

 Using manually curated signalling models of the IIS/TOR pathways, transcriptional feedback is shown to modulate ageing in worms and flies.
Using manually curated signalling models of the IIS/TOR pathways, transcriptional feedback is shown to modulate ageing in worms and flies.
Abstract
Several components have been previously identified, that modulate longevity in several species, including the target of rapamycin (TOR) and the Insulin/IGF-1 (IIS) signalling pathways. In order to infer paths and transcriptional feedback loops that are likely to modulate ageing, we manually built a comprehensive and computationally efficient signalling network model of the IIS and TOR pathways in worms. The core insulin transduction is signalling from the sole insulin receptor daf-2 to ultimately inhibit the translocation of the transcription factor daf-16 into the nucleus. Reduction in this core signalling is thought to increase longevity in several species. In addition to this core insulin signalling, we have also recorded in our worm model the transcription factors skn-1 and hif-1, those are also thought to modulate ageing in a daf-16 independent manner. Several paths that are likely to modulate ageing were inferred via a web-based service NetEffects, by utilising perturbed components (rheb-1, let-363, aak-2, daf-2;daf-16 and InR;foxo in worms and flies respectively) from freely available gene expression microarrays. These included “routes” from TOR pathway to transcription factors daf-16, skn-1, hif-1 and daf-16 independent paths via skn-1/hif-1. Paths that could be tested by experimental hypotheses, with respect to relative contribution to longevity, are also discussed. Direct comparison of the IIS and TOR pathways in both worm and fly suggest a remarkable similarity. While similarities in the paths that could modulate ageing in both organisms were noted, differences are also discussed. This approach can also be extended to other pathways and processes.
1. Introduction
The ageing process, defined as a decrease in the ability of organisms to respond to environmental stimuli, stress, decline in physiological functions and inevitable death, is a pliable biological process. Interventions, such as dietary restriction (DR, reduction in calorie intake without malnutrition), have been previously shown to robustly extend lifespan in a range of species, from yeast to mammals and possibly primates.1 Inevitably, there is a general interest in interventions and mechanisms that underlie or affect longevity in humans, despite the restrictions on experimental interventions or genetic manipulations. Because of long life expectancy, the use of primates as a proxy to humans, to investigate potential mechanisms of ageing is a lengthy process.1 However, some of the mechanisms are conserved over large evolutionary distances2 and therefore the use of short-lived model organisms is a fruitful and convenient choice.
Several components and biological pathways have been identified that modulate the ageing process, including nutrient sensing pathways, the target of rapamycin (TOR)3,4 and the insulin/insulin-like growth factor (IIS) signalling.5 Moreover, the first pathway discovered which can regulate ageing is the IIS pathway in the roundworm Caenorhabditis elegans (reviewed in Kenyon et al. 20116). Following these observations, genetic variants in human orthologues of several worm IIS components, among others the forkhead box class O transcription factor (FOXO, daf-16 in worms), have been found to be associated with exceptional longevity in genome-wide association studies in humans (for a list of genes and studies see Kenyon et al. 20116).
One of the most widely used techniques to study the role of component proteins in a biological pathway in model organisms is the overexpression or knockout of specific genes, followed by experimental determination of the effects that such perturbations cause. Numerous perturbations in the IIS signalling pathway have been reported to affect lifespan in several species. In worms and flies these include, along with many others, the insulin receptor InR 7/daf-2,8,9 chico,10 skn-1,11 hif-1,11 Pi3K/age-1,12 foxo/daf-16,13,14 pha-4 15 and several components of the TOR pathway.16 The insulin signalling pathway is a neuroendocrine pathway that among other functions monitors nutrients on the whole organism level.17 Similarly, the TOR pathway is also a nutrient-sensing pathway, although it monitors the levels of intracellular nutrients.
The basic observation in multiple organisms is that a reduction in insulin signalling activity (either by interfering with components of the pathway or partly by DR) extends lifespan. This process is thought to proceed by increasing the translocation of the FOXO transcription factor (daf-16 and foxo in worms and flies) into the nucleus. In turn, FOXO modulates the expression of a multitude of genes18 and the side effect is extended lifespan. So, predicting the effects of gene interference is very complex.
Following several lines of evidence, multiple signals that affect the nutrient sensing properties within the IIS and TOR pathways modulate ageing. In this paper we have made an attempt to capture the current knowledge of the IIS and TOR pathways in worm and how their modulation affects longevity, in data that we have collected in a consistent manner. This gave the ability to programmatically explore and differentiate between potential signalling “routes” within the IIS and TOR pathways that ultimately lead to changes in longevity. We utilised publicly available genome-wide gene expression microarray experiments that perturb genes within the IIS and TOR pathways. We also made inferences about paths, including transcriptional feedback loops, which support or contradict the observed ageing phenotype (short and long-lived) in these experiments. Furthermore, we have attempted a direct comparison between the effects of perturbation of orthologous genes in the worm and fly with respect to longevity phenotype recorded in both organisms. These experiments comprised daf-2;daf-16, rheb-1, let-363, aak-2 in worms and InR;foxo in Drosophila. Using our model and these publicly available microarray studies we are able to draw inferences with respect to several functionally important paths within the IIS and TOR pathways. These included paths to longevity via skn-1, daf-16/foxo and hif-1.
