

Abstract
Domoic acid (DomA) is a potent marine neurotoxin. By activating α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid/kainate receptors, DomA induces oxidative stress–mediated apoptotic cell death in neurons. The effect of prolonged (10 days) exposure to a low, nontoxic concentration (5nM) of DomA on acute (intermediate concentration) neurotoxicity of this toxin was investigated in cerebellar granule neurons (CGNs) from wild-type mice and mice lacking the glutamate cysteine ligase (GCL) modifier subunit (Gclm ˗/˗). CGNs from Gclm ˗/˗ mice have very low glutathione (GSH) levels and are very sensitive to DomA toxicity. In CGNs from wild-type mice, prolonged exposure to 5nM DomA did not cause any overt toxicity but reduced oxidative stress–mediated apoptotic cell death induced by exposure to an intermediate concentration (100nM for 24h) of DomA. This protection was not observed in CGNs from Gclm ˗/˗ mice. Prolonged DomA exposure increased GSH levels in CGNs of wild-type but not Gclm ˗/˗ mice. Levels of GCLC (the catalytic subunit of GCL) protein and mRNA were increased in CGNs of both mouse strains, whereas levels of GCLM protein and mRNA, activity of GCL, and levels of GCL holoenzyme were only increased in CGNs of wild-type mice. Chronic DomA exposure also protected wild-type CGNs from acute toxicity of other oxidants. The results indicate that CGNs from Gclm ˗/˗ mice, which are already more sensitive to DomA toxicity, are unable to upregulate their GSH levels. As Gclm ˗/˗ mice may represent a model for a common human polymorphism in GCLM, such individuals may be at particular risk for DomA-induced neurotoxicity.
Key Words: domoic acid, neurotoxicity, glutathione, glutamate-cysteine ligase.
Domoic acid (DomA) is a potent neurotoxin, which was identified as the toxic agent responsible for an outbreak of poisoning due to consumption of contaminated shellfish in Eastern Canada in 1987 (Perl et al., 1990). Neuronal loss, particularly in the hippocampus and the amygdala, was observed in the four individuals who died in this incident (Teitelbaum et al., 1990), and similar brain lesions have been seen in primates, rats, and mice following administration of DomA (Pulido, 2008). The pattern of brain damage observed in humans and animals following exposure to DomA resemble that observed after administration of kainic acid (Teitelbaum, 1990). DomA is indeed a structural analog of kainic acid and exerts its toxicity by activating the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid)/kainate subtype of glutamate receptors (Berman and Murray, 1997; Berman et al., 2002; Hampson and Manalo, 1998; Novelli et al., 1992). In vitro studies have shown that in mouse cerebellar granule neurons (CGNs), DomA causes necrotic or apoptotic cell death, depending on its concentration (Giordano et al., 2006, 2007, 2009). Necrosis is observed at higher concentrations of DomA (10µM) and involves activation of both AMPA/kainate and NMDA receptors, the latter activated by DomA-induced glutamate release (Giordano et al., 2006). In contrast, apoptotic damage predominates at lower concentrations of DomA (e.g., 100nM); DomA-induced apoptosis is solely due to activation of AMPA/kainate receptors (Giordano et al., 2007).
Both necrosis and apoptosis have been shown to be mediated by oxidative stress, as evidenced by the following observations (Giordano et al., 2006, 2007): (1) DomA causes a concentration-dependent increase of reactive oxygen species (ROS); lipid peroxidation is also increased; (2) various antioxidants inhibit DomA-induced neurotoxicity; (3) glutathione (GSH) levels modulate the necrotic and apoptotic effects of DomA. In particular, neurotoxicity of DomA is enhanced in CGNs from Gclm ˗/˗ mice, which lack the modifier subunit of glutamate-cysteine ligase, the first and rate limiting step in the synthesis of GSH, and as a consequence display very low levels of GSH (Yang et al., 2002). Depletion of GSH in CGNs from wild-type mice also significantly increases DomA neurotoxicity (Giordano et al., 2006).
While acute exposure to high doses of DomA (e.g., 2–4mg/kg) is clearly neurotoxic in humans (see references in Costa et al., 2010), less is known on the potential neurotoxicity of repeated low-level exposures to this toxin. Indeed, only few studies have investigated the effect of repeated exposure to DomA, at doses below those inducing overt clinical symptoms. In vivo observations in rodents and primates suggest that subsymptomatic doses of DomA do not increase sensitivity or enhance neurotoxicity (Truelove et al., 1996, 1997). A study in mice similarly indicates that repeated asymptomatic doses of DomA did not result in any clinical sign of toxicity, and upon repeated symptomatic doses, symptoms did not increase (Peng et al., 1997). Similarly, although a single dose of DomA (1 or 2mg/kg) caused memory impairment in mice, repeated exposures appeared to cause a lesser effect (Clayton et al., 1999). These findings may be due to toxicokinetic factors, i.e., to the fact that DomA is rapidly cleared and does not accumulate (Truelove et al., 1996, 1997; and references in Costa et al., 2010). In vitro studies have provided conflicting results; in one study, pre-exposure with lower doses of DomA produced tolerance to the effect of higher concentrations, which was not related to receptor downregulation (Kerr et al., 2002), whereas in another study, a similar pretreatment enhanced the neurotoxicity of a subsequent DomA exposure (Qiu et al., 2006).
Different investigators or agencies have determined values of tolerable daily intake (TDI) for DomA ranging between 0.018 and 0.1mg/kg (Costa et al., 2010). Limits of 20 µg DomA/g shellfish meat have been adopted all around the world based on the findings of the 1987 Canadian outbreak. This limit appears to be protective of the human population because no other known human poisonings due to DomA have been reported since 1987. However, food consumption data from the United States and the European Union indicate that at this limit or even at lower, more realistic levels of contamination, the TDI level may be exceeded (Costa et al., 2010). Thus, chronic, repeated low-level exposures to DomA may indeed occur. Upon oral administration, DomA is poorly absorbed, has a short half-life in blood, and poorly permeates the blood-brain barrier (Preston and Hynie, 1991; Truelove and Iverson, 1994). These characteristics may limit access of low levels of DomA in the central nervous system (CNS). However, the situation may differ during development, a period of suggested particular sensitivity to DomA neurotoxicity (Costa et al., 2010). In pregnant rats, a study by Maucher and Ramsdell (2007) indicated concentrations of DomA in blood in the high nanomolar range, lower levels in amniotic fluid, and comparable levels (low-mid nM) in the brain of the dam and the fetuses. More recent studies by the same authors (Maucher Fuquay et al., 2012a, b) have also shown that levels of DomA in amniotic fluid and brain remain elevated for longer periods, indicating a different kinetic from plasma.
