 a small molecule inhibitor of HIV-IN that uses a disubstituted hydroxypyrimidinone as the MBG (Fig. 3). Diketo acids were identified early on as potent HIV-IN inhibitors from high-throughput screening (HTS) of more than 250,000 molecules (Fig. 3). Diketo acids were the first compounds found to show high specificity for HIV-IN and display antiviral activity that was highly correlated with HIV-IN inhibition.15 Attempts to improve on the properties of the diketo acids led to the discovery of Raltegravir, which replaces the diketo acid MBG with a 5-hydroxy-3-methylpyrimidin-4(3H)-one MBG to bind the binuclear metal site in HIV-IN.14 The hydroxypyrimidinone group was also discovered by HTS and was found to be a suitable diketo acid replacement with improved activity and pharmacokinetics.16 Like the diketo acids, the hydroxypyrimidinone chelator provides three nearly co-planar oxygen donor atoms to bind and bridge the Mg(II) ions in the dinuclear HIV-IN active site. The coordination mode of Raltegravir has recently been confirmed by co- crystallization with an HIV-IN homologue (Fig. 3).17 As anticipated, the complex between Raltegravir and the PFV-IN (PFV = prototype foamy virus) shows that three oxygen atoms (shown in bold, Fig. 3) of Raltegravir generate four bonds to the Mg(II) ions in the dinuclear active site.
Fig. 3.

Structure and IC50 values of Raltegravir and other HIV-IN inhibitors (top).19 Proposed MBGs are indicated in bold. Structure of Raltegravir bound to the PFV-IN (bottom, PDB: 3OYA).17 Inhibitor and metal-binding protein residues are colored by atom. Coordinate bonds between the inhibitor and active site are shown as dashed bonds.
Raltegraviresistant HIV strains have already appeared due to rapid virus mutation;18, 19 therefore, other HIV-IN inhibitors are already being developed. These include the compounds Elvitegravir and GSK364735 (Fig. 3),20 which have been advanced to clinical trials. Both inhibitors contain similar heteroatom triads for binding the dinuclear Mg(II) center; however, the MBGs are distinct from that of Raltegravir, with different donor atoms and bite angles between the donor atoms. This indicates that different MBGs, with donor atoms in distinct relative orientations, can serve as scaffolds for HIV-IN inhibitors.
Inhibitors of ACE, HDACs, and HIV-IN represent just a small slice of the metalloprotein inhibition field. However, they serve to illustrate the scope of molecules, targets, and pathologies that are encompassed by this growing area of research. The examples also illustrate the range of MBGs that have been employed to provide key metal-ligand interactions between the inhibitors and protein active sites. These MBGs include thiols, carboxylates, hydroxamic acids, diketo acids, hydroxypyrimidinones, and others. In the following sections of this perspectives article, several studies will be presented that delve deeper into the importance and role of the MBG in the development of metalloprotein inhibitors. In addition, new approaches to the discovery of MBGs and methods to exploit MBG-metalloprotein interactions for prodrug development will also be described.
The Importance of the MBG: MetAP Metalloform-specific Inhibitors
Methionine aminopeptidase (MetAP) enzymes catalyze removal of the N-terminal methionine group from newly synthesized peptides.21, 22 There are two forms of MetAP, termed Type I and II, that are found in both prokaryotes and eukaryotes; however, a given prokaryote will contain only either Type I or II, while eukaryotes possess both forms of the enzyme.23 MetAPs have garnered attention as targets for antifungal, antibacterial, and anticancer therapies.24
All MetAPs are metalloenzymes with a dinuclear active site, but the specific metal composition for any given MetAP from a specific organism has been elusive. This is because many MetAPs are quite active when reconsistuted with any of several different divalent metal ions, including Mn(II), Fe(II), Co(II), Ni(II), or Zn(II). The crystal structure of E. coli MetAP Type I (EcMetAP1) reconstituted with Co(II) shows that the active site metal ions are coordinated by two aspartates, two glutamates, and one histidine residue. The Co(II) ions are 2.9 Å apart and bridged by the carboxylate oxygen atoms of Asp108 and Glu235.25 Of the different active ‘metalloforms’ of EcMetAP1, the Co(II) reconsituted form of the enzyme has been most frequently used for the screening of inhibitors. However, an intriguing series of studies show that the various metalloforms of MetAP are inhibited by very different types of small molecules.
In an attempt to discover metalloform-selective MetAP inhibitors, Ye et al. used a library of 43,736 drug-like molecules (ChemBridge, San Diego, CA) and screened it against Co(II)- and Mn(II)-reconstituted EcMetAP1.26 This high-throughput screen (HTS) produced 786 initial hits against the Co(II) metalloform, while the Mn(II) form generated 512 hits. From these initial hits, the IC50 value of the top hits against each EcMetAP1 metalloform were obtained. The structures of the compounds with the best IC50 values against each metalloform displayed a fascinating trend.
The most potent compounds against Co(II) EcMetAP1 contained a common thiazol-2-yl-oxalamide moiety, as exemplified by compound 1 in Fig. 4. In contrast, the most effective compounds against Mn(II) EcMetAP1 possessed a 5- phenylfuran-2-carboxylic acid scaffold (compound 2, Fig. 4). Despite screening against the same recombinant protein, replacing the metal ion in the active site resulted in different classes of inhibitors being identified.
Fig. 4.

Structure and IC50 values of inhibitors found to be selective for the Co(II)- (1), Mn(II)- (2), and Fe(II)- (3) metalloforms of EcMetAP1.26, 28 Proposed MBGs are indicated in bold for compounds 2 and 3. Note that the three inhibitors possess both different metal-binding groups and entirely different overall scaffolds. Structure of inhibitor 4 bound to Mn(II) EcMetAP1 (bottom, PDB: 1XNZ).26 Inhibitor and metal-binding protein residues colored by atom. Coordinate bonds between the inhibitor and active site are shown as dashed bonds.