2. Materials and methods
2.1. A signalling network model of the insulin and TOR pathways in C. elegans
In order to explore “routes” or paths within the IIS and TOR pathways that could potentially modulate longevity, a comprehensive knowledge of the genes part of the two pathways and their connectivity was required. Such knowledge, not only needed to be organised in a computationally efficient way of representing the different components, but also had to be visually portrayed in a manner that humans can understand. Thus, we built a signalling network model of the IIS and TOR pathways by using GraphML, a modified version of the Extensible Markup Language (XML), supported in the program yEd (http://www.yworks.com). These allowed the required formal interpretation and at the same time a graphical representation of the pathways.
Building the model, i.e. genes and their interactions/connections, of the two pathways required extensive literature review of the existing knowledge by utilising at first several review articles.5,6,19–22 In addition, primary sources based on PubMed literature searches (; http://www.ncbi.nlm.nih.gov/pubmed/), utilising a permutations of the insulin and TOR pathways names and gene symbols already recorded in the model, were also followed. Evidence suggesting components and their interactions were always followed to their primary source and interactions between components that were only suggestive or hypothetical were omitted. Thus, only connections that were shown as derived from experimental evidence were included. Protein–protein interactions, based on yeast two-hybrid system or other screens were also not included, due to the lack of directionality of the connections.
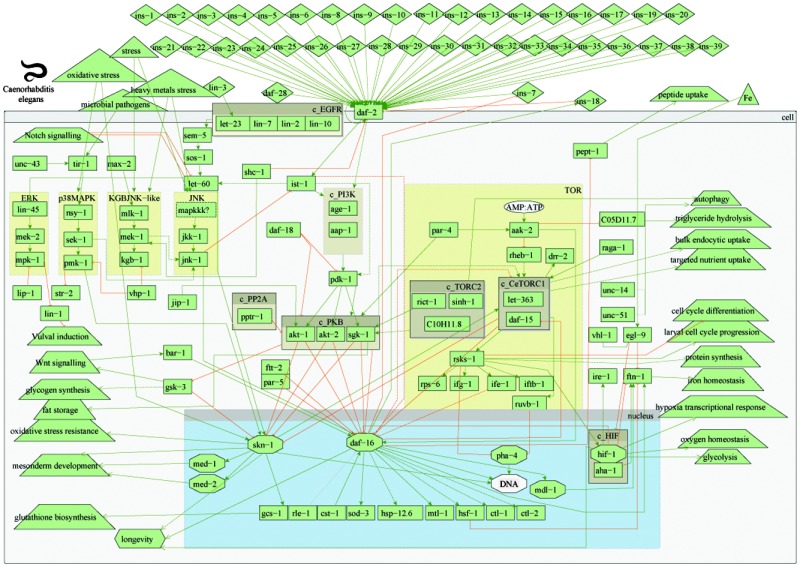
An overview of the model of the worm IIS and TOR signalling pathways in C. elegans and their interactions is presented in Fig. 1. The graph comprises activation and inhibition relationships between the protein components of the signalling network in the IIS and TOR pathways. A detailed description of the signalling is given in 3.1. In addition, all gene names and associated WormBase/Ensembl identifiers, part of the model of IIS and TOR pathways in worms, are listed in Table S1 (ESI†). All references used to build the worm connections/interactions within the TOR and IIS pathways are listed in Table S2 (ESI†).
Fig. 1. Overview of worm insulin and TOR pathways. Legend: rectangles represent genes; diamonds – molecules; triangles – environmental effects; trapezoids – other than IIS or TOR pathways; octagons – transcription factors; green arrow lines represent activation; red t-shaped lines represent inhibition; brown boxes starting with c_ represent complexes.

2.2. Whole-genome expression datasets
Having built a comprehensive model of the IIS and TOR pathways, we wanted to explore potential transcriptional feedback and paths that could modulate longevity. The most widely used technique that provides information on the effect of components in biological pathways is gene knockout or overexpression of specific genes. This is followed by microarray gene expression experiments to study the effect of these perturbations. For this reason all gene names and symbols, part of the two worm pathways, were used to search for available experiments in ArrayExpress23 and GEO24 databases. These databases are a comprehensive source of functional genomics experiments, including gene expression studies. Since microarray experiments are the most widely used technique to study the effect of genes in a particular biological pathway, we concentrated on microarray gene expression studies to try to maximise available experiments. In order to achieve consistent gene expression profiles across experiments and across species, data on such experiments needed to be in raw format to allow unified data analysis. Moreover, only experiments that had recorded longevity as phenotype were considered.
As a result, only four experiments were found during these searches. In total there were three worm studies comprising four whole worm microarray expression profiles and one whole fly experiment. Visual representation of the perturbed components can be found in Fig. 3. The identifiers, perturbed genes, references and phenotype outcome can be found in Table 1. The four worm microarray experiments comprised RNAi (i stands for double-stranded RNA interference) inhibition of the rheb-1 and let-363 genes, part of the TOR pathway, and loss-of-function mutations in daf-2 and daf-16, part of the core IIS pathway. An overexpression of aak-2 (TOR pathway) was also included. The whole fly experiment comprised loss-of-function mutations in InR and foxo.
Fig. 3. Perturbed components in the worm and fly experiments.

