

Introduction
Over the past decade since amyloid-modifying therapeutic agents have entered Alzheimer’s disease (AD) clinical trials, the occurrence of magnetic resonance imaging (MRI) abnormalities has required careful consideration by academic investigators, pharmaceutical companies and regulatory authorities. MRI signal changes, thought to represent “vasogenic edema” (VE) and cerebral microhemorrhages (mH), were first observed in trials of a monoclonal antibody against amyloid β [1–3], and have since been associated with other amyloid-modifying therapies [4]. In response to guidance issued by the U.S. Food and Drug Administration (FDA) to various sponsors on the conduct of clinical trials of amyloid-modifying agents for the treatment of AD, the Alzheimer’s Association Research Roundtable convened a Workgroup in July 2010.
The Workgroup was composed of academic and industry representatives identified on the basis of their expertise and interest in this area, and was tasked with the objective of providing expert advice regarding the FDA’s concerns related to MRI abnormalities, including signal changes thought to represent “vasogenic edema” (VE) and microhemorrhages (mH) and the relationship of these MR abnormalities to experimental treatment with amyloid-modifying therapies. As VE and mH are typically detected on different MRI sequences, and appear to represent a spectrum of image abnormalities which may share some common underlying pathophysiological mechanisms, both in the natural history of AD and in the setting of amyloid-modifying therapeutic approaches, the Workgroup suggests referring to this spectrum as Amyloid Related Imaging Abnormalities (ARIA). Despite the likelihood of shared underlying mechanisms, there may be instances in which it is useful to describe specific phenomena, thus the Workgoup further refined the terminology: ARIA-E refers to the MR signal alterations thought to represent VE and related extravasated fluid phenomena. ARIA-H refers to the MR signal alterations on attributable to mH and hemosiderosis.
The Workgroup reviewed the relevant publicly available information, including natural history studies and spontaneous occurrence of these adverse events in aging and AD, their occurrence in the setting of trials of amyloid lowering agents for AD and similar clinical conditions from which parallels might be drawn; and existing animal models which may elucidate the underlying mechanisms. The Workgroup sought to develop specific recommendations regarding the conduct of AD clinical trials in the setting of ARIA including inclusion/exclusion criteria, safety monitoring and potential areas of research which might help increase our understanding of these events.
The phenomenon formerly known as “VE”: ARIA-E alterations
Although early animal work had reported evidence of mH with anti-amyloid immunotherapy [5], an unexpected type of MRI signal alteration was first observed in the single dose-ascending Phase I trial of a monoclonal antibody against amyloid-β. Three out of ten patients in only the highest dose group (5mg/kg) developed transient signal abnormalities on T2 weighted/fluid attenuation inversion recovery (FLAIR) sequences, approximately 4–6 weeks after a single dose of bapineuzumab [1]. Additional cases were reported in the Phase II study [2, 3]. Initially the appearance and transience of the MRI abnormalities were thought to be reminiscent of posterior reversible encephalopathy syndrome (PRES) observed in hypertensive patients and pre-eclampsia, but as additional cases in AD clinical trials became apparent, this new entity was initially described as “vasogenic edema”, based on the MRI characteristics.
The term “vasogenic edema” or “VE” for this entity evolved from the observation that the increased MR signal on FLAIR sequences was usually transient, and was not associated with evidence of restricted diffusion, tissue necrosis, or other sequelae associated with cytotoxic edema. At a cellular level, vasogenic edema is thought to represent an increase in extracellular fluid volume due to increased permeability of brain capillary endothelial cells to serum proteins, as opposed to cytotoxic edema, which is increased intracellular fluid, thought to be related to high intracellular osmolality from cellular damage (clinically most often seen in acute infarction where the mechanism is felt to be failure to maintain a homeostatic Na/K gradient across the cell membrane). There is very limited histopathological evidence available to determine whether the signal changes observed on MRI are in fact related to underlying vasogenic edema. The term “VE” has also sometimes been applied to other MR alterations observed in the setting of amyloid-modifying therapy that vary in signal characteristics on different MR sequences and in location within the intracranial space. In particular, increased signal intensity on FLAIR images has been reported in the leptomeningeal or sulcal space with anti-amyloid treatment, and may represent leakage or effusion of proteinaceous fluid from meningeal vessels. Thus we refer to the signal hyperintensities seen in the parenchyma and leptomeninges more specifically as “ARIA-E” to cover the MRI alterations thought to represent edema in the gray and white matter, and effusion or extravasated fluid in the sulcal space (see Figures 1 and 2).
Figure 1.

ARIA-E as seen on FLAIR images from a monoclonal antibody trial demonstrating increased MR signal in multiple regions of the right hemisphere affecting both gray and white matter.
Figure 2.
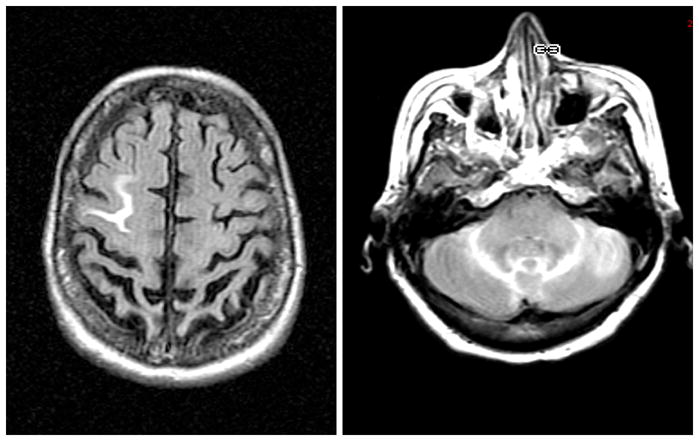
ARIA-E detected on FLAIR images from a monoclonal antibody trial study demonstrating increased MR signal in sulci, thought to represent proteinaceous fluid tracking in the leptomeninges and sulcal spaces.
Characteristics of ARIA-E observed in amyloid modifying therapeutic trials
ARIA-E most commonly manifests as increased MR signal intensity on FLAIR or other T2-weighted sequences in the parenchyma and/or leptomeninges in the parietal, occipital, and frontal lobes, but has also been observed in the cerebellum and brainstem [3]. It is not yet clear whether the edema begins in grey matter in some cases, as associated gyral swelling is sometimes apparent, with edema tracking into the underlying white matter, or whether there may be separate processes that affect grey and white matter. The parenchymal signal abnormalities can be quite subtle in a single region, multi-focal, or nearly pan-hemispheric (see Figure 1 below).