To evaluate the metalloform selectivity, the top hits were cross-screened against EcMetAP1 reconsituted with Mn(II), Fe(II), Co(II), and Ni(II).26 Both 1 and 2 proved to be remarkably selective. Compound 1 and related hits best inhibited the Co(II) form of EcMetAP1, with >350-fold selectivity versus the Mn(II) and Fe(II) forms of the enzyme. However, 1 was less selective between the Co(II) and Ni(II) forms of enzyme (~20-fold), consistent with earlier studies on MetAP inhibitors.27 Perhaps even more impressive was the specificity of 2, which displayed an IC50 value of 63 nM against the Mn(II) metalloform and >1000-fold selectivity against the other three metalloforms of EcMetAP1 tested.26
The selectivity of 1 for the Co(II) form and 2 for the Mn(II) form of EcMetAP1 is consistent with the known preference of these metals for soft (nitrogen or sulfur) and hard (oxygen) donor atoms, respectively. Clearly, the metal-ligand interactions of these inhibitors influences the types of compounds that are effective against a given metalloform. This dictates not only what MBG the inhibitors should possess, but also influences the entire molecular scaffold that is required, including the parts of the small molecule that do not directly interact with the active site metal ions. This striking result shows that identifying the optimum metal-ligand interactions can have a pronounced affect on the discovery and overall structure of metalloprotein inhibitors.
To understand the binding of the 5-phenylfuran-2- carboxylic acids (compound 2 and related hits), the structure of Mn(II) EcMetAP1 complexed with inhibitor 4 (Fig. 4) was determined by X-ray crystallography to 1.5 Å resolution.26 Prior to this study, the structures of MetAP enzymes had only been determined with Co(II) in the active site. The carboxylate MBG of 4 coordinates to both metal ions with one of the oxygen atoms in a μ-bridging fashion between the two ions. The bridging coordination results in the carboxylate group binding to one of the Mn(II) ions in a bidentate fashion, while only binding as a monodentate ligand to the second Mn(II) ion (Fig. 4). The rest of the inhibitor resides in a hydrophobic substrate binding pocket (S1) and interacts with several amino acid side chains. Importantly, there are essentially no major structural changes in the active site between the Mn(II) metalloform and previously determined Co(II) metalloforms,29 indicating that the selectivity of compounds such as 2 and 4 is not due to large rearrangements of the active site upon inhibitor binding. Rather, these findings further implicate the importance of the coordination chemistry (i.e. Lewis acid-base hard-hard matching) in the selectivity and potency of these inhibitors.
Building on these initial findings, the same group used HTS of 74,976 compounds (ChemBridge and ChemDiv, San Diego, CA) to identify inhibitors of Fe(II) EcMetAP1 for which no selective inhibitors were known at the time.28 Among the top ~300 hits found in the initial screen, a catechol-containing compound was identified that was further screened for selectivity against other EcMetAP1 metalloforms (compound 3, Fig. 4). Selection of this hit for further analysis was based on the well known affinity of the catechol moiety for iron, as exemplified by many bacterial siderophores. Indeed, inhibitor 3 showed an IC50 value of 13 μM against the Fe(II) enzyme, but showed no activity against the Co(II) or Mn(II) forms of EcMetAP1 at a concentration of 100 μM.28
The catechol moiety in compound 3 was determined to be essential for inhibition, based on a structure-activity relationship (SAR) study.28 Several derivatives were examined for their ability to inhibit the Fe(II) form of EcMetAP1. As shown in Fig. 5, the compounds examined (5–10) are structurally very similar to 3, but in every case the metal-chelating ability of 3 is disrupted by replacement (5–8), misspacing (9), or removal of one of the donor atoms altogether (10). All of the compounds in Fig. 5 show no inhibition of Fe(II) reconsititued EcMetAP1 at 100 μM, confirming the requirement for the catechol ligand.
Fig. 5.

Derivatives of compound 3 showing no activity against Fe(II) EcMetAP1 at a concentration of 100 μM (top).28 Structure of a catechol-based inhibitor 11 bound in the active site of Mn(II) EcMetAP1 (bottom, PDB: 3D27).28 Inhibitor 11 was found to be selective for the Fe(II) form of EcMetAP1. Inhibitor and metal-binding protein residues are colored by atom. Coordinate bonds between the inhibitor and active site are shown as dashed bonds.
Unambiguous confirmation for the mode of binding was obtained from the crystal structure of a related compound (11) with Mn(II) reconstituted EcMetAP1 (solved to 2.2 Å resolution).28 The Mn(II) metalloform of the enzyme was used for crystallization studies, in lieu of the Fe(II) metalloform, due to oxidation problems when trying to crystallize the latter form. As shown in Fig. 5, the catecholate moiety coordinates the dinuclear metal site in a bridging configuration quite similar to compound 4. Indeed, an overlay of the structures of 4 and 11 bound to the Mn(II) metalloform of EcMetAP1 shows little difference in the protein backbone (RMSD 0.23 Å) and reveals that the coordinating atoms from the inhibitors are in very similar positions on the metal ions.28 The observation that binding of the catechol-based inhibitor does not cause significant structural changes in the active site strongly suggests that the selectivity of the compound originates from a good match of the coordination chemistry between the MBG and the active site metal ions.