The aim of this study was to determine whether a prolonged exposure to low levels of DomA would result in neurotoxicity and/or in altered susceptibility to a subsequent DomA exposure in cultured mouse CGNs. Neurons from wild-type mice and from Gclm ˗/˗ mice, already known to be highly susceptible to DomA neurotoxicity (Giordano et al., 2006, 2007), were compared for this purpose.
MATERIALS AND METHODS
Materials.
Neurobasal-A, Hibernate A, and B27 Minus AO media, Hank’s balanced salt solution, GlutaMax, Superscript reverse transcriptase III, dispase, fetal bovine serum, and gentamycin were from Invitrogen (Carlsbad, CA). DomA, poly-D-lysine, horseradish peroxidase–conjugated anti-mouse IgG, horseradish peroxidase–conjugated anti-rabbit IgG, dimethylsulfoxide (DMSO), 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), hydrogen peroxide (H2O2), and 2,3-dimethoxy-1,4-naphtoquinone (DMNQ) were purchased from Sigma Chemical Co. (St Louis, MO). Mouse anti-β-actin antibody was from ABCAM (Cambridge, MA). Hoechst DNA binding dye and 2,7′-dichlorofluorescin diacetate (DCF-DA) were from Molecular Probes (Eugene, OR). Monobromobimane (MBB) was from EMD Biosciences, Inc. (San Diego, CA). The bicinchoninic acid assay for protein determination was from Pierce (Rockford, IL). Tissue culture plastic ware was from Corning Costar (Cambridge, MA).
Generation of Gclm-null mice and genotyping.
All procedures for animal use were in accordance with the National Institute of Health Guide for the Use and Care of Laboratory Animals and were approved by the University of Washington Animal Care and Use Committee. Gclm null (Gclm ˗/˗) mice were derived by homologous recombination techniques in mouse embryonic stem cells, as previously described in detail by Giordano et al. (2006) and McConnachie et al. (2007). Pups were genotyped as described by Giordano et al. (2006).
Cultures of CGNs and cell treatments.
CGNs were chosen for these studies as these cells have been extensively utilized for in vitro studies on the neurotoxicity of DomA (Berman and Murray, 1997; Berman et al., 2002; Giordano et al., 2006, 2007; Novelli et al., 1992). CGN cultures were prepared from 7-day-old mice, as described by Giordano et al. (2006, 2007). Neurons were grown for 10–12 days before treatments. All compounds were dissolved in Locke’s solution. For prolonged low-dose exposure, DomA (5nM) was added on day 1, and cultures were left untouched for 10 days. For acute intermediate-dose exposure, naïve or DomA-pretreated CGNs were incubated with DomA (100nM) for 1h in Locke’s buffer, followed by washout, and an additional 23-h incubation (Giordano et al., 2007).
Assessment of cytotoxicity.
Cell viability was quantified by a colorimetric method utilizing the metabolic dye MTT (Giordano et al., 2006).
Measurement of apoptosis.
To visualize nuclear morphology following DomA treatment, cells were fixed with methanol and stained in 10 μg/ml Hoechst DNA binding dye for 15min. All cells exhibiting either classical apoptotic bodies or shrunken, rounded nuclei were scored as being apoptotic. Five fields placed across the diameter of each well were assessed using a 20× objective on an inverted fluorescence microscope (Giordano et al., 2007).
Assay of ROS formation.
ROS formation was determined by fluorescence using DCF-DA, as previously described (Giordano et al., 2006, 2007). Upon entering cells, DCF-DA is de-esterified to DCFH, which is then oxidized by ROS to form the fluorescent DCF. In a typical experiment, cells were first washed with Locke’s solution and then preincubated for 30min (37ºC) with DCF-DA (50 nmol/mg cell protein) in Locke’s solution. DCF-DA was added from a stock solution in DMSO; the quantity of DMSO never exceeded 0.1% and was also added to the blank. Cells were then washed with Locke’ solution to remove extracellular DCF-DA, and fluorescence was immediately read using a fluorescence microplate reader (excitation 488nm, emission 538nm).
Immunoblotting analysis.
Neurons were scraped in lysis buffer (Tris 50mM, pH 7.5, 2mM EDTA, 0.5mM dithiothreitol, 0.5mM PMSF, 10 μg/ml leupeptin, and 2 μg/ml aprotinin, 1mM sodium orthovanadate, 1mM NaF, 1% Triton, and 0.1% SDS), and whole homogenates were subjected to SDS-PAGE and immunoblotting using rabbit anti-GCLC (1:2000), anti-GCLM (1:1000), or mouse anti-β-actin (1:2000) antibodies. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes and incubated with the above antibodies. Membranes were rinsed in Tris-buffered saline Tween and incubated with horseradish peroxidase–conjugated anti-rabbit IgG or with horseradish peroxidase–conjugated anti-mouse IgG (for actin) at the appropriate dilutions (1:2000, 1:5000, respectively).
Measurement of GSH levels.
CGNs were homogenized in Locke’s buffer, and an aliquot was taken to measure the protein concentration, whereas a second aliquot was diluted (1:1) in 10% 5-sulfosalicylic acid (SSA) for the measurement of GSH (Giordano et al., 2006). The SSA fraction was centrifuged for 5min at 4°C, and the supernatant was used for GSH determinations. Aliquots from the SSA fraction were added to a black flat-bottomed 96-well plate, and pH was adjusted to 7 with 0.2M N-ethylmorpholine/0.02M KOH. Oxidized GSH was reduced by adding 10 µl of 10mM tris (2-carboxyethyl)-phosphine hydrochloride (TCEP) for 15min at room temperature. The pH was then adjusted to 12.5 by using 0.5N NaOH before derivatizing the samples with 10mM naphthalene dicarboxaldehyde for 30min. Finally, the samples were analyzed on a spectrofluorometric plate reader (λEX = 472 and λEM = 528nm). After incubation, the total amount of GSH in the sample was expressed as nanomoles per milligram of protein.
Measurement of GCL activity.
GCL activity was assayed by a high-performance liquid chromatography (HPLC)–based method, as previously described (White et al., 1999). CGNs were scraped and sonicated on ice in TES buffer (20mM Tris, 1mM EDTA, 250mM sucrose, pH 7.4) containing 20mM serine, 1mM boric acid, and peptidase inhibitors. Protein levels were measured in the supernatant by the bis-cinchoninic acid method (Pierce). Samples were dissolved in Tris/EDTA buffer containing 21.3mM L-glutamate, 10.67mM ATP, 10.7mM MgCl2, and 10mM cysteine at 37°C. The reaction was stopped by ice-cold SSA solution, and baseline GSH levels were determined. Precipitated proteins were removed, and the supernatant was analyzed after MBB derivatization by HPLC. Gamma-glutamylcysteine (γ-GC) and GSH peaks were identified in the chromatographs; GCL activity level was normalized to protein concentrations and expressed as nanomoles of γ-GC formed per milligram protein per minute.