In some cases, the increased MR signal is primarily seen in what appears to be the leptomeningeal space (see Figure 2 below). This leptomeningeal involvement may be seen in isolation or near associated grey matter alterations. This focal increased signal has on occasion been misinterpreted as subarachnoid hemorrhage, but neither cerebrospinal fluid (CSF) studies nor susceptibility weighted imaging have revealed evidence of blood products in these cases. It remains unknown whether this increased signal in leptomeningeal compartments might represent other proteinaceous collections of fluid.
Cerebral amyloid angiopathy (CAA) with associated vascular or perivascular inflammatory infiltrates [6, 7] is another condition that has clinical and neuroimaging features which appear similar to ARIA associated with amyloid-modifying therapy. This spontaneously occurring syndrome presents on neuroimaging with a range of involvement of leptomeninges, grey matter and white matter [8, 9] similar to those reported with amyloid therapy-associated VE. CAA related inflammation appears pathologically to be driven by a spontaneous inflammatory response to the vascular amyloid deposits. The CSF pattern in inflammatory CAA (elevated protein, mostly without elevated WBCs) is similar to the CSF pattern seen in some treatment related VE with bapineuzumab [3]. This supports the assertion that focal CAA inflammation need not be associated with elevated CSF cell count. Following its resolution either spontaneously or with steroids, inflammatory CAA findings also reverse. In addition, both CAA related inflammation and “VE” with amyloid therapy have a shared association with ApoE ε4 [9]. There has been at least one report of similar pattern of leptomeningeal involvement in CAA [10]. A potential connection between inflammatory CAA and immunotherapy-associated ARIA was most recently suggested by identification of anti-Aβ autoantibodies in the CSF of a patient with the spontaneously occurring syndrome [11]. A similar spectrum of MR abnormalities as described in ARIA, involving both grey and white matter, has been reported in PRES syndrome [12, 13], although prominent leptomeningeal signal hyperintensities are not common.
Issues of Ascertainment
Patients identified with CAA related ARIA or PRES typically present with symptoms, while the majority of treatment associated ARIA-E cases reported in the literature have been asymptomatic and identified on “per-protocol” safety MRIs. A small number of ARIA-E cases in the bapineuzumab trials were detected on off-protocol MRI, prompted by change in clinical status or symptoms. These symptomatic cases have generally occurred within 4 to 8 weeks after infusion, suggesting that ARIA-E is not occurring early (at peak of antibody levels in blood). The timing of ARIA-E may provide a clue regarding potential pathophysiological mechanisms in bolus-dose immunotherapy, but it remains unclear whether there will be a temporal relationship between dosing and ARIA-E that may occur in other amyloid-lowering therapeutic approaches.
To our knowledge, there are no reports in the literature regarding the incidence of spontaneous ARIA-E in community based samples, but it is likely that there has not been systematic surveillance outside of clinical trials. The preliminary reports of rare cases of ARIA like phenomenon being detected in large cohorts being screened for clinical trials suggest that spontaneous ARIA-E may occur rarely in the natural history of AD, and perhaps more commonly in patients with genetic risk factors for high vascular amyloid burden and/or presumed CAA [4, 14]. The low incidence of spontaneous ARIA-E observed in clinical trial screening might reflect a bias towards recruiting subjects who have less vascular disease and vascular risk factors. A substantial proportion of clinical AD patients harbor some evidence of CAA changes at autopsy, particularly in ApoE ε4 carriers [15].
Clinical course associated with ARIA-E
There are currently very limited publicly available data regarding the clinical course associated with ARIA-E occurring in the setting of clinical trials of amyloid modifying therapies. Thus the Workgroup reviewed the data from bapineuzumab trials, but it is unknown whether ARIA seen in other amyloid-modifying therapies will have similar clinical course. In the Phase I bapineuzumab study, two out of three ARIA cases were asymptomatic at time of detection on per protocol MRI. In retrospect one of these patients had a reported period of transient confusion a few weeks prior to ARIA-E. The third patient had acute drop in MMSE and confusion prompting an off-protocol MRI 4 weeks after the infusion, revealing ARIA-E. This patient improved after several weeks without treatment [1].
In the first reports of the double-blind Phase II multiple dose study, 10 out of 12 ARIA-E cases were detected on per protocol MRI [2, 3]. Six of these 10 were asymptomatic even after retrospective review of all adverse events of the 30 days prior to or following detection of ARIA-E. Four of 10 were found to have reported transient symptoms of headache, confusion, visual disturbance in 30 days prior to or following detection of ARIA-E. The remaining 2 cases were symptomatic prompting off-protocol MRI, including confusion, headache, and gait difficulties. One of these subjects underwent treatment with IV steroids with resolution of symptoms.
There are very limited data on the long term clinical course of individuals with asymptomatic ARIA-E, but preliminary analyses of the asymptomatic cases of ARIA-E from the Phase II study did not reveal significant impact on MMSe compare to non-ARIA-E treated group [3]. Additional analyses on neuropsychological and functional outcomes in asymptomatic cases of ARIA-E are ongoing, as well as a more systematic central review of all MRI scans from the Phase II bapineuzumab studies.
Risk factors for ARIA-E
The pathophysiological mechanisms underlying vasogenic edema remain to be elucidated: however, the bapineuzumab Phase II data have provided some insight into the risk factors associated with the appearance of ARIA-E [3]. The most significant risk factor was dose of bapineuzumab, with 11 out of 12 cases occurring in the 1 or 2mg/kg dose groups. The Phase III bapineuzumab non-carrier study terminated the highest dose arm due to the number of ARIA-E cases observed in this dose cohort.