Having developed Co(II), Mn(II), and Fe(II) metalloform selective inhibitors, these compounds were used to elucidate the relevant metal ion in vivo for EcMetAP1.28 MetAP are essential enzymes for bacteria and therefore growth inhibition assays with two E. coli strains (as well as two Bacillus strains to examine activity against a Gram-positive organism) in the presence of metalloform-specific inhibitors 1–3 were performed. At concentrations as high as 1 mM, compounds 1 and 2 showed no growth inhibition on the two different E. coli bacterium. In contrast, the Fe(II)-specific inhibitor 3 showed growth inhibition at concentrations as low as 5.6 μM against one of the E. coli lines. Other derivatives of the catechol platform showed broad-spectrum micromolar level activity against both the Gram-negative and Gram-positive strains. These studies suggest that the relevant metalloform of MetAP in these organisms is the Fe(II) form.
Investigations on MetAP highlight the importance of the MBG on the activity of a metalloprotein inhibitor. The compounds were identified from HTS efforts, and clearly show that selectivity can be obtained with different chelating groups. In the case of MetAP, even within the context of an identical active site, the metal ions it contains has a pronounced effect on the types of MBGs and inhibitor scaffolds that are identified.26, 28 Metalloformspecific inhibitors were identified, unambiguously showing that the nature of the MBG plays a critical role in achieving selective inhibition. Furthermore, these selective inhibitors proved useful tools for elucidating the functional metal ions in vivo for MetAP.28, 30
Leading with the MBG: Using Coordination Chemistry to Discover New DXR Inhibitors
The studies on MetAP provide strong evidence that the choice of MBG used in a metalloprotein inhibitor is critical for achieving potent and selective inhibition. However, the number of different MBGs that have been explored in the development of metalloprotein inhibitors has been somewhat limited. As discussed in several reviews, certain moieties including hydroxamic acids, carboxylic acids, thiols, and a handful of others are the predominant MBGs found in inhibitors of metalloenzymes.10, 11, 31 However, several recent reports have made more deliberate efforts to explore, identify, and optimize new MBGs for use in metalloprotien inhibitors.
A recent study explored the use of different MBGs in the development of inhibitors of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR).32 DXR is a Mg(II)-dependent enzyme in the non-mevalonate biosynthesis pathway that is an attractive antibiotic target.33 Fosmidomycin is a naturally occuring and potent DXR inhibitor with an IC50 of 80 nM, yet it suffers a very short half life (90 min) and limited cellular uptake.34 Fosmidomycin is comprised of a retrohydroxamic acid, a propyl chain, and a terminal phosphonate group (Fig. 6). The crystal structure of fosmidomycin bound to E. coli DXR shows that the retrohydroxamic acid binds the Mg(II) ion in a bidentate fashion as found in many hydroxamate-containing inhibitors of metalloenzymes. The phosphonate group in fosmidomycin is anchored in a neighboring pocket by several hydrogen bonds. For many years, efforts to improve the activity of fosmidomycin focused on either the propyl chain or the phosphonate group because any changes to or removal of the MBG always resulted in a drastic loss of activity.35 The highly polar phosphonate group has been blamed for the limited cellular uptake observed with fosmidomycin, but substitution by sulfonic acids, carboxylic acids, or other groups results in decreased of activity.
Fig. 6.
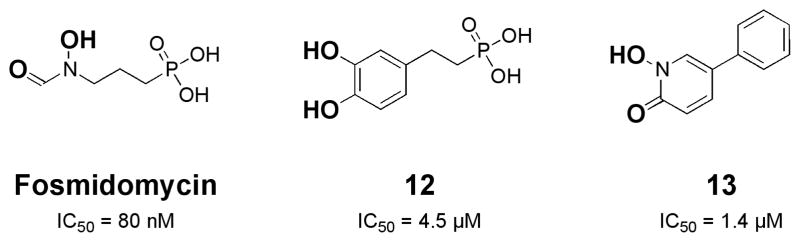
Structure and IC50 values of DXR inhibitors.32 Replacement of the reverse hydroxamic acid group in fosmidomycin with a catechol MBG led to compound 12. Subsequent exploration of hydrophobic derivatives of 12 led to compound 13, which contains a 1-hydroxypyridin-2-one MBG. Proposed MBGs are indicated in bold.
In a recent report by Deng et al., several new DXR inhibitors were reported in an attempt to break free of the fosmidomycin scaffold.32 Key to this approach was focusing on the coordination chemistry of Mg(II) and a deliberate effort to replace the retrohydroxamic acid found in fosmidomycin. Being a hard Lewis acid, Mg(II) would be expected to form stable complexes with hard Lewis base oxygen donor ligands; therefore, several compounds that employed a catechol MBG were examined as potential inhibitors. A catechol analogue of fosmidomycin yielded a promising lead with an IC50 value of 4.5 μM (12, Fig. 6). Using this lead as a basis for developing new inhibitors, specifically those that could do away with the polar phosphonate group, a series of hydrophobic compounds with different hard Lewis base, bidentate MBGs were explored. From a small library of compounds an even more potent fragment (IC50 value of 1.4 μM) that utilized a 1-hydroxypyridin-2-one MBG was discovered (13, Fig. 6). Even though this compound is about 16-fold less potent than fosmidomycin, the hydroxypyridinone inhibitor shows improved activity against gram positive and gram negative bacteria and improved lipophilicity and bioavailibility.32 The results of this study clearly demonstrate that exploration of new MBGs and an attention to coordination chemistry can reveal alternative scaffolds for metalloprotein inhibitor design.