Measurements of GCLC and GCLM levels and of GCL holoenzyme.
GCLC and GCLM, as well as GCL holoenzyme (GCLholo), were measured by Western blot as described by Lee et al. (2006). Standard curves were generated by loading incremental amounts of purified mouse GCLC and GCLM (kindly donated by Dr Christopher C. Franklin) on to each gel. Aliquots of supernatant containing 40 μg of total soluble proteins were separated by one-dimensional SDS/PAGE using 10% (wt/vol) gels. SDS/PAGE was run under nonreducing conditions without adding DTT or heat before loading. The presence of complex was detected adding increasing amounts of GCLM to GCLC and followed the formation of holoenzyme by nondenaturing PAGE. Upon the addition of GCLM to GCLC, a more slowly migrating complex was formed (data not shown), representing GCLholo; antibodies to either GCLM or GCLC recognized this complex and confirmed its identity.
Measurement of Gclc and Gclm mRNA levels.
Levels of mRNA of Gclc and Gclm were determined by quantitative RT-PCR, using a fluorogenic 5′ nuclease-based assay developed by the Functional Genomics Laboratory at the University of Washington. Briefly, reverse transcription was performed according to the manufacturer’s established protocol using total RNA and the SuperScript III First-Strand Synthesis System (Invitrogen). For gene expression measurements, 4 µl of cDNA was included in a PCR reaction (25 µl final volume) that also consisted of the appropriate forward and reverse primers at 360nM each, 80nM TaqMan probe, and TaqMan Gene Expression Master Mix (Applied Biosystems Inc., Foster City, CA). The PCR primers and the dual-labeled probes (6-carboxy-fluorescein and 6-carboxy-tetramethyl-rhodamine) for all genes were designed using ABI Primer Express v.1.5 software (Applied Biosystems Inc.). Amplification and detection of PCR amplicons were performed with the ABI PRISM 7900 system (Applied Biosystems Inc.) with the following PCR reaction profile: 1 cycle of 95°C for 10min, 40 cycles of 95°C for 30 s and 62°C for 1min. Beta-actin amplification plots derived from serial dilutions of an established reference sample were used to create a linear regression formula in order to calculate expression levels, and beta-actin gene expression levels were utilized as an internal control to normalize the data.
Statistical analysis.
Data are expressed as the mean ± SD of at least three independent experiments. Statistical analysis was performed by one-way ANOVA followed by Bonferroni’s Multiple Comparison Test.
RESULTS
Prolonged exposure to a low level of DomA (5nM for 10 days) caused no overt neurotoxicity, as evidenced by similar percentage of apoptotic cells in control and treated CGNs cultures of wild-type and Gclm ˗/˗ mice (Fig. 1A). Acute intermediate-dose exposure to DomA (100nM for 1h, followed by washout and by additional 23-h incubation) caused significant apoptotic cell death in CGNs from wild-type mice (25%) and an expected greater apoptosis (40%) in CGNs from Gclm ˗/˗ mice (Fig. 1A). Pretreatment of neurons for 10 days with 5nM DomA significantly reduced the acute toxicity of DomA in wild-type mice (by ~50%) but not in CGNs from Gclm ˗/˗ mice (Fig. 1A). Figure 1B shows representative fields of CGNs from Gclm +/+ mice upon different treatments, evidencing apoptotic cells.
Fig. 1.
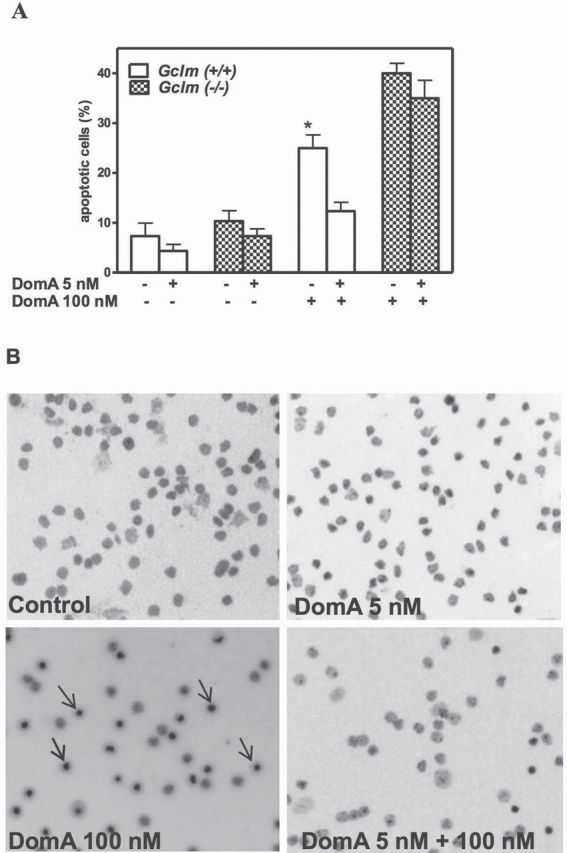
Prolonged exposure to low-level DomA protects CGNs of Gclm +/+ mice from acute DomA neurotoxicity. CGNs were prepared from Gclm +/+ and Gclm ˗/˗ mice. Forty-eight hours after seeding, 5nM DomA was added to the culture medium. After 10 days, cultures were challenged with 100nM DomA, and apoptosis was measured by Hoechst staining. (A) Summary of results (mean ± SD) of three separate experiments. Values that are significantly different from neurons pretreated with 5nM of DomA are indicated (*p < 0.05). (B) Pictures of apoptotic CGNs from Gclm +/+ mice under different experimental conditions.
DomA-induced neurotoxicity and apoptotic neuronal death have been previously reported to involve the induction of oxidative stress (Giordano et al., 2006, 2007). We, thus, measured oxidative stress induced by acute exposure to DomA (100nM) in CGNs of both genotypes without or with a pre-exposure to a low dose of DomA (5nM for 10 days). As shown in Figure 2A, acute DomA-induced ROS were significantly lower in CGNs from Gclm +/+ mice after chronic low-dose exposure to DomA. In contrast, oxidative stress induced by acute DomA in CGNs from Gclm ˗/˗ mice, which was higher than in wild-type neurons, was not altered by prior DomA exposure (Fig. 2B).
Fig. 2.