Table 1. Identifiers for the worm and fly experiments.
| Perturbed components (Fig. 3) | ArrayExpress ID | GEO ID | Reference (PubMed ID) | Background | Experiment | Longevity |
| 1 | E-GEOD-1762 | GSE1762 | 1530866368 | daf-2 (e1370,m577) | daf-2(e1370,m577);daf-16(df-50) | Short lived |
| 2 | E-GEOD-9682 | GSE9682 | 1907923943 | N2 (RNAi) | rheb-1 (RNAi) | Long lived |
| 3 | N2 (RNAi) | let-363 (RNAi) | Long lived | |||
| 4 | E-GEOD-25513 | GSE25513 | 2133104469 | N2 | aak-2 (overexpression) | Long lived |
| 5 | E-TABM-757 | NA | 2169471918 | daGAL4>UAS-dInR DN | dfoxo Δ/Δ daGAL4>UAS-dInR DN | Short lived |
Analysis of differentially expressed genes for each experiment was performed using the R programming language. Initial quality control was performed using Relative Log Expression (RLE) values and Normalized Unscaled Standard Errors (NUSE) boxplots, part of the affyPLM package.25 Thus, arrays for a particular experiment were excluded from further analysis if the interquartile range (IQR) in the NUSE boxplot were outside +/–1.05 and the IQR in the RLE boxplot exceeded +/–0.2. Raw data were then normalised and summarised using the rma function,26–28 part of the affy package,26,29 followed by a quantile normalisation, part of the limma package.30 To identify differentially expressed genes, linear models and the empirical Bayes moderated t-statistic were used, as implemented in the limma package. Differentially expressed genes were identified as exhibiting an adjusted p-value of <0.005 for all worm experiments and <0.001 for the fly experiment (correction applied: Benjamini and Hochberg or “BH” in the limma package). Only differentially expressed genes that are part of the IIS and TOR pathways were considered in both worm and fly experiments.
2.3. NetEffects
In order to identify “routes” or paths that could potentially modify the longevity phenotype in the abovementioned experiments we used NetEffects as a tool for inferring the impact of differential gene expression on the IIS and TOR pathways and relating these inferences on the longevity phenotype. It has been previously developed and tested to identify paths related to longevity changes from fly mutants of the insulin/insulin-like growth factor signalling pathway.31 NetEffects employs Answer Set Programming (ASP), a method for declarative programming that supports logic-based inferences. NetEffects was adapted to analyse the IIS and TOR pathways in C. elegans in the same fashion as the equivalent pathway for the fly described in Papatheodorou et al. 2012.31 In addition, paths were classified as “primary” or “secondary” by using the perturbed or differentially expressed component/gene as starting point respectively. The web-service for the C. elegans version of the IIS pathway is available via; http://www.ebi.ac.uk/thornton-srv/software/NetEffects/worm_path.php.
3. Results and discussion
3.1. A signalling network model of the Insulin and its interaction with the TOR pathway
The single insulin receptor (daf-2) can be activated by at least 40 different insulin-like (ins) peptides,32–34 following signals from olfactory and chemosensory neurons. The activation of daf-2 leads to the recruitment of the insulin receptor substrate (ist-1),35 which in turn can activate the phosphoinositide-3-kinase (PI3K) complex. The PI3K complex comprises the age-1 (PI3K-like catalytic subunit) and aap-1 (PI3K-like adaptor subunit) proteins and can also be directly activated by daf-2. 35 Phospholipid products of PI3K (phosphatidylinositol(3,4,5) triphosphate – PIP3) can activate phosphoinositide-dependent kinase-1 (pdk-1), leading to the activation of AKT/protein kinase B-like proteins (akt-1 and akt-2) and serum and glucocorticoid-inducible kinase (sgk-1). AKT-1, AKT-2 and SGK-1 kinases, separately and in the form a complex (protein kinase B – PKB), antagonise the activity of the forkhead transcription factor daf-16. 36
This core signal transduction pathway receives input and is modulated by a number of other proteins. These include, among others, the worm orthologue of the human PTEN tumour suppressor gene, daf-18 (ref. 37) that dephosphorylates the phospholipid PIP3, thus limiting the activation of downstream AKT-1/2 kinases. AKT kinases activity is also modulated by the PP2A regulatory subunit (pptr-1)38,39 and c-Jun N-terminal kinase (JNK)-interacting protein 1 (jip-1).40 Furthermore, activated daf-16 can inhibit the transcription of at least one of the insulin-like peptides (ins-7)33 and activate another (ins-18).41 These positive and negative feedback loops are in effect a self-regulation of the core insulin signal transduction. However, it has to be considered that transcriptional feedback operates on much slower timescale compared to signalling processes.