Identifying New MBGs: Fragment-based Drug Discovery
Recent efforts from our laboratory have been focused on vastly expanding the range of MBG scaffolds available for the development of metalloprotein inhibitors. With this goal in mind, we have taken a fragment-based drug discovery (FBDD) approach and have developed chelator fragment libraries for screening against metalloproteins. FBDD, sometimes referred to as fragment-based lead discovery (FBLD),36 is an increasingly popular approach to the discovery of small molecule therapeutics and is largely viewed as an alternative to HTS. In HTS, as used in the previously described MetAP studies,26, 28 large libraries (>10,000) of relatively complex, drug-like compounds (MW >300) are screened against a target of interest. Lead compounds produced from the screen are then improved using medicinal chemistry approaches. In FBDD, relatively small libraries (100–1000) of small molecular fragments (MW <300) are screened against a target. Fragment hits are then matured into more drug-like compounds by a variety of strategies, including fragment linking or growing.36 Some of the proposed advantages of FBDD include the use of smaller libraries and a more efficient exploration of chemical diversity.
The FBDD strategy was applied very early on to metalloproteins, namely MMP-3, where the MBG and backbone substituents were treated as two fragments that could be connected with a linker (so-called fragment linking approach).37, 38 However, in these early applications of FBDD to metalloproteins, the MBG fragment was generally not varied. Except for one notable report,39 in most other cases the commonly used hydroxamic acid group was employed as the MBG, and a fragment library was screened to identify novel backbone substitutents. Despite the very early application of FBDD to metalloprotein inhibitors, the screening of diverse libraries of MBGs against metalloproteins has been largely overlooked. Only recently have fragment libraries comprised of MBGs been described and screened against a diverse panel of metalloprotein targets.
The overall scheme used for developing a FBDD approach to metalloprotein drug discovery is outlined in Fig. 7. In order to screen a wide range of chelating motifs, a fragment library of consisting of 96 MBGs possessing between 2 and 4 donor atoms was assembled, largely from commercially available compounds. This chelator fragment library (CFL) was initally screened against an MMP (MMP-2). At a fragment concentration of 1 mM, 31 fragments were classified 5 as hits (>50% inhibitory activity). From this screen, two compounds, 3-hydroxy-1,2-dimethylpyridine-4(1H)-thione and 3-hydroxy-1,2-dimethylpyridine-4(1H)-one, were selected for preparation of a focused library (extended library or sublibrary) containing 87 molecules. This sublibrary utilized these MBGs in combination with small hydrophobic substituents to generate compounds that might serve as more advanced scaffolds for lead compounds (Fig. 7). This approach is often referred to as a ‘fragment growth approach’ within the context of FBDD. The sublibrary was screened against several Zn(II)-dependent metalloenzymes, including three MMPs and the anthrax lethal factor (LF). Collectively, more than 50 hits were obtained against MMP-2, MMP-3, MMP-9 and LF at a fragment concentration of 50 μM. Moreover, out of the 10 hits identified against LF, one produced an IC50 value of 1.8 μM and a ligand efficiency (LE, a measure of the binding affinity as a function of fragment size)40 of 0.37 kcal/mol which surpasses many of the previously reported LF inhibitors identified through HTS. These findings suggest that CFLs can provide potent hits using much smaller libraries than traditional HTS, consistent with the goals of a true FBDD strategy.
Fig. 7.

(1) Screening of a chelator fragment library gives a hit, resulting in (2) the synthesis of a focused sublibrary (fragment growth strategy), which upon (3) screening of the sublibrary produces a potent lead compound against the Zn(II)-dependent anthrax LF.42
Following this initial screening of the CFL against a handful of Zn(II)-dependent metalloproteins, a subsequent study from our laboratory more broadly examined the applicability of the library against a larger panel of metalloproteins.41 Screening against nine different metalloproteins was performed using a slightly-modified version of the 96-member CFL (a few compounds were replaced due to poor solubility). The nine metalloproteins examined included five different MMPs (MMP-1, -2, -3, -8, -9), anthrax LF, 5-lipoxygenase (5-LO), inducible nitric oxide synthase (iNOS), and tyrosinase (TY). Unlike our initial study, in this report we screened against zinc-dependent metalloproteins (MMPs, LF), as well as iron-dependent (5- LO), heme-dependent (iNOS), and copper-dependent (TY) metalloenzymes. In addition, TY is a dinuclear copper active site while all the other metalloproteins examined have mononuclear active sites. In this way, preferences by different types of metalloenzymes for different MBGs could be revealed.
Indeed, upon screening the CFL against this panel of metalloproteins, several interesting trends emerged. The screening results clearly show that although all 96 fragments are avid metal binders (i.e. MBGs), they do not exhibit non- specific, broad spectrum inhibition against all nine metalloproteins. Rather, inhibitory activity is dependent on the fragment structure and is clearly not solely due to indiscriminate metal binding to any metalloprotein active site.41 Unmistakable patterns of selectivity are observed, as is expected for a true FBDD approach. The results of this study suggest that by judicious selection of the MBG, inhibitors with improved affinity and selectivity for a given metalloprotein target can be realized.
Although the 96 fragments did not produce non-specific metalloprotein inhibition, high hit rates were observed, ranging from 29–43% for the five MMPs, 24% for anthrax LF, 49% for 5-LO, and 60% for TY. In contrast, few fragments inhibited iNOS, which is consistent with the fact that fragments chosen for the CFL are chelators designed to bind metals through two or more donor atoms. iNOS, being a heme-dependent metalloenzyme with only one axial coordination site accessible for binding, rendered most of the fragments in the CFL ineffective due to the lack of a second vacant coordination site through which to chelate the metal center (~4% hit rate against iNOS).41
A number of other significant trends were found from these screening experiments. First, IC50 values were obtained for several hits, which showed that the LE values of the MBGs were excellent (0.4–0.8 kcal/mol).41 Second, the MMPs all generally elicit the same MBGs as hits from the CFL; hits against unrelated metalloenzymes (LF, 5-LO, TY) vary considerably. Finally, fragments with either very broad (inhibiting nearly all the metalloproteins in the panel) or selective (inhibiting only 1–2 metalloproteins in the panel) activity were found.