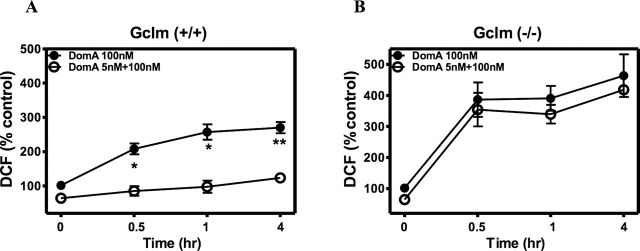
Prolonged exposure to low-level DomA prevents the increase in ROS induced by acute intermediate-dose DomA in CGNs from Gclm +/+ mice. (A) Time course of ROS production induced by acute DomA (100nM) in CGNs from Gclm +/+ control mice and after 10 days of exposure to 5nM DomA. (B) Time course of ROS production in CGNs from Gclm ˗/˗ mice, treated as in (A). Results are expressed as the mean (± SD) of three separate experiments. Values that are significantly different from neurons pretreated with 5nM DomA are indicated (*p < 0.05; **p < 0.01).
As GSH has been shown to be a major determinant of neuronal susceptibility to DomA-induced apoptosis (Giordano et al., 2007), we measured GSH levels in CGNs following chronic DomA exposure. As expected, GSH levels in CGNs from wild-type mice were significantly higher than those present in CNS from Gclm ˗/˗ mice (Fig. 3). Indeed, in the absence of GCLM, the efficiency of the catalytic subunit GCLC is greatly diminished (Giordano et al., 2006). After 10 days of exposure to 5nM DomA, GSH levels were significantly increased in CGNs from wild-type mice (by ~75%) but not in neurons from Gclm ˗/˗ mice (Fig. 3).
Fig. 3.
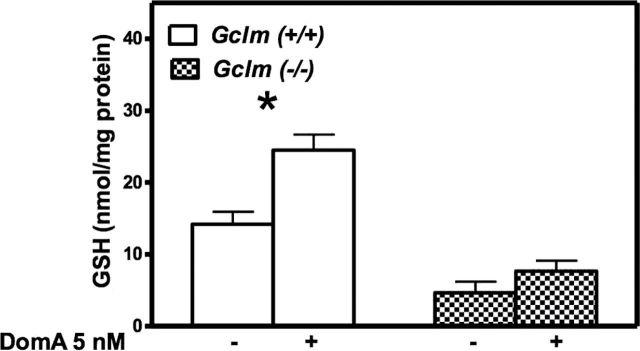
Prolonged exposure to low-level DomA increases intracellular GSH level in CGNs from Gclm +/+ mice. Results represent the mean (± SD) of three separate experiments. Values that are significantly different from the respective control are indicated (*p < 0.05).
Measurements of levels of GCLC protein in control CGNs indicated that they were higher in Gclm ˗/˗ mice than in Gclm +/+ mice (Fig. 4A). This confirms previous findings (Giordano et al., 2006) and may represent an attempt to compensate for the lack of GCLM. Chronic exposure to low-level DomA increased GCLC levels in CGNs of both mouse genotypes (Figs. 4A and C). GCLM was obviously absent in CGNs from Gclm ˗/˗ mice (Fig. 4B). In neurons from wild-type mice, prolonged exposure to 5nM DomA also increased the levels of GCLM protein (Figs. 4B and C). The observed effects of chronic DomA exposure on the two GCL subunits were likely due to enhanced transcription, as levels of mRNA of Gclc and Gclm were altered by chronic low-dose DomA exposure, similar to what was observed for GCLC and GCLM proteins (Figs. 4D and E).
Fig. 4.
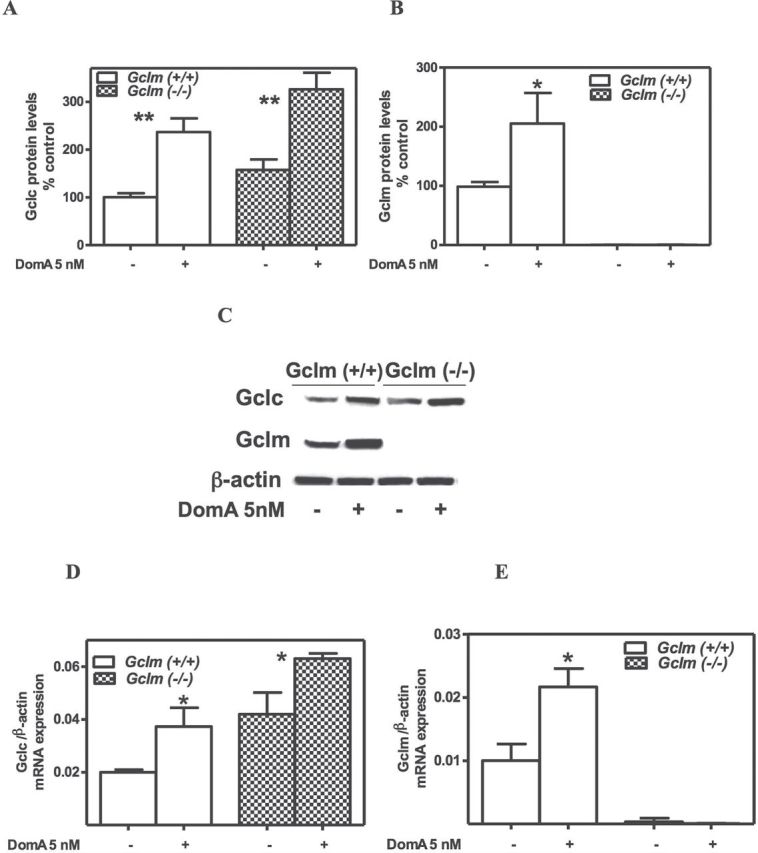
Prolonged exposure to low-level DomA increases GCLC and GCLM protein and mRNA levels in CGNs from Gclm +/+ mice. Shown are levels of GCLC (A) and GCLM (B) protein levels, as determined by Western blot after normalization with β-actin. Results represent the mean (± SD) of three separate experiments. In (C), a representative blot is shown. Levels of mRNA for Gclc (D) and Gclm (E), as determined by quantitative RT-PCR are shown, after normalization with β-actin. Values represent the mean (± SD) of three separate determinations. Values that are significantly different from control are indicated (*p < 0.05; **p< 0.01).