The insulin pathway is highly connected and interacts with several other pathways, such as ERK, p38MAPK, KGB, JNK, RAS, notch and wnt signalling. In addition, it is also well connected with the target-of-rapamycin (TOR) pathway, mainly via TORC2 and CeTORC1 complexes. The energy sensing AMP-activated protein kinase (aak-2), part of the TOR pathway, is activated by decreased energy or glucose levels (i.e. low AMP to ATP ratio),42 which in turn inhibits GTPase rheb-1, an upstream activator of CeTOR (let-363).43 The CeTORC1 and TORC2 complexes are thought to regulate fat metabolism, feeding, larval development and growth.44,45
3.2. Paths that modulate longevity in worms
We built a model of the worm IIS and TOR pathways following a literature review of the current (as of time of writing) knowledge of the different components, part of the two pathways. Connections (i.e. transcriptional inhibitions and activations) were compiled solely from experimental evidence, thus allowing inferences of the effect of perturbed genes to reproduce as closely as possible the paths that could be operational in vivo. Despite the worm pathway being as comprehensive as possible, enzymatic kinetics, mRNA levels/half lives and post-transcriptional modifications have not been included in the model. This was mainly due to a lack of comprehensive and consistent data. Moreover, correlative studies between mRNA and protein abundance suggest a relatively medium degree of correlation.46–48 Thus, although mRNA expression studies are somewhat easier to perform than protein identification and quantification, they are only correlative to the product of interest, i.e. proteins. An example of such discrepancy within the IIS pathway is a transcriptional feedback from FOXO to the insulin receptor InR, in Drosophila 49 and mammals.50 In these experiments transcriptionally active FOXO activates transcription of InR itself, but the two-fold increase in the InR mRNA does not account for the five-six fold increase in protein abundance.
Despite the basic pitfalls of this research, several important paths and transcriptional feedback loops are relatively worthy of further examination and these are described below. Although there were other paths and subpaths that are likely to have an effect on longevity, the inferred effects were considered too speculative. Therefore, such paths are described in the supplementary results (ESI†), along with graphs that illustrate the paths.
Several paths, parallel to the core insulin transduction signalling, within the worm experiments considered, were found to support or contradict the observed phenotype. If these are operational within the cells, they could enhance or diminish the longevity effect of core insulin signalling via daf-16. At least according to our model, these paths converge on two transcription factors, i.e. hif-1 and skn-1.
3.2.1. Longevity path via hif-1
Lifespan extension of stabilised hypoxia-induced transcription factor hif-1 appears to be IIS independent51 and deletions of hif-1 have an effect on longevity in a temperature-dependent manner52 and are daf-16 dependent. Four of the experiments (daf-2 vs. daf-2;daf-16, N2 vs. aak-2 oe, N2 vs. let-363i and N2 vs. rheb-1i) exhibited a common subpath in their primary effect (path starts from the perturbed component/gene). This path comprised an activation of the ribosomal protein S6 kinase (rsks-1) by CeTORC1 complex, which in turn activates the hypoxia-induced factor (hif-1) and a potential hif-1 mediated increase in longevity (Fig. S1, S6 and S14, ESI†). The signalling in this particular common path in some of the experiments was likely increased (daf-2 vs. daf-2;daf-16) and in others decreased (N2 vs. aak-2 oe, N2 vs. let-363i and N2 vs. rheb-1i), but in all of the four experiments this path contradicted the observed phenotype.
Thus in the long-lived experiments (rheb-1i and let-363i) a decreased activity of the CeTORC1 complex is likely to lead to a decreased activity of hif-1 and a potential hif-1-mediated decrease in longevity. It has to be said that although an inhibition from hif-1 to daf-16 is recorded in the worm pathway model, the current version of NetEffects only displays the shortest path from the perturbed components to longevity. While not displayed in NetEffects, the likely outcome from a reduced hif-1 activity would be a decreased inhibition of daf-16 and possible daf-16-mediated lifespan extension, as previously shown under normoxic conditions.52 In the short-lived daf-2;daf-16 experiment, a path comprising an activated CeTORC1 complex and a stabilised hif-1 (Fig. S1, ESI†), following a knock-out of daf-16, is likely to oppose the observed reduction in longevity. It would be interesting to investigate if removing such a path (e.g. hif-1 RNAi), would result in further reduction in lifespan. Additionally, for this particular experiment a likely skn-1 mediated increase in longevity was also observed, due to the down-regulation of akt-1/2 kinases, although a compensatory gsk-3 inhibition of skn-1 was also likely to be increased (Fig. S4, ESI†).
3.2.2. Longevity paths via skn-1
The transcription factor skn-1 is a stress-response gene that activates Phase 2 detoxification response53 and mainly exerts its effect in the intestine.54 Several lines of evidence suggest that the effect of activated skn-1 could potentially modulate ageing and this could be achieved in daf-16 independent manner. Our results suggest that a primary path that leads to an increased longevity for two of the experiments (N2 vs. let-363i and N2 vs. rheb-1i) involves skn-1. This path exhibits a decreased activity of the CeTORC1 complex, subsequent decrease in the inhibitory link to skn-1 and daf-16 and an increase in longevity, mediated by daf-16 and/or skn-1 (Fig. S5 and S9, ESI†). It is not obvious if the increased longevity in both experiments is due to the synergistic nucleic accumulation of both transcription factors. The sek-1 kinase, part of the p38 mitogen-activated protein kinase (MAPK), has been shown to be required for skn-1 activation.11 Thus, a lifespan analysis of double mutants (rheb-1i;sek-1i and let-363i;sek-1 as compared to single mutants of rheb-1i and let-363i) is likely to shed some light on the relative importance of skn-1 and daf-16 with respect to the effect on longevity from decreased activity of the TOR pathway. Furthermore, two kinases sek-1 and jnk-1, part of the p38 and JNK MAPKs, were up-regulated in both experiments, suggesting a transcriptional feedback from TOR pathway to JNK. These up-regulated kinases led to several paths that were likely responsible for the skn-1 and daf-16 mediated increase in longevity, although this does not preclude the possibility of other TOR-mediated independent of skn-1 and daf-16 longevity mechanisms, such as a reduction of mRNA translation, increased autophagy or via the germline.55–57 An experiment of inhibited activity of sek-1 kinase would also likely reduce the activation of skn-1 by pmk-1 58 and the activation of daf-16 by jnk-1. 59 If such experiments are performed, according to our worm IIS and TOR pathways model, the increased longevity phenotype of rheb-1i and let-363i, would only result from a decreased inhibition of daf-16 by the TOR pathway and a survival analysis would point the relative contribution of skn-1. In support of such hypothesis comes a study by Tullet et al. 2008 that has suggested an independent of daf-16 pro-longevity phenotype of skn-1. Although, as both transcription factors compete for binding to the negative regulators akt-1, akt-2 and sgk-1, elimination of either one could result in availability of these negative regulators.11
In these two experiments (rheb-1i and let-363i), several paths that contradicted the increase in longevity were also revealed (Fig. S8 and S12, ESI†). Some were due to a likely increased IIS signalling by up-regulated insulin-like peptides. As the observed phenotype was an increase in longevity, these paths were found to contradict this phenotype. It would be interesting to investigate the relative roles of increased IIS signalling and decreased TOR signalling, with respect to the increase in longevity. Similarly to the previous experiments proposed, a double (daf-2;rheb-1 and daf-2;let-363) mutant could help delineate the relative contribution of the increased IIS signalling and reduced TOR signalling, with respect to the longevity phenotype.