CFLs allow for screening a broad range of MBGs against any metalloprotein target of interest. However, when knowledge of coordination chemistry is applied to FBDD, it is possible to predict which MBGs will likely have a better affinity for a specific metalloenzyme based on the nature of the active site metal ion. For example, we recently prepared focused libraries targeted against Zn(II)-dependent metalloenzymes based on the MBGs of several well known small molecule Zn(II) ion sensors.43 Zinquin is a thoroughly investigated Zn(II) ion sensor based on a 8-sulfonamidoquinoline core (Fig. 8).44 Similarly, the 2-sulfonamidophenylbenzimidazole MBG has been used in a related family of ratiometric fluorescent Zn(II) ion sensors (Fig. 8).45, 46 Using these known scaffolds for selective Zn(II) ion binding, two sublibraries containing a combined ~80 compounds were generated based on the 8-sulfonamidoquinoline and 2-sulfonamidophenylbenzimidazole cores with variable sulfonamide substituents (Fig. 8). These sublibraries were then screened against MMP-2, -3, -8, -9, and anthrax LF. These screens identified fragments with low micromolar activity against both MMP and LF targets. In addition, differences in target selectivity were observed for 8-sulfonamidoquinoline and 2-sulfonamidophenylbenzimidazole fragments possessing identical sulfonamide substituents. Thus the nature of the MBG can apparently play a role in target selectivity. Similar to the previously described study on DXR inhibitors,32 our findings on these sulfonamide-based inhibitors illustrate that insight into the relevant coordination chemistry can facilitate rational design of metalloprotein inhibitors. In the present case, rationally designed, focused libraries were devised for screening against certain metalloprotein targets. These libraries were based on the known coordination chemistry of Zn(II) metal ion sensors, some of which had been in the literature more than a decade, but never made the transition from bioinorganic metal ion sensing to metalloprotein inhibition. This kind of library development provides a complimentary approach to screening large numbers of chelators via CFLs.
Fig. 8.

Structure of Zn(II) metal ion sensors (top), including the coordination mode of Zinquin (left). Two focused libraries (QSL = quinoline sulfonamide library; BISL = benzimidazole sulfonamide library) for inhibiting Zn(II)-dependent metalloproteins were developed based on these metal ions sensors (bottom).43 Proposed MBGs are indicated in bold.
Exploiting the Metal-Ligand Interaction: A Case for Metalloprotein Prodrugs
It is estimated that 5–7% of all drugs approved worldwide can be classified as prodrugs. Prodrugs are an important class of therapeutics that are metabolized or otherwise activated in vivo to generate a bioactive compound.47, 48 Prodrugs are attractive for their potential to provide targeted activity thereby reducing side effects. Several widely prescribed prodrugs include Vasotec® (ACE inhibitor), Zocor® (statin), and Clarinex® (antihistamine). Despite their clinical importance, the development of prodrugs that target metalloproteins is surprisingly small. Blocking of the MBG of a metalloprotein inhibitor can provide a route for the development of prodrugs for inhibiting metalloproteins. Prodrugs can have several advantages including improved pharmacokinetics, enhanced oral availability, and spatially and temporally localized activity (the latter of which may help reduce systemic toxicity).
To date, a very limited number of metalloprotein-targeted prodrugs have been studied, with the majority of studies focused on histone deacetylase (HDAC), matrix metalloproteinase (MMP), carbonic anhydrase (CA), and ACE prodrugs. In the case of ACE, as described earlier, carboxylate-based inhibitors are clinically employed as prodrugs. Ionization of the carboxylate MBG leads to unfavorable oral availability; however, this can be overcome by transforming the inhibitor into an ester prodrug. The ester group is hydrolyzed in vivo by esterases to give the active compound containing a carboxylate MBG. This concept was discovered more than 20 years ago and since that time all new FDA approved ACE inhibitors have been manufactured as prodrugs.1
Recently, our laboratory has initiated a research program broadly focused on the development of metalloprotein prodrugs. As demonstrated by ACE inhibitors, blocking of the MBG is an effective strategy for devising metalloprotein-targeted prodrugs. Using our experience with the synthesis and evaluation of MMP inhibitors, we have focused on these compounds for testing various approaches to blocking the MBG in order to generate prodrugs. Taking inspiration and adapting strategies from the work of Orvig,49–51 Franz,52–54 and others on ‘prochelators,’ we have recently demonstrated that blocking of the MBG can produce metalloprotein prodrugs. We have found that both enzymatic and chemical stimuli can be used to trigger these inhibitors in vitro.
In our first study, a glucose group was used to block the MBG of an MMP inhibitor.55 Such glycoside prodrugs have the added advantage of enhanced water solubility due to the hydrophilic sugar moiety. A previous study had reported on a glycoside-protected MMP inhibitor prodrug; however, the reported system did not release the desired inhibitor upon enzymatic activation.56 For our studies, a previously reported MMP inhibitor, 1,2-HOPO-2 was employed (Fig. 9). 1,2- HOPO-2 utilizes a 1-hydroxypyridin-2-one MBG and displays IC50 values of <100 nM against select MMPs.57 The cleaveage of the glucose group from prodrug 13 was achieved by the action of β-glucosidase, which cleanly produced 1,2-HOPO-2. The cleavage reaction could be readily monitored by either UV-Visible spectroscopy or HPLC. Importantly, the prodrug 14 was stable in aqueous solution for >24 h, indicating that the glycosidic protecting group was not readily hydrolyzed under ambient conditions.
Fig. 9.