The presence of Apolipoprotein E ε4 allele was also found to be a significant risk factor for the development of ARIA-E, with 6 of 18 (33%) ε4/4 homozygotes; 4 of 56 (7.1%) x/ε4 heterozygotes; and only 2 of 47 (4.3%) non-carriers developing ARIA-E in the Phase II double-blind study. These findings led to the plans for separate ε4 Carrier and Non-Carrier Phase III protocols with different dose levels of bapineuzumab. Preliminary data from ARIA-E seen in trials with other amyloid-modifying therapies also suggest that ApoE ε4 carrier status may be a risk factor [16]. Similarly, the preliminary reports of the rare cases of ARIA detected in AD patients being screened for clinical trials have been in ApoE ε4 carriers, suggesting vascular A-β pathology, such as CAA, as a common underlying mechanism [14]
ARIA-H: MRI findings thought to represent hemosiderin deposits and microhemorrhages
Even the first reports of the ARIA cases raised the possibility of a relationship between FLAIR abnormalities thought to represent ARIA-E and the appearance of alterations on long echo time gradient refocused echo (T2*-GRE) sequences, thought to represent hemosiderin deposits, including microhemorrhages (mH) and superficial siderosis[1]. mH typically manifests as a focal, round, very low intensity (relative to adjacent brain) lesion in the brain parenchyma, detected on an appropriately weighted (T2 or T2*) MRI sequence, such as gradient refocused echo (GRE) sequences. Additional susceptibility weighting imaging (SWI) may be imparted by post processing to improve mH visualization. mH are small deposits of iron in tissue in the form of hemosiderin and are felt to represent residua of a small leakage of blood from a vessel into adjacent tissue parenchyma. Size criteria have been recommended, with mH defined by a cut off of ≤ 10 mm diameter in some and ≤ 5 mm diameter in other studies. However, employing size criteria to define mH is problematic without specifying the technical features of image acquisition linked to the size criteria because the apparent size of the low intensity lesion depends on features of image acquisition as outlined below[17]. In addition, the apparent size of a mH on MRI is generally greater than the size of the histologically defined area of hemosiderin deposited in tissue. This is a well-established phenomenon in MRI referred to as the “blooming effect” and is due to the fact that the microscopic field gradient causing signal loss extends spatially beyond the histologically defined hemosiderin deposit.
Curvilinear low intensities on an appropriately weighted (T2 or T2*) sequence that lie adjacent to the surface of brain are referred to as superficial siderosis. Similar to mH, siderosis is attributed to deposition of iron in the form of hemosiderin and is felt to represent residua of a leakage of blood from a vessel into the adjacent subarachnoid space or the peri adventitial compartment (as opposed to leakage into parenchyma to form a mH) [18].
Technical issues of image acquisition for ARIA-H
The conspicuity of mH and superficial siderosis can be enhanced or diminished by specific attributes of the image acquisition. MRI sequences that enhance signal loss due to micro gradients in tissue are generally used for detection of mH and superficial sidersosis. Two general approaches have been used, T2*-weighted gradient echo sequences (GRE) and SWI:
T2* gradient echo sequences (GRE)
T2 represents the loss of signal due to the intrinsic dephasing of spins moving randomly in the local tissue environment. T2′ represents signal loss due to the dephasing of spins that is attributable to eddy currents, macroscopic susceptibility effects or to focal microscopic magnetic susceptibility effects that would result from a mH or other sources of iron or mineralization. T2* represents the sum of T2 and T2′ effects. T2 weighted sequences employ a spin echo while T2* employ a gradient recalled echo. T2* GRE images are gradient echo sequences with a long TE, which enhances spin dephasing and hence focal loss of signal in the immediate vicinity of a mH. T2* GRE sequences can be executed in either 2D or 3D mode. An advantage of 3D is generally thinner slices and hence reduced partial volume averaging effects [19]. A major disadvantage however is that manufacturer available product 3D T2* GRE sequences are a purchasable option that is not available on all MRI systems. The majority of MRI systems in a typical clinical trial based on community recruitment centers would not have access to this technology at this time.
Susceptibility weighted imaging (SWI)
SWI is essentially a T2* gradient echo sequence that has added susceptibility weighting. This is accomplished by forming both a magnitude and a phase image, enhancing the phase image, and then multiplying the magnitude image by the enhanced phase image [20]. SWI was originally developed as a method to improve visualization of cortical veins but is also a more sensitive technique for detection of mH than T2* GRE images [21] (see Figure 4 below)
Figure 4. Conspicuity Depends on Technique. SWI vs T2* GRE.
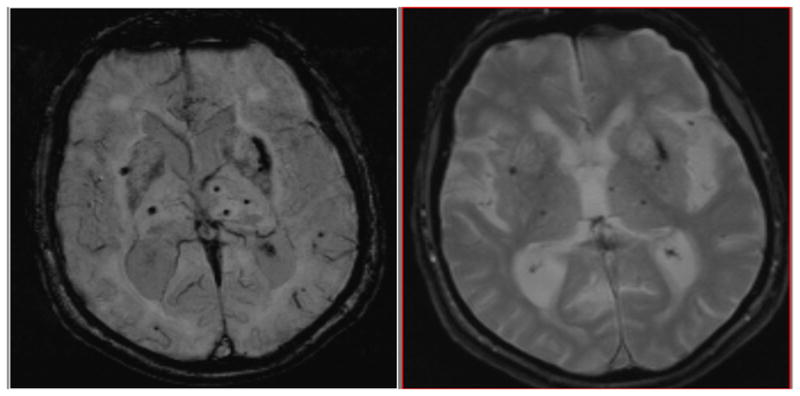
SWI image (left) compared to T2* image of same patient on same day on the right. Note, there are 4 more MB detected on the SWI image
The conspicuity of mH and hence sensitivity to detection is also increased by higher field strength[22], longer TE, lower read out bandwidth, and asymmetric centering of the echo with respect to the read out gradient. Small voxel sizes (i.e. higher resolution) will decrease partial volume averaging of small mH thus increasing conspicuity; however, all else being equal, smaller voxels produce a proportional reduction in image signal to noise (SNR). Low SNR images incur a penalty in diagnostic accuracy. The image geometry parameter that varies most from protocol to protocol in determining voxel size is slice thickness; in-plane resolution typically is not greatly different across different imaging protocols. Thinner slices increase resolution but decrease SNR and all else being equal; systems with superior SNR performance will have greater diagnostic sensitivity. The two major hardware features that improve SNR are field strength (SNR scales linearly with field strength), and receiver RF coil type (multi channel arrays have greater SNR than single channel volume coils).