In CGNs from Gclm +/+ mice, the increase in GCLC and GCLM (Fig. 4) due to chronic low-level DomA should also result in an increase in GCL activity, which would be responsible for the increased synthesis of GSH (Fig. 3). This is indeed the case, as shown in Fig. 5A. In contrast, GCL activity was not increased in CGNs from Gclm ˗/˗ mice (Fig. 5A). This was not unexpected as GCL activity has been shown to be regulated not only by changes in the amount of GCLC but also mostly by changes in the k cat for the enzyme due to formation of the holoenzyme (GCLholo) between GCLC and GCLM (Chen et al., 2005; Lee et al., 2006). Indeed, the catalytic efficiency of GCLholo is more than fourfold higher than that of GCLC (Chen et al., 2005). Figure 5B shows that levels of GCLholo were increased in neurons from wild-type mice; as expected, GCLholo was not present in CGNs from Gclm ˗/˗ mice (Fig. 5B).
Fig. 5.

Prolonged exposure to low-level DomA increases GCL activity and GCLholo formation in CGNs from Gclm +/+ mice. (A) Activity of GCL measured after 10 days of exposure to 5nM DomA. Results represent the mean (± SD) of three separate experiments. Significantly different from control, *p < 0.05. (B) Representative blot of GCL holoenzyme in CGNs from both mouse genotypes after 10 days of exposure to 5nM DomA. Shown is the shift of GCLC from monomer to the holoenzyme form. The experiment was repeated thrice with similar results.
In addition to protecting neurons of wild-type CGNs from the acute toxicity of DomA, the increase in GSH induced by chronic low-dose DomA exposure also provided protection against other oxidative stressors. As shown in Table 1, the toxicity of H2O2 and DMNQ was significantly lower (by ~threefold) in CGNs from wild-type mice previously exposed to DomA (5nM for 10 days) than in control CGNs. In contrast, in CGNs from Gclm ˗/˗ mice, already highly susceptible to oxidant toxicity because of low GSH levels (Table 1), chronic DomA exposure did not provide any protection against these agents.
Table 1 .
Prolonged Exposure to Low-Level DomA Protects CGNs From Gclm +/+ Mice From Toxicity of Oxidants
| Compound | Control | Chronic DomA | Ratio |
|---|---|---|---|
| CGNs from Gclm +/+ mice | |||
| H2O2 | 6.1±0.5 | 19.0±1.3* | 3.1 |
| DMNQ | 9.4±1.8 | 24.7±1.5** | 2.6 |
| CGNs from Gclm ˗/˗ mice | |||
| H2O2 | 1.5±0.2 | 2.2±0.3 | 1.5 |
| DMNQ | 1.8±0.5 | 2.4±0.4 | 1.3 |
Notes. CGNs from Gclm +/+ or Gclm ˗/˗ mice were treated for 10 days with 5nM DomA. After washout, cells were treated for 24h with different concentrations of H2O2 or DMNQ. Cell viability was measured by the MTT assay as described in Materials and Methods section. Results indicate values of IC50 (µM), determined from concentration-response curves using 3–4 concentrations of each compound in duplicate, and represent the mean (± SD) of three separate experiments.
Significantly different from control, *p < 0.05; **p < 0.01.
DISCUSSION
Results of this study show that prolonged exposure to low-level DomA in vitro provides protection against toxicity of a higher acute exposure of the same neurotoxin and other toxicants. The underlying mechanism appears to be related to the ability of chronic low-dose DomA to upregulate GSH synthesis though additional mechanisms cannot be ruled out. Genetic conditions leading to low GSH levels, which already increase susceptibility to DomA neurotoxicity, could potentially negate such protective mechanisms.
DomA is a potent human and animal neurotoxin, and its mechanisms of neurotoxicity at the cellular level have been characterized (Berman and Murray, 1997; Berman et al., 2002; Giordano et al., 2006, 2007; Liu et al., 2001; Novelli et al., 1992; Puttfarcken et al., 1993). At relatively high concentrations (> 1µM), DomA induces neuronal cell death which is primarily necrotic in nature. By activating AMPA/kainate receptors, DomA causes an increase in [Ca2+]i, which then results in a release of L-glutamate. This released glutamate in turn activates NMDA receptors and promotes further glutamate release. This combined action causes a rapid accumulation of [Ca2+]i, promotes GSH efflux, and causes a concomitant decrease in intracellular GSH. High [Ca2+]i and low GSH lead to production of ROS, which, because of the insufficient GSH for scavenging, increases lipid peroxidation and leads to neuronal cell death (Giordano et al., 2006). In contrast, at lower concentrations (100nM or less), DomA causes primarily apoptotic cell death of neurons (Giordano et al. 2007). Apoptosis is only due to activation of AMPA/kainate receptors as these lower concentrations of DomA do not stimulate L-glutamate release (Giordano et al., 2007). A small and transient increase in [Ca2+]i is followed by sequestration of calcium in the mitochondria, which leads to a loss of mitochondrial membrane potential, an increase in mitochondrial oxidative stress, and the opening of the permeability transition pore. This is then followed by the release of cytochrome c, activation of caspase-3, and degradation of poly (ADP-ribose) polymerase (Giordano et al., 2007). Both DomA-induced necrosis and apoptosis, thus, involve oxidative stress, are inhibited by antioxidants, and are more pronounced in GCNs from Gclm ˗/˗ mice, which have very low GSH levels (Giordano et al., 2006, 2007).
The first important finding of this study is that prolonged exposure of mouse CGNs to low-level DomA (5nM applied on day 1 and left for 10 days) protects neurons from a subsequent insult by a high concentration of DomA itself and by other oxidants as well. The mechanism of such protection appears to be related to the ability of chronic low DomA to increase the levels of the two subunits of GCL (GCLC and GCLM) and those of GCLholo, leading to increased GCL activity and GSH synthesis. An alternative mechanism would be represented by downregulation of AMPA/kainate receptors or of high-affinity GTPase activity (Hesp et al., 2004). However, these possibilities appear unlikely as they would be expected to occur in CGNs from both Gclm +/+ and Gclm ˗/˗ mice, where instead different results were obtained (Fig. 1).
The regulation of GCL has been the subject of several investigations (Bea et al., 2003; Dasgupta et al., 2007; Franklin et al., 2002; Huang et al., 2000; Hudson and Kavanagh, 2000; Iles and Liu, 2005; Krejsa et al., 2010; Krzywanski et al., 2004; Moellering et al., 1999; Moinova and Mulcahy, 1999; Mulcahy and Gipp, 1995; Mulcahy et al., 1997; Soltaninassab et al., 2000; Thompson et al., 2009; Toroser et al., 2006; Wild et al., 1999). Increases in GCL activity are often due to a transcriptional mechanism, involving an increased level of Gclc, and in some cases of Gclm mRNAs. Transcription of GCL subunits occurs through the sequence-specific binding of NF-E2-related factor to antioxidant response elements present in the promoters of these two genes (Bea et al., 2003; Huang et al., 2000; Hudson and Kavanagh, 2000; Moinova and Mulcahy, 1999; Mulcahy and Gipp, 1995; Mulcahy et al., 1997; Wild et al., 1999; Yang et al., 2001; Zipper and Mulcahy, 2000). GCL activity has been shown to be regulated both by changes in the amount of GCLC and by changes in the k cat for the enzyme due to formation of the holoenzyme (GCLholo) between GCLC and GCLM (Chen et al., 2005; Lee et al., 2006). The catalytic efficiency of GCLholo is more than fourfold higher than that of GCLC (Chen et al., 2005), providing an explanation for the low activity of GCL in Gclm ˗/˗ mice, despite an upregulation of GCLC (Giordano et al., 2006; Yang et al., 2002).