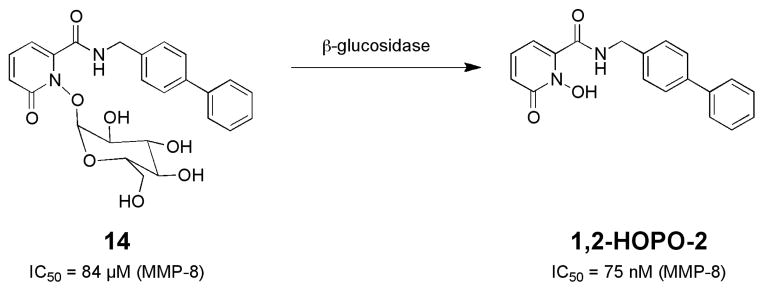
Activation of a glucose-protected MMP inhibitor prodrug.58 Note the substantial improvement in IC50 values upon conversion of the prodrug (14) to the active inhibitor (1,2-HOPO-2).
The ability of prodrug 14 to inhibit MMPs was greatly attenuated relative to 1,2-HOPO-2. The IC50 value of 14 against MMP-8 was determined to be 84 μM, compared to 75 nM for 1,2-HOPO-2.58 The ratio of these values, the quotient IC50 value (QIC50), is >1000, indicating a very substantial difference in activity between the prodrug and free inhibitor. The ability of 14 to be activated by β-glucosidase was evaluated in a cell-free MMP assay. In the absence of β-glucosidase, prodrug 14 (at a concentration of 150 nM) displayed little inhibition of MMP-8. However, upon activation with β-glucosidase, 33% inhibition of MMP-8 activity was observed.58 Hence, this study demonstrated that blocking the MBG with a glycoside group was a viable strategy for producing metalloprotein inhibitor prodrugs.
In two subsequent studies, our group reported the preparation of MMP inhibitor prodrugs that are activated by the presence of reactive oxygen species (ROS), specifically 59, 60 Activation of MMP inhibitor prodrugs by ROS H2O2. could prove to be particularly relevant to the treatment of ischemic injury, such as that observed in the case of stroke. It has been shown that MMPs can be activated by ROS during ischemia, and that both ROS and ROS-activated MMPs break down the blood brain barrier (BBB), leading to ischemic injury. Therefore, ROS-triggered MMP inhibition could greatly reduce ischemic brain injury directly at the site of a stroke by improving the spatial and temporal delivery of the MMP inhibitor to the site of injury.
In one study, we examined the use of ROS-sensitive sulfonate esters to prepare MMP prodrugs.59 As with our study on glucose-protected MMP inhibitors, the semi- selective 1,2-HOPO-2 inhibitor was utilized. Sulfonate esters have been reported as ROS-labile groups for use in small molecule fluorescent sensors to detect ROS. Coupling of 1,2-HOPO-2 with several sulfonyl chlorides containing different substituents gave a series of sulfonate ester MMP prodrugs (Fig. 10). Prodrugs 15a–b were rapidly cleaved in the presence of H2O2, with rates dependent on the nature of the sulfonate ester substituent. Although these prodrugs were relatively easy to synthesize and did respond to ROS as desired, they ultimately proved to have several substantial shortcomings. The most serious limitation of sulfonate ester prodrugs 15a–b is that they were quite susceptible to hydrolysis in aqueous solution. Hence compounds 15a–b were not stable and even in the absence of ROS, the sulfonate ester groups would dissociate to produce 1,2-HOPO-2.59 Due to this and other limitations, we turned our efforts to other ROS-sensitive protecting groups, in order to establish a better performing prodrug platform.
Fig. 10.

Structure of ROS-activated MMP inhibitor prodrugs based on sulfonate ester (top)59 and boronate ester self-immolative (bottom)60 protecting groups.
As in the sulfonate ester strategy, we were inspired by work on ROS-sensitive fluorophores. Studies by Chang and co- workers,61–63 as well as the work on self-immolative linkers by Shabat and co-workers,64, 65 directed our attention to a boronate ester strategy for developing ROS-activated MMP prodrugs. After exploring several synthetic approaches, we found that protecting the MBG with a benzylic ether that contained a boronate ester trigger group gave a very well behaved prodrug conjugate (Fig. 10).60 Upon cleavage of the boronate ester with H2O2, the benzylic ether undergoes a 1,6-benzyl elimination, liberating the MBG and producing an active MMP inhibitor. As per our other studies on prodrugs, the MMP inhibitor 1,2-HOPO-2 was used as a model compound (Fig. 10). As expected, activation of prodrug 15 with excess H2O2 rapidly produced 1,2-HOPO-2, and control experiments with other ROS (e.g. superoxide) showed that cleavage of the boronic ester was specific to H2O2. More importantly, the boronate ester self-immolative prodrugs were very stable in buffer, as 16 was unchanged over a 24 h period in the absence of H2O2. Compound 16 was assayed against MMP-9 and MMP-12, which showed IC50 values >1 mM and 18 μM, respectively (Fig. 10). Upon activation by H2O2, producing 1,2-HOPO-2, more than a 100-fold improvement in IC50 value was observed (i.e. QIC50 value of >100).60 These findings demonstrated an effective, general route to the preparation of ROS-activated metalloprotein-targeted prodrugs. In addition, by changing the triggering element from a boronate ester to other cleavable groups, it is anticipated that this approach will be useful for preparing metalloprotein prodrugs that can be activated by a wide range of biological and chemical stimuli.