Ascertainment
To our knowledge, reliable automated algorithms do not exist for identification of mH or superficial siderosis from medical images. Ascertainment by visual reading of scans by trained experts is the only way, but counting is not as exact as desired since it is dependant on several things: the level of training of a reader; where a particular reader falls on the Receiver Operating Characteristic curve (i.e. is he/she an under caller or an over caller); features of image acquisition discussed above; artifacts in the images themselves. Despite this, inter-and intraobserver agreement has been satisfactory with reported kappas for inter observer agreement ranging from .33 to .78 [17]
Common interpretative difficulties are: (1) motion artifacts (2) bulk susceptibility effects that produce regional signal loss (e.g. skull base, sinuses, and metallic foreign bodies like dentures), (3) partial volume effects, (4) distinguishing true mH from vessel flow voids which also appear as punctuate low intensity areas when the vessel transects the imaging plane of section perpendicularly, (5) distinguishing true mH from physiological mineralization in the basal ganglia. (6) mH number, for example the tendency exists to exclude a dubious mH in a patient without other mHs, in contrast to a similar lesion in the context of many mHs that will tend to receive the benefit of the doubt. On the other end of the spectrum, when mHs are very numerous difficulties with counting may arise as well, especially when lesions become confluent. Each of these may contribute to false positive or negative diagnostic readings.
Many mH cannot be ruled in or out with absolute confidence on a single scan. In some individuals, areas of the brain exist where mH can never be detected, for example bulk susceptibility artifact in areas next to paransal sinuses or at the base of the brain in subjects with dentures, and it can sometimes be difficult to differentiate mH from vascular structures in a single plane (see Figure 3). There are instances when artifacts on a baseline MRI scan preclude identification of a mH that becomes apparent only when (side-by-side) comparisons are made to follow-up MRI exams
Figure 3.
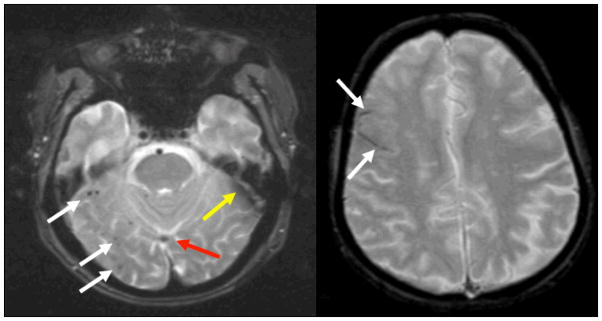
Microhemorrhage and superficial siderosis
Left: White arrows indicate multiple 1–3mm dark foci in the right inferior temporal and occipital lobes, typical of the appearance of mH. Red arrow indicates inferior sagittal sinus, and yellow arrow indicates susceptibility artifact, as vascular structures and artifacts can sometimes mimic the appearance of mH and siderosis. Right: White arrows indicate curvilinear dark sulci in the right frontal lobe, typical of the appearance of superficial siderosis. Both images were acquired at 1.5T with a 2D long TE (30ms) GRE sequence.
Etiology and clinical significance of mH
mH are generally attributed to one of two etiologies: small vessel angiopathy and CAA. In the stroke literature mH, attributed to small vessel angiopathy, increase in prevalence with age and vascular risk factors, especially hypertension [23]. mH also occurs in patients who undergo cardiac by-pass and are attributed to micro emboli [24]. In AD, mH and superficial siderosis are attributed to leakage of blood from CAA vessels [25]. CAA is felt to weaken the vessel wall, increasing the risk of micro leaks of blood into adjacent brain, forming mH. Some data suggests that the locations of mH are related to etiology, with lobar mH more often due to CAA while deep central grey and brain stem mH are more often due to hypertensive angiopathy [26].
mH is, in part, an age-related phenomenon that does occur even in neurologically healthy individuals (see Table below). Prevalence increases with age [19, 27, 28]. Additionally, a greater number of mH at baseline confers greater risk of subsequent incident mH [29]. The prevalence of mH is significantly increased in elderly individuals with cardiovascular risk factors and/or evidence of a previous cerebrovascular event [30]. The incidence of new mHs in a longitudinal study of ischemic stroke patients over a mean 5.5 year follow-up was 23%. In patients with cerebrovascular disease, the presence of mH has been linked to worse cognitive outcomes; however it is uncertain whether mH themselves cause impairment or are simply a marker of cerebrovascular disease [31, 32]. Indeed, although the point prevalence of mH in a healthy (no cerebrovascular disease) elderly population is ~ 6%, this value rises to ~ 50–80% in elderly individuals with cerebrovascular disease [33]. Thus, in a patient population with a high prevalence of cardiovascular risk factors, an association with a significant proportion of individuals with mH would be expected. In the setting of AD and presumed CAA, it is not established that mH themselves cause neurological symptoms [34]. A recent study from a memory disorders clinic noted that 12 % of these patients showed incident mHs during an average follow-up of 1.9 years [30]with no clinically apparent symptoms attributable to these incident mHs. However, mHs are felt to be an indicator of CAA and hence identify individuals at risk of a more serious complication of CAA – lobar hemorrhage. In individuals who present with an initial lobar hemorrhage, the number of mH increases risk of a subsequent lobar hemorrhage[35, 36]. The prevalence of superficial siderosis is much less common than mH (only 0.7% in the Rotterdam population based study), and also is felt to be an indicator of CAA [18, 37].
Association of ARIA with pre-existing white matter disease or other abnormalities
The cross-sectional association between mH and other MRI markers of small-vessel disease has been repeatedly reported. For example, one cross-sectional study of mild-moderate probable AD subjects, compared to normal controls, reported that patients with mH had higher white matter disease scores using a standardized rating scale (the Age-Related White Matter Changes (ARWMC) score [48], particularly in the frontal and parietal-occipital regions [34]. Also, the number of mH correlated significantly with the ARWMC score, particularly in the parieto-occipital regions of the AD subjects, and in the occipital region of controls [34].
Confluent periventricular white matter hyperintensities may relate to leakage from the deep medullary veins, due to venous collagenosis, a veno-occlusive disease of aging, exacerbated by vascular risk factors [49]. This may interfere with cerebral interstitial fluid circulation and drainage of protein solutes from the brain, and could encourage amyloid deposition in the perivascular spaces of the penetrating arterioles of the cerebral cortex. Clearance of amyloid from the brain may thereby be reduced, possibly exacerbating amyloid angiopathy and amyloid deposition[50]. Further studies are required to better understand this apparent association between white matter disease, mH’s and amyloid clearance.
Moreover, a large prospective population-based study associated mHs in deep locations (basal ganglia, thalami, and infratentorial) with arterial hypertension, white matter hyperintensities and lacunar infarcts [19] This is in line with the cross-sectional and temporal relation reported between new nonlobar mHs and progression of white matter hyperintensities and lacunes. In the paper by Goos et al (2010) [30], no association was found between lobar incident mHs and progression of white matter hyperintensities. In line with these findings, a former longitudinal study in patients with CAA did not find a relation between incident lobar mHs and progression of WmH [51]. These findings might further support the notion that etiology of mHs may differ according to mH location, with deep mH caused by hypertensive vasculopathy, more closely associated with vascular risk factors and other markers of small-vessel disease and lobar mHs caused by CAA and potentially related to APOE ε4 allelic status.