The effect observed with DomA in wild-type CGNs resembles the phenomenon of preconditioning, which is considered part of hormesis and has been defined as the situation in which “exposure to a low dose of an agent that is toxic at high doses induces an adaptive, potentially beneficial effect on the cell or organism if exposed to a subsequent more massive dose of the same or related stressor agent” (Calabrese et al., 2007). Low levels of DomA may thus elicit a mild degree of oxidative stress, particularly in mitochondria (Giordano et al., 2007), which would lead to increased transcription of Gclc and Gclm, ultimately leading to increased GSH synthesis and neuroprotection. This mild oxidative stress may also favor the formation of GCLholo (Krejsa et al., 2010), which would also be expected to increase GCL activity and GSH synthesis.
As shown in this study, the protective effect of prolonged low-level DomA is not apparent in CGNs from Gclm ˗/˗ mice. Gclm ˗/˗ mice lack GCLM, the modifier subunit of GCL, the first and rate-limiting enzyme in the synthesis of GSH (Yang et al., 2002). In the absence of GCLM, the ability of the catalytic subunit GCLC to synthesize GSH is drastically reduced (Dalton et al., 2004; Giordano et al., 2006; McConnachie et al., 2007). Indeed, in CGNs from Gclm ˗/˗ mice, GCLM is absent, and GSH levels are only 20% of those present in CGNs from wild-type animals, despite an upregulation of GCLC (Giordano et al., 2006). Chronic exposure to low-dose DomA, which by itself does not elicit any toxicity, upregulates GCLC (Fig. 4). However, in the absence of GCLM, this increase is not sufficient to increase GSH synthesis.
The Gclm ˗/˗ mouse used in this study may represent a model for a relatively common polymorphism (C588T) in the 5′-flanking region of the GCLM gene, which has been associated with low plasma levels of GSH (Nakamura et al., 2002). If individuals with the T allele display lower levels of GSH also in the central nervous system, they may be more susceptible to the neurotoxicity of DomA, as well as to other excitatory amino acids, including glutamate itself, that activate AMPA/kainate and NMDA receptors. The present findings suggest that these individuals may also be unable to mount a hormetic response, i.e., to “build up” protection upon chronic exposure. We had previously shown that a mechanism for counteracting DomA neurotoxicity, i.e., inhibition of apoptosis by increased signalling of endogenous acetylcholine-activated M3 muscarinic receptors, was also only partially effective in CGNs from Gclm ˗/˗ mice (Giordano et al., 2009). Thus, individuals with genetic polymorphisms of Gclm (1) may be more susceptible to the acute neurotoxic effects of DomA (Giordano et al., 2006, 2007), (2) may be unable to provide protection through other antiapoptotic pathways (e.g., acetylcholine and muscarinic receptors; Giordano et al., 2009), and (3) may also be unable to build up protection upon chronic exposure to DomA (this study). Of much interest would also be an investigation of the effects of low DomA exposure in CGNs from Gclm +/˗ mice. These cells have GSH levels only slightly lower than wild-type animals (Giordano et al., 2006) but may be less capable of compensatory reactions upon repeated exposures to oxidative stress, as shown in vivo in Gclm heterozygote mice (Weldy et al., 2011).
The findings of this study may be potentially relevant in “real-life” situations, particularly during development. Due to the rapid serum clearance and elimination of DomA in humans, no information is available on potential blood levels upon exposure in adults (Costa et al., 2010). Similarly, in nonhuman primates and rodents, DomA has been shown to have a very short half-life (Truelove and Iverson, 1994). In pregnant rats, a study by Maucher and Ramsdell (2007) indicated concentrations of DomA in blood in the high nanomolar range, lower levels in amniotic fluid, and comparable levels (low-mid nM) in the brain of the dam and the fetuses. Moreover, recent studies by the same authors (Maucher Fuquay et al., 2012a, b) have shown that levels of DomA in brain and amniotic fluid remain elevated for longer periods, indicating a different kinetic from plasma. Thus, the present findings may be most relevant in humans when exposure occurs in utero; indeed, it has been shown that women of child-bearing age are often exposed to DomA levels that approximate or exceed the recommended safety limits (Costa et al., 2010; Tsuchiya et al., 2008).
The dose-response curve for DomA in animals and humans is very steep. For example, in humans a dose of 0.3mg/kg is asymptomatic, whereas exposure to 2mg/kg results in frank neurotoxicity (Todd, 1993). As said earlier, the proposed TDI levels range from 0.018 to 0.1mg/kg/day, and these values are often exceeded in populations consuming DomA-contaminated foods at the current limit (20 ppm) (Costa et al., 2010; EFSA, 2009; Tsuchiya et al., 2008). Thus, the low nanomolar levels of repeated exposure to DomA used in this study are within realistic exposure scenarios, and individuals with genetic polymorphisms of GCLM may be particularly at risk for the neurotoxic effects of DomA.
FUNDING
National Science Foundation and the National Institute for Environmental Health Sciences (ES012762/NSF-OCE-0434087, NSF-OCE-0910624, P30ES07033).
ACKNOWLEDGMENTS
We thank Dr Fred Farin and Jasmine (Hue-Wen) Wilkerson (Functional Genomics Laboratory, University of Washington) for measurements of Gclm and Gclc mRNA levels and Dr Christopher C. Franklin (University of Colorado at Denver) for kindly donating purified GCLC and GCLM.