3.3. A comparison with fly IIS and TOR signal transduction pathways (InR vs. InR;foxo)
A direct comparison between the IIS and TOR pathways in both flies and worms reveals that the two pathways are in general evolutionarily conserved. The core components of the IIS are present in both organisms, including the insulin/IGF-like receptor, PI3K, PDK, AKT signal transduction kinases and the forkhead box O (foxo) transcription factor. Differences are also noticeable. There are at least 40 different insulin-like peptides in the worm as compared to only eight in the fly. These insulin-like peptides agonise the sole insulin/IGF receptor (InR) in the fly, where at least one insulin-like peptide (ins-18) in worms has been shown to antagonise daf-2 41 and transcriptional feedback loops from daf-16 to daf-2 have been shown. The majority of components part of the TOR pathway are also relatively conserved across the worm and fly. The TORC1/2 complexes are both present, although there is not a worm orthologue of the tuberous sclerosis complex (TSC).
As previously mentioned, the IIS and TOR pathways are evolutionarily conserved in fly and worm.6,19 However, having experiments of orthologous components in both the fly and worm, now allowed us to compare the differences in the two pathways and the effect on longevity.
In the fly experiment (InR vs. InR;foxo) all possible paths (i.e. primary and secondary) led only to a decrease in longevity, supporting the observed phenotype (Fig. S17 and S18, ESI†). This was to be expected, as in the IIS/TOR model in flies, longevity is recorded as only modulated via foxo and foxo is down-regulated in the experiment (Fig. 2).
Fig. 2. Overview of the fly insulin and TOR pathways. Legend: rectangles represent genes; diamonds – molecules; triangles – environmental effects; trapezoids – other than IIS or TOR pathways; octagons – transcription factors; green arrow lines represent activation; red t-shaped lines represent inhibition; brown boxes starting with c_ represent complexes.

On the other hand, in our worm signalling model there are several possible inputs into longevity. These comprise daf-16 (orthologue of the fly foxo), skn-1 and hif-1. Thus, other paths that decrease longevity in the worm were observed, apart from the daf-16/foxo mediated decrease in longevity, which was observed in both experiments.
Several paths from the TOR pathway and the interaction with IIS were observed to complement the observed decrease in longevity (Fig. S17 and S18, ESI†). These paths were mainly down-regulating S6k, followed by a decreased inhibition of chico and increased IIS signalling. While an interaction between the IIS and TOR pathways was shown in the worm experiment (reduced inhibition of CeTORC1 complex by daf-16, Fig. S1, S3 and S4, ESI†), this was not via rsks-1, the worm orthologue of S6k. Furthermore, in the worm, the transcription factors skn-1 and hif-1 were part of paths that were likely to lead to an increase in longevity as compared to paths that would result only in decrease in longevity in the fly experiment.
3.4. Transcriptional feedback in insulin signalling
The primary target of the worm and fly insulin-like peptides is the sole insulin receptor34 InR in flies and daf-2 in worms,34 part of the core IIS signal transduction pathway to ultimately inhibit the nuclear translocation of the transcription factor daf-16/foxo.
For several of the microarray experiments, a consistent transcriptional feedback to the insulin-like peptides was evident. In the long-lived experiments (rheb-1i and let-363i), there was a substantial number of up-regulated insulin-like peptides (seven and nine in the let-363 and rheb-1 respectively), as compared to the wild-type (N2). In these two worm experiments out of the 40 there were altogether nine up-regulated insulin-like peptides and seven of those were found in both experiments. The overall effect of the up-regulated insulin-like peptides would be an increase in the core insulin signal transduction, thereby leading to an overall inhibition of daf-16 and/or skn-1, followed by a potential daf-16 and/or skn-1 mediated decrease in longevity.