Association between components of ARIA
There are very limited data available to date regarding the relationship between mH and/or hemosiderosis detected on FLAIR images (ARIA-H) and “vasogenic edema” and/or sulcal effusions/exudates detected on GRE/T2* images (ARIA-E). Preliminary analyses of the Phase II Bapinezumab trial initially noted baseline mH as a risk factor for developing ARIA-E (Baseline mH, 6/17 (35%) vs. No baseline mH, 6/107 (6%))[3]. This finding may be confounded by the overlapping risk due to ApoE ε4 carrier status, as all of the ARIA-E cases with baseline mH were also ε4 carriers.
Similarly, there are limited publicly available data on incident mH in the setting of ARIA-E associated with amyloid-modifying therapies. One out of 3 VE cases in Phase I bapineuzumab [1] and four out of 12 VE cases in one Phase II (201- double blind) were reported to have incident mH [2, 3]. It also appears that ARIA-H can occur in some regions with ARIA-E and not others (see Figure 5). It will be important to study the relationship between these two phenomena in the setting of amyloid-lowering clinical trials, and in particular, to understand whether these entities are related to any change in clinical outcome or response to therapy.
Figure 5.

Relationship between ARIA-E and ARIA-H. Left: FLAIR image demonstrating ARIA-E with increased signal in white and gray matter and sulcal effacement in right frontal and parietal regions. Right: GRE image showing mH in right parietal region only.
Common pathophysiological processes underlying ARIA
The limited findings reported to date suggest several potential pathophysiological mechanisms that might underlie and tie together the various components of ARIA. The relationship to dose level in the bapineuzumab studies suggests that ARIA may be related to increased clearance of parenchymal plaque with transient increase in vascular amyloid. This hypothesis is supported by the published autopsy results from the AN-1792 (active immunization) trial [52, 53]. It remains unclear whether rapid movement of amyloid from parenchymal plaques into perivascular space might result in a “drainage back up” leading to excess fluid shifts. It is also possible that movement of amyloid into cerebral vessel walls might result in increased vascular friability and increased permeability. This mechanism might also relate to increased incident mH, if the vessel wall integrity is sufficiently impaired to permit small amounts of red blood cell passage.
Indeed the various FLAIR and T2*-GRE abnormalities observed in ARIA might be conceptualized as related to altered vascular permeability according to the location of the vessels and the nature of the material allowed to leak through the vessels (see Table 2 below).
Table 2.
Categorization of ARIA components
| Primary Imaging Sequence | Location of Increased Vascular Permeability | |||
|---|---|---|---|---|
| Parenchyma | Leptomeninges | |||
| Nature of leakage products | Proteinaceous fluid | FLAIR for ARIA-E | “Vasogenic edema” | Sulcal Effusion/Exudate |
| Heme products | T2* GRE/SWI for ARIA-H | Microhemorrhage | Superficial Hemosiderosis | |
The increased risk of ARIA in ApoE ε4 carriers in the bapineuzumab trials and the reports of spontaneous ARIA in CAA are also suggestive of vascular amyloid burden as a common pathophysiologic mechanism underlying these phenomena [8, 9, 54]. ApoE ε4 carriers are known to have higher vascular amyloid burden than non-carriers [15, 55]. PET amyloid imaging studies in patients with CAA have suggested that mHs are more likely to occur in regions with high amyloid burden as assessed by 11C-PiB [56, 57].
It is possible that direct removal of amyloid from the vessel wall would be associated with compromise in the vascular integrity. Alternatively, there may be amyloid related endothelial cell dysfunction resulting in increased vascular permeability, which might explain the similarity to increased permeability as described above, and as seen in PRES. It is also possible that there is a focal inflammatory component that would result in both ARIA-E and ARIA-H, as suggested by the pathology reports from patients with CAA. The cerebrospinal fluid results from a small number of participants identified with ARIA in the Phase II bapineuzumab studies are not consistent with a widespread inflammatory process. Normal CSF has also been reported in inflammatory CAA, and it is possible that focal amyloid-related vascular inflammation may play a role in some cases of ARIA. Thus far, it does not appear that ARIA cases show evidence of meningo-encephalitis, as was reported the early trials of active immunization with AN-1792 [58]. It also remains unknown whether different forms of immunotherapy or specific antibodies are more or less likely to be associated with ARIA [59].
As there are very limited data regarding ARIA in non-immunotherapeutic approaches for lowering A-β, it is difficult to speculate on more general additional mechanisms that might be associated with ARIA. Preliminary reports of ARIA occurrence in therapeutic strategies aimed at decreasing production of specific A-β peptides suggest that decreasing A-β1–42 or altering the ratio of various A-β species might change the dynamics of amyloid production and clearance, resulting in ARIA through similar mechanisms discussed above [14, 60]. The preliminary reports of spontaneous ARIA in AD patients detected at screening or occurring rarely during treatment with placebo, suggest that ARIA may represent the byproduct of the natural A-β clearance processes in the aging brain [14]. Similar to the CAA cases described in the literature, it is likely that specific genetic factors, such as the presence of ApoE ε4 alleles, influence the likelihood of spontaneous ARIA occurrence. Perhaps the common link with treatment associated ARIA across several therapeutic approaches and the rare spontaneous ARIA cases may be that we are tipping the balance of “Mother Nature’s” clearance mechanisms, but additional data are clearly needed to elucidate the underlying pathophysiology.
In order to further elucidate the potential pathophysiological mechanisms underlying the appearance of ARIA in both amyloid-modifying trials and in the natural history of AD, the working group also researched the limited literature on the histopathology of various aspects of ARIA and potentially relevant conditions.
Histopathological correlates of ARIA
To date, no neuropathologic descriptions of ARIA-E have been reported, perhaps due to the transient nature of ARIA-E. Our working understanding of the pathology and pathophysiology underlying this condition is therefore based on extrapolation from conditions with similar clinical and neuroimaging features.
As described above, ARIA shares some features with posterior reversible encephalopathy syndrome (PRES) disorders, which are also characterized by subacute clinical symptoms and reversible T2 hyperintensities [61, 62]. Neuropathologic descriptions of PRES [63, 64] are sparse and somewhat difficult to interpret because of the heterogeneity of the underlying processes that can trigger PRES, including hypertension, pre-eclampsia, and medications. From the limited available data, PRES-associated T2 hyperintensities appear to be generated by rarefaction and vacuolation of the white matter, without reported tissue infarction.