REFERENCES
- Bea F., Hudson F. N., Chait A., Kavanagh T. J., Rosenfeld M. E. (2003). Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ. Res. 92, 386–393 [DOI] [PubMed] [Google Scholar]
- Berman F. W., LePage K. T., Murray T. F. (2002). Domoic acid neurotoxicity in cultured cerebellar granule neurons is controlled preferentially by the NMDA receptor Ca(2+) influx pathway. Brain Res. 924, 20–29 [DOI] [PubMed] [Google Scholar]
- Berman F. W., Murray T. F. (1997). Domoic acid neurotoxicity in cultured cerebellar granule neurons is mediated predominantly by NMDA receptors that are activated as a consequence of excitatory amino acid release. J. Neurochem. 69, 693–703 [DOI] [PubMed] [Google Scholar]
- Calabrese E. J., Bachmann K. A., Bailer A. J., Bolger P. M., Borak J., Cai L., Cedergreen N., Cherian M. G., Chiueh C. C., Clarkson T. W, et al. (2007). Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 222, 122–128 [DOI] [PubMed] [Google Scholar]
- Chen Y., Shertzer H. G., Schneider S. N., Nebert D. W., Dalton T. P. (2005). Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 280, 33766–33774 [DOI] [PubMed] [Google Scholar]
- Clayton E. C., Peng Y. G., Means L. W., Ramsdell J. S. (1999). Working memory deficits induced by single but not repeated exposures to domoic acid. Toxicon 37, 1025–1039 [DOI] [PubMed] [Google Scholar]
- Costa L. G., Giordano G., Faustman E. M. (2010). Domoic acid as a developmental neurotoxin. Neurotoxicology 31, 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton T. P., Chen Y., Schneider S. N., Nebert D. W., Schertzer H. G. (2004). Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Rad. Biol. Med. 37, 1511–1526 [DOI] [PubMed] [Google Scholar]
- Dasgupta A., Das S., Sarkar P. K. (2007). Thyroid hormone promotes glutathione synthesis in astrocytes by up regulation of glutamate cysteine ligase through differential stimulation of its catalytic and modulator subunit mRNAs. Free Radic. Biol. Med. 42, 617–626 [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Agency) (2009). Marine biotoxins in shellfish-domoic acid. Scientific opinion of the panel on contaminants in the food chain. EFSA J. 1181, 1–61 [Google Scholar]
- Franklin C. C., Krejsa C. M., Pierce R. H., White C. C., Fausto N., Kavanagh T. J. (2002). Caspase-3-Dependent Cleavage of the Glutamate-L-Cysteine Ligase Catalytic Subunit during Apoptotic Cell Death. Am. J. Pathol. 160, 1887–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G., Klintworth H. M., Kavanagh T. J., Costa L. G. (2008). Apoptosis induced by domoic acid in mouse cerebellar granule neurons involves activation of p38 and JNK MAP kinases. Neurochem. Int. 52, 1100–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G., Li L., White C. C., Farin F. M., Wilkerson H. W., Kavanagh T. J., Costa L. G. (2009). Muscarinic receptors prevent oxidative stress-mediated apoptosis induced by domoic acid in mouse cerebellar granule cells. J. Neurochem. 109, 525–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G., White C. C., McConnachie L. A., Fernandez C., Kavanagh T. J., Costa L. G. (2006). Neurotoxicity of domoic Acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Mol. Pharmacol. 70, 2116–2126 [DOI] [PubMed] [Google Scholar]
- Giordano G., White C. C., Mohar I., Kavanagh T. J., Costa L. G. (2007). Glutathione levels modulate domoic acid induced apoptosis in mouse cerebellar granule cells. Toxicol. Sci. 100, 433–444 [DOI] [PubMed] [Google Scholar]
- Hampson D. R., Manalo J. L. (1998). The activation of glutamate receptors by kainic acid and domoic acid. Nat. Toxins 6, 153–158 [DOI] [PubMed] [Google Scholar]
- Hesp B. R., Wrightson T., Mullaney I., Kerr D. S. (2004). Kainate receptor agonists and antagonists mediate tolerance to kainic acid and reduce high-affinity GTPase activity in young, but not aged, rat hippocampus. J. Neurochem. 90, 70–79 [DOI] [PubMed] [Google Scholar]
- Huang Z. A., Yang H., Chen C., Zeng Z., Lu S. C. (2000). Inducers of gamma-glutamylcysteine synthetase and their effects on glutathione synthetase expression. Biochim. Biophys. Acta 1493, 48–55 [DOI] [PubMed] [Google Scholar]
- Hudson F. N., Kavanagh T. J. (2000). Cloning and characterization of the proximal promoter region of the mouse glutamate-L-cysteine ligase regulatory subunit gene. Biochim. Biophys. Acta 1492, 447–451 [DOI] [PubMed] [Google Scholar]
- Iles K. E., Liu R. M. (2005). Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic. Biol. Med. 38, 547–556 [DOI] [PubMed] [Google Scholar]
- Kerr D. S., Razak A., Crawford N. (2002). Age-related changes in tolerance to the marine algal excitotoxin domoic acid. Neuropharmacology 43, 357–366 [DOI] [PubMed] [Google Scholar]
- Krejsa C. M., Franklin C. C., White C. C., Ledbetter J. A., Schieven G. L., Kavanagh T. J. (2010). Rapid activation of glutamate cysteine ligase following oxidative stress. J. Biol. Chem. 285, 16116–16124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywanski D. M., Dickinson D. A., Iles K. E., Wigley A. F., Franklin C. C., Liu R. M., Kavanagh T. J., Forman H. J. (2004). Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch. Biochem. Biophys. 423, 116–125 [DOI] [PubMed] [Google Scholar]
- Lee J. I., Kang J., Stipanuk M. H. (2006). Differential regulation of glutamate-cysteine ligase subunit expression and increased holoenzyme formation in response to cysteine deprivation. Biochem. J. 393,(Pt 1), 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Liu R., Chun J. T., Bi R., Hoe W., Schreiber S. S., Baudry M. (2001). Kainate excitotoxicity in organotypic hippocampal slice cultures: Evidence for multiple apoptotic pathways. Brain Res. 916, 239–248 [DOI] [PubMed] [Google Scholar]
- Maucher J. M., Ramsdell J. S. (2007). Maternal-fetal transfer of domoic acid in rats at two gestational time points. Environ. Health Perspect. 115, 1743–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maucher Fuquay J., Muha N., Wang Z., Ramsdell J. S. (2012a). Toxicokinetics of domoic acid in the fetal rat. Toxicology 294, 36–41 [DOI] [PubMed] [Google Scholar]
- Maucher Fuquay J., Muha N., Wang Z., Ramsdell J. S. (2012b). Elimination kinetics of domoic Acid from the brain and cerebrospinal fluid of the pregnant rat. Chem. Res. Toxicol. 25, 2805–2809 [DOI] [PubMed] [Google Scholar]