Several insulin-like peptides were down-regulated in the daf-2;daf-16 double mutant experiment as compared to daf-2 background (ins-33 and ins-35). Hence, the down-regulated insulin-like peptides in this short-lived experiment would ultimately reduce the core insulin signalling transduction. However, since daf-2 and daf-16 are inhibited in this experiment, the transcriptional feedback would appear dysfunctional.
The down-regulated insulin-like peptides could potentially be explained by a positive feedback loop from daf-16. This has been shown for ins-18,41 but crucially not for any of the other up-regulated peptides pertinent to the results obtained in this work.
In addition, the two of the perturbed components (i.e. rheb-1 and let-363) are part of the TOR pathway. Therefore, if a transcriptional feedback from daf-16 to the insulin-like peptides is indeed active then the effect of the perturbed components (i.e. rheb-1 and let-363) must converge on daf-16. Indeed, several paths within these two experiments show a decreased inhibition of daf-16 by the CeTORC1 complex. Thus, a transcriptionally active daf-16 could provide a positive feedback loop to the insulin-like peptides.
Conversely, a negative transcriptional feedback in the daf-2;daf-16 double mutant experiment that contradicted the observed phenotype, was also observed. The ins-7 was up-regulated and the likely outcome would be an overall increase in the core insulin signalling and a likely daf-16 and/or skn-1 mediated decrease in longevity. A previously suggested negative transcriptional feedback from daf-16, could explain the observed up-regulation of ins-7. 33
In the fly experiment two of the Ilps were also down-regulated (Ilp3 and Ilp6), nonetheless there is a paucity of experimental evidence to suggest a transcriptional feedback from the IIS similar to the worm. Still, it has been suggested that a positive feedback loop from fly foxo could result in the up-regulation of Ilp3 and Ilp5 60 and Ilp3 was found to be up-regulated in the fly InR;foxo experiment.
Despite several lines of evidence, suggesting a transcriptional feedback loop from daf-16/foxo, it is also possible that a signal other than daf-16 or foxo could provide a transcriptional feedback loop to the insulin-like peptides. Several of the paths examined intersect on the transcription factor skn-1, shown to inhibit one of insulin-like peptides (ins-7)33,61 and skn-1 suppression coupled with a daf-28 induction has been suggested as a negative transcriptional feedback.62 Furthermore, both ins-7 and daf-28 were found to be up-regulated in 2 of the experiments, i.e. rheb-1i and let-363i. As a result, it is possible that differential expression of at least some of the insulin-like peptides in these experiments is due to skn-1.
As previously argued, the effects of the inhibition of rheb-1 and let-363 RNAi experiments, could converge on daf-16 and/or skn-1, hence providing positive or negative feedback to the insulin-like peptides. This could explain the up- and down-regulated insulin-like peptides in the double mutant daf-2;daf-16. However, ins-7, daf-28 and several other insulin-like peptides were found to be down-regulated in the rheb-1 and let-363 experiments. By this means if, ins-7 and daf-28 are indeed under negative transcriptional control of daf-16/skn-1 and the others under positive control, both up and down-regulated insulin-like peptides would be expected, similarly to the double mutant daf-2;daf-16.
Therefore, other mechanisms could also be responsible for the differential insulin-like peptides regulation, similar to a gene dosage effect of ins-18 over other insulin-like genes.34
4. Conclusions
Drosophila melanogaster and Caenorhabditis elegans diverged ∼990 million years ago.63 Thus, it is not surprising that several cellular mechanisms and physiological processes exhibit substantial differences between flies and worms. Flies have relatively well differentiated brain, a heart, vascular system, vision and hearing, where worms do not. In terms of cellular processes, phagocytosis in C. elegans for example, does not seem to play a role in microorganism clearance. On the other hand apoptotic pathway defence response in flies has not yet been reported within the innate immune response.64 The sensory system in flies is substantially more complex than in worms. It has been proposed that there are at least two sensory-lifespan pathways present in flies with its genes expressed in different sensory neurons, in contrast to worms (for a detailed review see Linford et al. 201165).
A general overview of the IIS and TOR pathways in the worm and fly suggests remarkable similarities. This signifies the importance of the nutrient sensing properties of the IIS and TOR pathways. Even though both species occupy distinct ecological niches, the molecular basis of nutrient sensing is similar over large evolutionary distance. The core IIS signal transduction pathway appears evolutionary conserved, although some differences are noticeable. A major difference is the absence of the Drosophila insulin receptor substrates (IRS) chico and Lnk and the absence of the TSC complex, part of the TOR pathway, although an equivalent IRS homolog, ist-1, is present in worms. Unfortunately, due to the absence of corresponding experiments in the fly we could not show similarities and/or differences in the paths leading to longevity, apart from the foxo-mediated effect on longevity. There were some indications of conserved transcriptional feedback mechanisms that are very similar in both model organisms. That is, a transcriptional feedback from foxo to the insulin-like peptides. One major difference that would greatly influence any such comparison is the lack of different factors recorded in our fly pathway model that could mediate longevity, apart from foxo. It is possible that some of these factors are worm-specific. The hypoxia-induced factor hif-1 is evolutionary conserved and present in worms, flies and mammals,66 thus it is very likely performing a homologous function similar to worms, although a hif mediated effect on longevity in flies remains to be seen. On the other hand, the stress response gene skn-1 in worms appears to be without a homolog in Drosophila, thus it is likely to be worm-specific, although functional counterparts in mammals have been previously described.67
The work presented here indicates that such approach is useful in analysing the effects of perturbed components within the IIS and TOR pathways, with respect to the effect on longevity. Furthermore, this approach could be extended, as new knowledge is accumulated, to other pathways that have an effect on lifespan or biological processes, such as autophagy.