CAA-related inflammation, a second condition which shares some of the neuroimaging features of ARIA (see Figure 6) similarities to VE[6, 8], as well as a shared association with apolipoprotein E ε4[2, 9]) demonstrates similar white matter pathology to that reported in PRES. CAA-associated inflammation appears to be driven by a spontaneous inflammatory response to the vascular amyloid deposits, with accumulations of leukocytes surrounding amyloid-laden vessel segments and multinucleated giant cells containing amyloid-immunoreactive material. White matter rarefaction is again the likely neuropathologic correlate of the observed T2 hyperintensities.[6] Following resolution of CAA-related inflammation, the underlying inflammatory pathology also appears to reverse.[6, 65]
Figure 6.

Spontaneous CAA-related inflammation. MR images show VE-like appearance (left) with resolution following course of corticosteroids (center). Pathology image (right) shows white matter rarefaction without necrosis.
The cases of ARIA in amyloid-modifying therapies do not appear to share the clinical or CSF findings of the meningoencephalitis reaction reported in clinical trial participants receiving active vaccination with the AN1792 formulation of Aβ42[58]; however, there are limited histopathological reports that may be relevant to understanding the pathophysiology. The post-mortem examination of two meningoencephalitis cases demonstrated advanced CAA,[52, 66] suggesting that active amyloid immunotherapy might trigger a response similar to that observed in spontaneous CAA-related inflammation. Examination of the white matter in the meningoencephalitis cases again disclosed vacuolation and rarefaction without infarction.[52]
There is a much more extensive histopathology literature, regarding ARIA-H. Cerebral mH are focal deposits of hemosiderin in the brain, typically associated with localized cell loss and gliosis. The hemosiderin can be found in macrophages, astrocytes and microglia. While most lesions are imaged or observed pathologically in the remote state (that is, having occurred in the past), acute lesions are occasionally found, with extravasation of red blood cells and associated reactive changes.
Cerebral mH are typically seen in association with diseases of the small arteries and arterioles, in particular hypertensive vasculopathy (often in the white matter) and CAA (restricted to the grey matter) [23, 67–69]. A recent study [57] found that among patients with probable CAA, cerebral mH are associated with increased local amyloid deposits as measured by Pittsburgh Compound B (PiB), a finding that highlights the spatial association between small vessel pathology and mH location.
Two features regarding the neuropathology of cerebral mH are worth highlighting. First, the lesions are indeed quite small, generally on the order of 1 to 2mm in diameter [69] (“blooming” to a larger size on T2*-weighted MRI sequences as described in section 1). Although there is associated gliosis and some neuronal loss at mH loci (Figure 7), there is little or no evident abnormality in the surrounding brain tissue.
Figure 7.

Cerebral microhemorrhages in association with advanced CAA
A second point is that aside from brains with florid vascular disease, mH are a rare neuropathologic finding. Systematic sampling of brain sections from both non-demented or demented individuals [70, 71] identifies many clinically silent ischemic microinfarctions, but only rare mH. The apparent discrepancy between the high prevalence of mH reported in MRI-based studies versus their low prevalence in neuropathologic series likely reflects the ability of MRI (but not neuropathologic examination) to sample the entire brain with high sensitivity. Based on the well characterized mathematical relationship between the abundance and size of mH versus the neuropathological “slicing” required to accurately sample them, [72] the standard sectioning and sampling of the brain at autopsy is predicted to be insensitive to the presence of a small number of lesions the size of mH. Thus the 1, 2, or 3 mH detected by MRI in a given individual may indeed represent close to that individual’s total lesion burden.
As evident from the above discussion, CAA may be a neuropathologic common denominator for both the treatment-related ARIA and the rare spontaneous cases reported in screening cohorts [4]. CAA—present at the time of study entry or perhaps exacerbated by amyloid immunotherapy[53, 73]—might thus be the underlying factor predisposing individuals to adverse treatment responses. It might be wrong to conclude, however, that anti-amyloid immunotherapy is necessarily deleterious for CAA. In a post-mortem of study of nine patients immunized with AN1792, two of the longest treated participants demonstrated essentially no CAA. [53] This and data from anti-amyloid antibody-treated transgenic mice [74, 75] suggest that amyloid can be successfully cleared from vessels without triggering substantial bleeding or vascular dysfunction. If achievable, clearance of CAA might reduce an individual’s risk both for future adverse treatment responses as well as for other manifestations of CAA such as intracerebral hemorrhage.
Insights from animal models
Given the paucity of human histopathological data in ARIA, the workgroup also explored relevant reports from animal models of AD and CAA, particularly those exposed to amyloid-modifying therapies. It is now roughly 15 years since the first successful transgenic mouse models of AD were developed; over the ensuing interval there have been additional models generated, yet there remain a relatively small number of models that are widely used by investigators [76, Davis, 2004 #2299, 77–82]. The development of these models drew on the identification of mutations in APP that are associated with familial forms of AD –those adjacent to the BACE cleavage site (APP-sw), those which are just past the γ-secretase site, those internal to the Aβ peptide sequence, or elaborate combinations of these mutations. Transgenes were expressed under the control of active, heterologous promoters (PRNP, PDGF-B chain, Thy1) at high levels in order to accelerate the accumulation of Aβ in the brains of animals [76, 78, 82, 83].
These models were then advanced by the inclusion of mutated forms of PS1 in order to preferentially shift the Aβ towards the 42 amino acid form; these combinations were generated either by crossing individual strains or by the co-injection of constructs during the derivation of the mouse lines [77, 79, 80]. The end-point for determination of an “acceptable” or “useful” mouse model was the presence of Aβ deposition in the brain, often accompanied by vascular wall deposits in the form of cerebral amyloid angiopathy. Many of these models were subsequently shown to have some of the other alterations associated with affected brain regions in AD, including gliosis and microglial activation.
Importantly, neither significant neuronal loss on par with the changes observed in AD nor the presence of tangles followed the overexpression of the APP and PS1 transgenes even in the face of significant Aβ deposition. Attempts have been made by the use of additional transgene constructs to provide a model in which both plaques and tangles appear, but in order to develop tangles it was necessary to use a mutant form of tau that is associated with frontotemporal lobar degeneration but not with AD [81].