- McConnachie L. A., Mohar I., Hudson F. N., Ware C. B., Ladiges W. C., Fernandez C., Chatterton-Kirchmeier S., White C. C., Pierce R. H., Kavanagh T. J. (2007). Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol. Sci. 99, 628–636 [DOI] [PubMed] [Google Scholar]
- Moellering D., Mc Andrew J., Patel R. P., Forman H. J., Mulcahy R. T., Jo H., Darley-Usmar V. M. (1999). The induction of GSH synthesis by nanomolar concentrations of NO in endothelial cells: A role for gamma-glutamylcysteine synthetase and gamma-glutamyl transpeptidase. FEBS Lett. 448, 292–296 [DOI] [PubMed] [Google Scholar]
- Moinova H. R., Mulcahy R. T. (1999). Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 261, 661–668 [DOI] [PubMed] [Google Scholar]
- Mulcahy R. T., Gipp J. J. (1995). Identification of a putative antioxidant response element in the 5’-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem. Biophys. Res. Commun. 209, 227–233 [DOI] [PubMed] [Google Scholar]
- Mulcahy R. T., Wartman M. A., Bailey H. H., Gipp J. J. (1997). Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J. Biol. Chem. 272, 7445–7454 [DOI] [PubMed] [Google Scholar]
- Nakamura S., Kugiyama K., Sugiyama S., Miyamoto S., Koide S., Fukushima H., Honda O., Yoshimura M., Ogawa H. (2002). Polymorphism in the 5’-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation 105, 2968–2973 [DOI] [PubMed] [Google Scholar]
- Novelli A., Kispert J., Fernández-Sánchez M. T., Torreblanca A., Zitko V. (1992). Domoic acid-containing toxic mussels produce neurotoxicity in neuronal cultures through a synergism between excitatory amino acids. Brain Res. 577, 41–48 [DOI] [PubMed] [Google Scholar]
- Peng Y. G., Clayton E. C., Means L. W., Ramsdell J. S. (1997). Repeated independent exposures to domoic acid do not enhance symptomatic toxicity in outbred or seizure-sensitive inbred mice. Fundam. Appl. Toxicol. 40, 63–67 [DOI] [PubMed] [Google Scholar]
- Perl T. M., Bédard L., Kosatsky T., Hockin J. C., Todd E. C., Remis R. S. (1990). An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N. Engl. J. Med. 322, 1775–1780 [DOI] [PubMed] [Google Scholar]
- Preston E., Hynie I. (1991). Transfer constants for blood-brain barrier permeation of the neuroexcitatory shellfish toxin, domoic acid. Can. J. Neurol. Sci. 18, 39–44 [DOI] [PubMed] [Google Scholar]
- Pulido O. M. (2008). Domoic acid toxicologic pathology: A review. Mar. Drugs 6, 180–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttfarcken P. S., Getz R. L., Coyle J. T. (1993). Kainic acid-induced lipid peroxidation: Protection with butylated hydroxytoluene and U78517F in primary cultures of cerebellar granule cells. Brain Res. 624, 223–232 [DOI] [PubMed] [Google Scholar]
- Qiu S., Pak C. W., Currás-Collazo M. C. (2006). Sequential involvement of distinct glutamate receptors in domoic acid-induced neurotoxicity in rat mixed cortical cultures: Effect of multiple dose/duration paradigms, chronological age, and repeated exposure. Toxicol. Sci. 89, 243–256 [DOI] [PubMed] [Google Scholar]
- Soltaninassab S. R., Sekhar K. R., Meredith M. J., Freeman M. L. (2000). Multi-faceted regulation of gamma-glutamylcysteine synthetase. J. Cell. Physiol. 182, 163–170 [DOI] [PubMed] [Google Scholar]
- Teitelbaum J. S., Zatorre R. J., Carpenter S., Gendron D., Evans A. C., Gjedde A., Cashman N. R. (1990). Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N. Engl. J. Med. 322, 1781–1787 [DOI] [PubMed] [Google Scholar]
- Thompson J. A., White C. C., Cox D. P., Chan J. Y., Kavanagh T. J., Fausto N., Franklin C. C. (2009). Distinct Nrf1/2-independent mechanisms mediate As 3+-induced glutamate-cysteine ligase subunit gene expression in murine hepatocytes. Free Radic. Biol. Med. 46, 1614–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd E. C. D. (1993). Domoic acid and amnesic shellfish poisoning-a review. J. Food Protect. 56, 69–83 [DOI] [PubMed] [Google Scholar]
- Toroser D., Yarian C. S., Orr W. C., Sohal R. S. (2006). Mechanisms of gamma-glutamylcysteine ligase regulation. Biochim. Biophys. Acta 1760, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truelove J., Iverson F. (1994). Serum domoic acid clearance and clinical observations in the cynomolgus monkey and Sprague-Dawley rat following a single i.v. dose. Bull. Environ. Contam. Toxicol. 52, 479–486 [DOI] [PubMed] [Google Scholar]
- Truelove J., Mueller R., Pulido O., Iverson F. (1996). Subchronic toxicity study of domoic acid in the rat. Food Chem. Toxicol. 34, 525–529 [DOI] [PubMed] [Google Scholar]
- Truelove J., Mueller R., Pulido O., Martin L., Fernie S., Iverson F. (1997). 30-day oral toxicity study of domoic acid in cynomolgus monkeys: Lack of overt toxicity at doses approaching the acute toxic dose. Nat. Toxins 5, 111–114 [DOI] [PubMed] [Google Scholar]
- Tsuchiya A., Hardy J., Burbacher T. M., Faustman E. M., Mariën K. (2008). Fish intake guidelines: Incorporating n-3 fatty acid intake and contaminant exposure in the Korean and Japanese communities. Am. J. Clin. Nutr. 87, 1867–1875 [DOI] [PubMed] [Google Scholar]
- Weldy C. S., White C. C., Wilkerson H. W., Larson T. V., Stewart J. A., Gill S. E., Parks W. C., Kavanagh T. J. (2011). Heterozygosity in the glutathione synthesis gene Gclm increases sensitivity to diesel exhaust particulate induced lung inflammation in mice. Inhal. Toxicol. 23, 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C. C., Krejsa C. M., Eaton D. L., Kavanagh T. J. (1999). HPLC-based assay for enzyme of glutathione biosynthesis. Curr. Protoc. Toxicol. 6.5.1–6.5.14 [DOI] [PubMed] [Google Scholar]
- Wild A. C., Moinova H. R., Mulcahy R. T. (1999). Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 274, 33627–33636 [DOI] [PubMed] [Google Scholar]
- Yang H., Wang J., Huang Z. Z., Ou X., Lu S. C. (2001). Cloning and characterization of the 5’-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem. J. 357(Pt 2),447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Dieter M. Z., Chen Y., Shertzer H. G., Nebert D. W., Dalton T. P. (2002). Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 277, 49446–49452 [DOI] [PubMed] [Google Scholar]
- Zipper L. M., Mulcahy R. T. (2000). Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem. Biophys. Res. Commun. 278, 484–492 [DOI] [PubMed] [Google Scholar]