Acknowledgments
We would like to thank Dr Michele Riesen and Prof. David Gems for useful discussions on building the worm IIS/TOR pathways and different paths and transcriptional feedback. We also thank the EMBL-EBI for providing computing resources. We thank all members, part of the Wellcome Trust Strategic Award at UCL (WT081394MA), Institute of Healthy Ageing for their guidance and advice.
Footnotes
References
- Colman R. J., Anderson R. M., Johnson S. C., Kastman E. K., Kosmatka K. J., Beasley T. M., Allison D. B., Cruzen C., Simmons H. A., Kemnitz J. W., Weindruch R. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge L., Gems D. Nat. Rev. Genet. 2002;3:165–175. doi: 10.1038/nrg753. [DOI] [PubMed] [Google Scholar]
- Hansen M., Taubert S., Crawford D., Libina N., Lee S. J., Kenyon C. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Kapahi P., Zid B. M., Harper T., Koslover D., Sapin V., Benzer S. Curr. Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kenyon C. Philos. Trans. R. Soc., B. 2011;366:9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M., Kopelman A., Epstein D., Tu M. P., Yin C. M., Garofalo R. S. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Kimura K. D., Tissenbaum H. A., Liu Y., Ruvkun G. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Clancy D. J., Gems D., Harshman L. G., Oldham S., Stocker H., Hafen E., Leevers S. J., Partridge L. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Tullet J. M., Hertweck M., An J. H., Baker J., Hwang J. Y., Liu S., Oliveira R. P., Baumeister R., Blackwell T. K. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D. B., Johnson T. E. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S., Paradis S., Gottlieb S., Patterson G. I., Lee L., Tissenbaum H. A., Ruvkun G. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Murphy C. T. Exp. Gerontol. 2006;41:910–921. doi: 10.1016/j.exger.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Panowski S. H., Wolff S., Aguilaniu H., Durieux J., Dillin A. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M., Kennedy B. K. Aging Cell. 2011;10:185–190. doi: 10.1111/j.1474-9726.2010.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S. D., Yen K., Tissenbaum H. A. Curr. Biol. 2009;19:R657–R666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alic N., Andrews T. D., Giannakou M. E., Papatheodorou I., Slack C., Hoddinott M. P., Cocheme H. M., Schuster E. F., Thornton J. M., Partridge L. Mol. Syst. Biol. 2011;7:502. doi: 10.1038/msb.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L., Kenyon C. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Honjoh S., Nishida E. J. Biochem. 2011;149:381–388. doi: 10.1093/jb/mvr026. [DOI] [PubMed] [Google Scholar]
- McCormick M. A., Tsai S. Y., Kennedy B. K. Philos. Trans. R. Soc., B. 2011;366:17–27. doi: 10.1098/rstb.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A., Kaarniranta K. Trends Mol. Med. 2009;15:217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Parkinson H., Kapushesky M., Kolesnikov N., Rustici G., Shojatalab M., Abeygunawardena N., Berube H., Dylag M., Emam I., Farne A., Holloway E., Lukk M., Malone J., Mani R., Pilicheva E., Rayner T. F., Rezwan F., Sharma A., Williams E., Bradley X. Z., Adamusiak T., Brandizi M., Burdett T., Coulson R., Krestyaninova M., Kurnosov P., Maguire E., Neogi S. G., Rocca-Serra P., Sansone S. A., Sklyar N., Zhao M., Sarkans U., Brazma A. Nucleic Acids Res. 2009;37:D868–D872. doi: 10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., Troup D. B., Wilhite S. E., Ledoux P., Evangelista C., Kim I. F., Tomashevsky M., Marshall K. A., Phillippy K. H., Sherman P. M., Muertter R. N., Holko M., Ayanbule O., Yefanov A., Soboleva A. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B. M., Collin F., Brettschneider J., Simpson K., Cope L., Irizarry R. A. and Speed T. P., Quality Assessment of Affymetrix GeneChip Data, Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Springer, New York, 2005. [Google Scholar]
- Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B. M., Irizarry R. A., Astrand M., Speed T. P. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Gautier L., Cope L., Bolstad B. M., Irizarry R. A. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Smyth G. K., Limma: linnear models for microarray data, Bioinformatics and Computational Biology Solutions using R and Bioconductor, Springer, New York, 2005. [Google Scholar]
- Papatheodorou I., Ziehm M., Wieser D., Alic N., Partridge L., Thornton J. M. PLoS One. 2012;7:e50881. doi: 10.1371/journal.pone.0050881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Kennedy S. G., Ruvkun G. Genes Dev. 2003;17:844–858. doi: 10.1101/gad.1066503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., Kenyon C. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Pierce S. B., Costa M., Wisotzkey R., Devadhar S., Homburger S. A., Buchman A. R., Ferguson K. C., Heller J., Platt D. M., Pasquinelli A. A., Liu L. X., Doberstein S. K., Ruvkun G. Genes Dev. 2001;15:672–686. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkow C. A., Munoz M. J., Riddle D. L., Ruvkun G. J. Biol. Chem. 2002;277:49591–49597. doi: 10.1074/jbc.M207866200. [DOI] [PubMed] [Google Scholar]