Conclusions
Metalloproteins represent a broad class of high-value medicinal targets. The vast majority of metalloprotein inhibitors, either under investigation or in clinical use, employ metal-binding groups for interacting with the active site metal ion. This provides a tremendous opportunity for bioinorganic chemists to harness their expertise in metal-ligand binding to address important problems in medicinal chemistry. Herein, we have highlighted just a handful of new approaches, some of which have originated in our laboratory, that have been applied to developing new metalloprotein inhibitors. We hope that the work highlighted here and related efforts will inspire more interactions between the bioinorganic and medicinal chemistry communities. We believe that such cross- pollination is the key to developing new ideas, achieving revolutionary breakthroughs, and ultimately producing new metalloprotein-targeted therapeutics for treating human 5 disease.
Acknowledgments
Studies in the area of metalloprotein inhibitors have been generously supported by the University of California, San Diego, the National Institutes of Health (R01 HL00049; R21 HL094571), the American Heart Association (0970028N), and the Center for AIDS Research (U.C. San Diego). The authors thank J.A. Jacobsen for compiling Table 1.
Notes and references
- 1.Brown NJ, Vaughan DE. Circulation. 1998;97:1411–1420. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E. New Engl J Med. 1991;325:351–353. doi: 10.1056/NEJM199108013250508. [DOI] [PubMed] [Google Scholar]
- 3.Ondetti MA, Rubin B, Cushman DW. Science. 1977;196:441–444. doi: 10.1126/science.191908. [DOI] [PubMed] [Google Scholar]
- 4.Patchett AA, Harris E, Tristram EW, Wyvratt MJ, Wu MT, Taub D, Peterson ER, Ikeler TJ, ten Broeke J, Payne LG, Ondeyka DL, Thorsett ED, Greenlee WJ, Lohr NS, Hoffsommer RD, Joshua H, Ruyle WV, Rothrock JW, Aster SD, Maycock AL, Robinson FM, Hirschmann R, Sweet CS, Ulm EH, Gross DM, Vassil TC, Stone CA. Nature. 1980;288:280–283. doi: 10.1038/288280a0. [DOI] [PubMed] [Google Scholar]
- 5.Patchett AA. Br J Clin Pharmac. 1984;18:201S–207S. doi: 10.1111/j.1365-2125.1984.tb02599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webb RL, Miller DM, Traina V, Gomez HJ. Cardiovasc Drug Rev. 1990;8:89–104. [Google Scholar]
- 7.Bolden JE, Peart MJ, Johnstone RW. Nature Rev Drug Dis. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 8.Paris M, Porcelloni M, Binaschi M, Fattori D. J Med Chem. 2008;51:1505–1529. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- 9.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 10.Whittaker M, Floyd CD, Brown P, Gearing AJH. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 11.Jacobsen FE, Lewis JA, Cohen SM. ChemMedChem. 2007;2:152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 12.Mai A, Massa S, Rotili D, Simeoni S, Ragno R, Botta G, Nebbioso A, Miceli M, Altucci L, Brosch G. J Med Chem. 2006;49:6046–6056. doi: 10.1021/jm0605536. [DOI] [PubMed] [Google Scholar]
- 13.Pommier Y, Johnson AA, Marchand C. Nat Rev Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 14.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Paz OG, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. J Med Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 15.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 16.Iwamoto M, Wenning LA, Petry AS, Laethem M, De Smet M, Kost JT, Merschman SA, Strohmaier KM, Ramael S, Lasseter KC, Stone JA, Gottesdiener KM, Wagner JA. Clin Pharmacol Ther. 2008;83:293–299. doi: 10.1038/sj.clpt.6100281. [DOI] [PubMed] [Google Scholar]
- 17.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Nature. 2010;464:232–237. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metifiot M, Maddali K, Naumova A, Zhang XM, Marchand C, Pommier Y. Biochemistry. 2010;49:3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Biochemistry. 2008;47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serrao E, Odde S, Ramkumar K, Neamati N. Retrovirology. 2009;6:25–39. doi: 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowther WT, Matthews BW. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 22.Lowther WT, Matthews BW. Chem Rev. 2002;102:4581–4607. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 23.Bradshaw RA, Brickey WW, Walker KW. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 24.Vaughan MD, Sampson PB, Honek JF. Curr Med Chem. 2002;9:385–409. doi: 10.2174/0929867023371102. [DOI] [PubMed] [Google Scholar]
- 25.Roderick SL, Matthews BW. Biochemistry. 1993;32:3907–3912. doi: 10.1021/bi00066a009. [DOI] [PubMed] [Google Scholar]
- 26.Ye OZ, Xie SX, Huang M, Huang WJ, Lu JP, Ma ZQ. J Am Chem Soc. 2004;126:13940–13941. doi: 10.1021/ja045864p. [DOI] [PubMed] [Google Scholar]
- 27.Li JY, Chen LL, Cui YM, Luo QL, Li J, Nan FJ, Ye QZ. Biochem Biophys Res Comm. 2003;307:172–179. doi: 10.1016/s0006-291x(03)01144-6. [DOI] [PubMed] [Google Scholar]
- 28.Wang WL, Chai SC, Huang M, He HZ, Hurley TD, Ye QZ. J Med Chem. 2008;51:6110–6120. doi: 10.1021/jm8005788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowther WT, Orville AM, Madden DT, Lim S, Rich DH, Matthews BW. Biochemistry. 1999;38:7678–7688. doi: 10.1021/bi990684r. [DOI] [PubMed] [Google Scholar]