It remains important to recognize that these mouse models of Aβ deposition capture only some aspects of the pathologic changes observed in brains of individuals with AD. In addition to being incomplete models, it is also likely that they do not replicate all of the anatomic findings observed in AD.
Microhemorrhages in animal models of treatment of AD/CAA
In addition to being useful models of AD for experimental analysis of disease mechanism and pathogenesis, these transgenic mice provide a fruitful testing arena for potential therapeutic interventions to slow, reverse or prevent the pathologic changes of AD. Again, it is necessary to emphasize that these models were engineered to develop elevated brain Aβ levels which progresses to plaque formation. Additionally, the models have gradually been enhanced in order to accelerate the process, as a major determining factor of the cost of animal experiments is the age of the animals under study (i.e., a study which can be performed with 4 month old mice will have roughly 25% the animal housing costs of a study which requires 16 month old mice).
There is no widely agreed upon individual animal model that consistently develops mH, and little evidence for consistent or prominent edema, either at baseline or after various therapeutic challenges. Rather, there has been a suggestion that, in mouse models, there are effects of mode of delivery of immunotherapeutics that may influence the development of mH, as well as an apparent dependence on the presence of CAA. There is not clear evidence from animal studies that there is an increased incidence of mH with non-immunologic approaches to alterations of brain Aβ levels.
Although not a feature of the earliest studies demonstrating the potential of anti-Aβ antibody based approaches to clearance of Aβ from the brains of transgenic mice [84], there have been subsequent studies (using a variety of mouse models, types of immunization, distinct antibodies/epitopes, duration and age of animals tests) in which mH were detected [5, 73, 75, 85–90]. As summarized in a series of recent reviews [91–93], these studies were mostly focused on clearance of existing deposits (which included both parenchymal and vascular Aβ). Because mH are associated, by definition, with an alteration in vascular integrity, there has been specific interest in the changes in CAA burden which follow the treatment and accompany the development of mH. It is not possible to determine, a priori, whether an increase in CAA burden or a decrease in CAA burden would be more likely to be responsible for mH – or even if the pre-existing Aβ burden on a vessel-by-vessel basis might play a role. Some of the studies which identified increased mH also found an increase in the vascular CAA burden, suggesting that increased vascular wall injury associated with ‘mobilization’ of parenchymal Aβ is at least partially responsible [85–87]. Another study, which compared intraventricular vs systemic antibody delivery observed that the approach associated with decreased CAA burden as well as parenchymal Aβ clearance (intraventricular) was also associated with decreased mH incidence [94]. Despite the evidence from the studies cited above regarding the association of mH with immunologically-based therapeutic approaches, it should be stated that these are by no means an absolute finding across models, types of immunization approaches, epitopes, etc (reviewed in [91–93].
When considering the translation of these findings on mH associated with therapeutic interventions that increase CAA Aβ burden while decreasing parenchymal burden to human subjects, it is necessary to recognize the fact that there is far greater vessel-to-vessel Aβ vascular burden heterogeneity in humans with AD than there is in mouse models. Given the current concept that Aβ departs the central nervous system along perivascular spaces of arterioles [95–100], in a human with patchy CAA there will be a significant number of perivascular pathways along which any deposited Aβ would represent an initiation event for local CAA – while in a uniformly and heavily affected mouse model, a comparable level of deposition might well be shifting the vessel over the threshhold from moderate to severe CAA, and thus increasing the risk of local hemorrhage. It is disappointing that there has not been a direct association between the level of local vascular Aβ burden and the risk of mH in these mouse models of immunotherapies, as such studies might have provided relevant answers to the question of “how much CAA can you have before clearing Aβ along perivascular spaces becomes a riskier proposition?”
In a recent paper, imaging was used to track the appearance of mH in Tg2576 mice treated with a series of antibodies in a passive immunotherapy protocol for 12 or 18 weeks [101]. This study found that the antibodies used did increase the number of MR-detected mH and that these correlated well with histologic observations at the end point of the experiment. Given the associated interest in ARIA-E, it is disappointing that there is no information regarding this other potential complicating feature of anti-Aβ therapy in the paper. Another recent paper described the combination of passive immunization and adaptive transfer of activated T cells [102]. The combination produced hemorrhage in animals with CAA, but neither alone did. This experiments highlights the possible complex interactions in immune responses that determine whether – and when – mH might occur in the setting of anti-amyloid therapies.
In humans, the presence of lobar predominant mH is a strong predictor of the presence of CAA. In the mouse models, the presence and timing of CAA is a well-characterized component of each specific model, although it varies across models [103–105]. Moreover, even in the animal models, CAA is not always tightly aligned with mH after therapeutics (which to date is focused almost exclusively on immunotherapy). The variability of detection of mHs in animal models may be, in part, because the condition does not occur uniformly in all animals, even within a specific age range of a specific transgenic background, and the likelihood of detecting mHs can vary substantially from experiment to experiment. Additionally, it may be influenced by the severity of CAA, which is more variable than plaque burden in some animal models.
Difficulty in modeling ARIA-E in transgenic animals
Other aspects of ARIA, in particular the phenomena known as “vasogenic edema” have not been reported in animal models: however, it is unclear whether systematic MR surveillance with sequences sensitive to ARIA-E has been performed longitudinally in a large number of animal models treated with amyloid-modifying therapies.
As mentioned above, cerebral edema is the increase in water content of the extravascular but intraparenchymal compartment of the brain. It includes water that is present in the extracellular space as well as within cellular components of the brain. In general, cerebral edema is divided into two types: cytotoxic (associated with altered handling of water and ion transport across cellular membranes of neurons and glia) and vasogenic (associated with alterations in the blood-brain barrier [BBB] either through direct actions on cerebrovascular endothelial cells or as mediated through the neurovascular unit).
In many disease processes, the etiology of cerebral edema is a combination of these cytotoxic and vasogenic edema. Mouse models of some human diseases, particularly the well-studied models of hypoxic/ischemic injury and brain tumors, include edema as a component of the pathologic process. This appropriately mimics the situation in human disease processes, by involving both vasogenic and cytotoxic edema. Vasogenic edema, as it is dependent on the integrity of the BBB, may be measured through the capacity of the cerebral vasculature to prevent the leakage into the brain of an agent typically retained within the intravascular compartment. Overall measurement of marked edema in model systems can also be performed through MR imaging methods, akin to clinically relevant studies in humans. Histologic methods are not well-suited to the detection and quantification of edema, particularly when focal and towards the less severe end of the disease spectrum.