- Hertweck M., Gobel C., Baumeister R. Dev. Cell. 2004;6:577–588. doi: 10.1016/s1534-5807(04)00095-4. [DOI] [PubMed] [Google Scholar]
- Ogg S., Ruvkun G. Mol. Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S., Mukhopadhyay A., Narasimhan S. D., Tesz G., Czech M. P., Tissenbaum H. A. Cell. 2009;136:939–951. doi: 10.1016/j.cell.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. C., Huang K. Y., Yang C. H., Yang Y. S., Lee W. Y., Chiang C. W. J. Biol. Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- Kim A. H., Sasaki T., Chao M. V. J. Biol. Chem. 2003;278:29830–29836. doi: 10.1074/jbc.M305349200. [DOI] [PubMed] [Google Scholar]
- Matsunaga Y., Gengyo-Ando K., Mitani S., Iwasaki T., Kawano T. Biochem. Biophys. Res. Commun. 2012;423:478–483. doi: 10.1016/j.bbrc.2012.05.145. [DOI] [PubMed] [Google Scholar]
- Greer E. L., Dowlatshahi D., Banko M. R., Villen J., Hoang K., Blanchard D., Gygi S. P., Brunet A. Curr. Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjoh S., Yamamoto T., Uno M., Nishida E. Nature. 2009;457:726–730. doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- Jia K., Chen D., Riddle D. L. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Soukas A. A., Kane E. A., Carr C. E., Melo J. A., Ruvkun G. Genes Dev. 2009;23:496–511. doi: 10.1101/gad.1775409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum D., Colangelo C., Williams K., Gerstein M. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Xiao P., Lei S., Deng F., Xiao G. G., Liu Y., Chen X., Li L., Wu S., Chen Y., Jiang H., Tan L., Xie J., Zhu X., Liang S., Deng H. Acta Biochim. Biophys. Sin. 2008;40:426–436. doi: 10.1111/j.1745-7270.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Puig O., Tjian R. Genes Dev. 2005;19:2435–2446. doi: 10.1101/gad.1340505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr, 2nd M. T., D'Alessio J. A., Puig O., Tjian R. Genes Dev. 2007;21:175–183. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R., Steinkraus K. A., Sutphin G. L., Ramos F. J., Shamieh L. S., Huh A., Davis C., Chandler-Brown D., Kaeberlein M. Science. 2009;324:1196–1198. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser S. F., Begun A., Kaeberlein M. Aging Cell. 2011;10:318–326. doi: 10.1111/j.1474-9726.2011.00672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J. H., Blackwell T. K. Genes Dev. 2003;17:1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop N. A., Guarente L. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Kapahi P., Chen D., Rogers A. N., Katewa S. D., Li P. W., Thomas E. L., Kockel L. Cell Metab. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfel M. N., Shamieh L. S., Kaeberlein M., Kennedy B. K. Biochim. Biophys. Acta. 2009;1790:1067–1074. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robida-Stubbs S., Glover-Cutter K., Lamming D. W., Mizunuma M., Narasimhan S. D., Neumann-Haefelin E., Sabatini D. M., Blackwell T. K. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H., Hisamoto N., An J. H., Oliveira R. P., Nishida E., Blackwell T. K., Matsumoto K. Genes Dev. 2005;19:2278–2283. doi: 10.1101/gad.1324805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. W., Mukhopadhyay A., Svrzikapa N., Jiang F., Davis R. J., Tissenbaum H. A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton S., Alic N., Slack C., Bass T., Ikeya T., Vinti G., Tommasi A. M., Driege Y., Hafen E., Partridge L. PLoS One. 2008;3:e3721. doi: 10.1371/journal.pone.0003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira R. P., Abate J. P., Dilks K., Landis J., Ashraf J., Murphy C. T., Blackwell T. K. Aging Cell. 2009;8:524–541. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong M. C., Na K., Kim H., Jeong S. K., Joo H. J., Chitwood D. J., Paik Y. K. J. Biol. Chem. 2011;286:7248–7256. doi: 10.1074/jbc.M110.189183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureta-Vidal A., Ettwiller L., Birney E. Nat. Rev. Genet. 2003;4:251–262. doi: 10.1038/nrg1043. [DOI] [PubMed] [Google Scholar]
- Mylonakis E., Aballay A. Infect. Immun. 2005;73:3833–3841. doi: 10.1128/IAI.73.7.3833-3841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linford N. J., Kuo T. H., Chan T. P., Pletcher S. D. Annu. Rev. Cell Dev. Biol. 2011;27:759–785. doi: 10.1146/annurev-cellbio-092910-154240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Uden P., Kenneth N. S., Webster R., Muller H. A., Mudie S., Rocha S. PLoS Genet. 2011;7:e1001285. doi: 10.1371/journal.pgen.1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J. H., Vranas K., Lucke M., Inoue H., Hisamoto N., Matsumoto K., Blackwell T. K. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16275–16280. doi: 10.1073/pnas.0508105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee J. J., Schuster E., Blanc E., Thomas J. H., Gems D. J. Biol. Chem. 2004;279:44533–44543. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- Mair W., Morantte I., Rodrigues A. P., Manning G., Montminy M., Shaw R. J., Dillin A. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.