- 30.Lu JP, Chai SC, Ye QZ. J Med Chem. 2010;53:1329–1337. doi: 10.1021/jm901624n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. Biochim Biophys Acta. 2010;1803:72–94. doi: 10.1016/j.bbamcr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Sundriyal S, Rubio V, Shi Zz, Song Y. J Med Chem. 2009;52:6539–6542. doi: 10.1021/jm9012592. [DOI] [PubMed] [Google Scholar]
- 33.Singh N, Chevé G, Avery MA, McCurdy CR. Curr Pharm Des. 2007;13:1161–1177. doi: 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]
- 34.Wiesner J, Borrmann S, Jomaa H. Parasitol Res. 2003;90:S71–S76. doi: 10.1007/s00436-002-0770-9. [DOI] [PubMed] [Google Scholar]
- 35.Deng L, Endo K, Kato M, Cheng G, Yajima S, Song Y. ACS Med Chem Lett. 2010 doi: 10.1021/ml100243r. ASAP contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Congreve M, Chessari G, Tisi D, Woodhead AJ. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 37.Hajduk PJ, Sheppard G, Nettesheim DG, Olejniczak ET, Shuker SB, Meadows RP, Steinman DH, Carrerea GM, Marcotte PA, Severin J, Walter K, Smith H, Gubbins E, Simmer R, Holzman TF, Morgan DW, Davidsen SK, Summers JB, Fesik SW. J Am Chem Soc. 1997;119:5818–5827. [Google Scholar]
- 38.Olejniczak ET, Hajduk PJ, Marcotte PA, Nettesheim DG, Meadows RP, Edalji R, Holzman TF, Fesik SW. J Am Chem Soc. 1997;119:5828–5832. [Google Scholar]
- 39.Hajduk PJ, Shuker SB, Nettesheim DG, Craig R, Augeri DJ, Detebenner D, Albert DH, Guo Y, Meadows RP, Xu L, Michaelides M, Davidsen SK, Fesik SW. J Med Chem. 2002;45:5628–5639. doi: 10.1021/jm020160g. [DOI] [PubMed] [Google Scholar]
- 40.Hopkins AL, Groom CR, Alex A. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 41.Jacobsen JA, Fullagar J, Miller MT, Cohen SM. J Med Chem. 2010 doi: 10.1021/jm101266s. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agrawal A, Johnson SL, Jacobsen JA, Miller MT, Chen LH, Pellecchia M, Cohen SM. ChemMedChem. 2010;5:195–199. doi: 10.1002/cmdc.200900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouffet M, de Oliveira CAF, Udi Y, Agrawal A, Sagi I, McCammon JA, Cohen SM. J Am Chem Soc. 2010;132:8232–8233. doi: 10.1021/ja101088j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fahrni CJ, O’Halloran TV. J Am Chem Soc. 1999;121:11448–11458. [Google Scholar]
- 45.Wu YG, Lawson PV, Henary MM, Schmidt K, Bredas JL, Fahrni CJ. J Phys Chem A. 2007;111:4584–4595. doi: 10.1021/jp068832s. [DOI] [PubMed] [Google Scholar]
- 46.Henary MM, Wu YG, Cody J, Sumalekshmy S, Li J, Mandal S, Fahrni CJ. J Org Chem. 2007;72:4784–4797. doi: 10.1021/jo070433l. [DOI] [PubMed] [Google Scholar]
- 47.Kratz F, Muller IA, Ryppa C, Warnecke A. ChemMedChem. 2008;3:20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- 48.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Nat Rev Drug Disc. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira CL, Bayly SR, Green DE, Storr T, Barta CA, Steele J, Adam MJ, Orvig C. Bioconjugate Chemistry. 2006;17:1321–1329. doi: 10.1021/bc060085e. [DOI] [PubMed] [Google Scholar]
- 50.Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, Thomas F, Allen DD, Lockman PR, Merkel M, Thompson KH, Orvig C. Angew Chem Int Ed. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]
- 51.Scott LE, Page BDG, Patrick BO, Orvig C. Dalton Trans. 2008:6364–6367. doi: 10.1039/b815404j. [DOI] [PubMed] [Google Scholar]
- 52.Charkoudian LK, Pham DM, Franz KJ. J Am Chem Soc. 2006;128:12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 53.Charkoudian LK, Pham DM, Kwon AM, Vangeloff AD, Franz KJ. Dalton Trans. 2007:5031–5042. doi: 10.1039/b705199a. [DOI] [PubMed] [Google Scholar]
- 54.Dickens MG, Franz KJ. ChemBioChem. 2010;11:59–62. doi: 10.1002/cbic.200900597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Major-Jourden JL, Cohen SM. Chem Commun. 2010;46:1241–1243. doi: 10.1039/b923302d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitchell MB, Whitcombe IWA. Tetrahedron Lett. 2000;41:8829–8834. [Google Scholar]
- 57.Agrawal A, Romero-Perez D, Jacobsen JA, Villarreal FJ, Cohen SM. ChemMedChem. 2008;3:812–820. doi: 10.1002/cmdc.200700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Major Jourden JL, Cohen SM. Chem Commun. 2010;46:1241–1243. doi: 10.1039/b923302d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daniel KB, Major Jourden JL, Cohen SM. J Biol Inorg Chem. 2010 doi: 10.1007/s00775-010-0727-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Major Jourden JL, Cohen SM. Angew Chem Intl Ed. 2010;49:6795–6797. doi: 10.1002/anie.201003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller EW, Chang CJ. Curr Opin Chem Biol. 2007;11:620–625. doi: 10.1016/j.cbpa.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang MCY, Pralle A, Isacoff EY, Chang CJ. J Am Chem Soc. 2004;126:15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- 64.Haba K, Papkov M, Shamis M, Lerner RA, Barbas CF, III, Shabat D. Angew Chem Int Ed. 2005;44:716–720. doi: 10.1002/anie.200461657. [DOI] [PubMed] [Google Scholar]
- 65.Sella E, Shabat D. Chem Commun. 2008:5701–5703. doi: 10.1039/b814855d. [DOI] [PubMed] [Google Scholar]