In the available imaging data from therapeutic trials for AD, there was evidence of transient imaging abnormalities in the white matter of some trial participants with the characteristics of ARIA-E in the absence of cytotoxic features. Despite this observation, there has been absence of evidence from studies of therapeutic candidates in mouse models of Aβ deposition in the forms of plaques and CAA for the development of edema (either cytotoxic or vasogenic). In general, these studies have been end-point studies that would not be well-suited to detect transient changes which might occur in the early stages of treatment. Additionally, these studies have used a variety of end-point measures, such as behavioral assays, histopathologic measurements (including plaque burden, inflammatory reactions, mH), and biochemical determination of Aβ in brain and CSF.
At present, no well established protocols or specific models are available to consistently model Aβ therapy induced ARIA-E. ARIA-H is observed to various extents in different models and with different therapeutic interventions. The emphasis of investigators to date has been on the validation of specific therapeutic approaches (alteration of Aβ generation, immunologic and non-immunologic clearance of Aβ, etc). There remains an opportunity for development and characterization of model systems in which therapeutic interventions aimed at modifying Aβ levels in the brain can be consistently associated with relevant potential side effects, particularly those with activation of the immune system.
Challenges to development of an animal model for ARIA
Could one develop a standardized animal model of ARIA, ideally a model that would exhibit all components of ARIA observed in AD patients? There are certain barriers: for ARIA-E, there is uncertainty if it exists in animal models at all. For example, there are no publications describing this, and unpublished studies of which the Workgroup is aware have been regularly negative. The absence of ARIA-E in mice might reflect a difference in animal model and human pathobiology, or technical difficulties in imaging timing, sequence optimization, etc but to date this component of ARIA is not described in animal models.
Although we endorse the idea of careful screening for mH complications with any anti-amyloid therapy, certain barriers exist in this context as well. There is uncertainty about the predictive value of mH in a mouse regarding likelihood of developing ARIA-E in patients (or even ARIA-H in patients). Barriers to establishing a common and uniform model of mH are also substantial – access to specific models are not universal, the specific model appropriate for each therapeutic approach may differ (eg the presence or absence of a PS1 mutation that accelerates pathology can impact age of animals; the specific APP mutation can affect the amount of CAA) and phenotypic drift within models. These factors will also potentially significantly confound the interpretation of studies undertaken by sponsors with different experimental models as they follow the FDA advice to conduct non-clinical studies investigating the potential for induction of ARIA.
Monitoring for ARIA in clinical trials
The Workgroup concluded that there is very limited information thus far regarding ARIA in the natural setting or publicly available information from ongoing clinical trial programs to make conclusive recommendations for clinical trial policies. In particular, there is limited data regarding the relationship between ARIA-H and ARIA-E. Additional information regarding whether the presence of baseline ARIA-H as a potential risk factor for ARIA-E, and ARIA-E as a risk factor for incident ARIA-H is needed to provide guidance for the conduct of these studies. Most importantly, we need information about whether specific components of ARIA have an impact on clinical course and response to treatment.
The limited data available thus far suggest that there may be common risk factors for the components of ARIA, in particular, ApoE genotype. This genetic risk factor, as well as limited reports from patients with CAA, suggest that vascular amyloid may be a pathophysiological mechanism for ARIA, but additional investigation is needed. As the ApoE ε4 allele is present in a large proportion of AD patients, and some degree of vascular amyloid is present in nearly all AD patients, it is critically important to gather additional information on the relationship of genotype to the incidence of ARIA in the setting of amyloid-lowering therapy.
From the limited data available thus far, it appears that amyloid-modifying treatment can be continued with careful monitoring and possible dose adjustment after the occurrence of ARIA. At this point, it seems premature to preclude continued therapy in the setting of incident ARIA, as trial participants with ARIA are currently being redosed in ongoing trials, without clear evidence of untoward effects. If ARIA is related to successful amyloid clearance, it might introduce significant bias in ongoing trials to terminate all of these trial participants. It will be important to gather systematic data regarding the clinical course associated with ARIA in the setting of amyloid-modifying therapy. In addition, it is important to better understand the frequency of spontaneous ARIA, the risk factors associated with this phenomenon, and its clinical course. Given the evidence that incident mH are relatively frequent in the course of AD, and the likelihood that spontaneous mH share some common aspects of pathophysiology with CAA, it is likely that additional spontaneous cases of ARIA will be identified in ongoing research studies.
It will be critically important to continue to monitor for ARIA in ongoing studies, and especially to relate the imaging features of ARIA to the clinical course of these patients. To that end, the Workgroup developed a set of recommendations for the FDA and the industry to consider in the conduct of clinical trials.
Recommendations
The Workgroup recognizes important issues both in the technical aspects of MR image acquisition to detect ARIA, as well as the interpretation of MRI findings suggestive of ARIA. In turn, the recommendations that are included in this report provide guidance on how technical consistency and a uniform neuroradiological approach to ARIA might be accomplished. In addition, the Workgroup provides some recommendations regarding exclusion from participation in clinical trials based on baseline or incident ARIA and for ongoing monitoring during clinical trials of amyloid modifying therapies.
1. MRI protocols for detection of ARIA within amyloid therapy trials
MRI Protocol minimum standards
A protocol which can be implemented in a wide variety of settings of care is suggested, particularly one that can be undertaken in an average community based setting. It might optimally include:
Scanner Field Strength: While high field strength scanners are likely to have greater sensitivity, the use of 1.5 T scanners is endorsed as a minimum standard, recognizing that the availability of higher field units is limited to certain centers. The implementation of more sensitive MRI measures to detect ARIA-H needs to be balanced by the clinical importance of such findings.
Scan Sequences: 2D T2*GRE, to identify ARIA-H, are presently available on any scanner worldwide and recommended.
Slice thickness of 5 mm or less;
TE = 20ms or greater.
T2 FLAIR sequence for identification of ARIA-E
2. Frequency and Intensity of Scanning Protocols within Clinical Development
In Phase I through early Phase II, more frequent scanning to ascertain the rates of these abnormalities is warranted. As new amyloid lowering therapies are introduced into early clinical development, knowledge regarding the frequency and timing of ARIA occurrence is likely to be limited.
Discussions of the scanning requirements for Phase III should be actively undertaken based on Phase II results, given the significant burden to patients and cost implications of frequent scanning, Additional monitoring for ARIA in the setting of clinical symptoms may be more appropriate than a priori fixed frequencies given the transience of ARIA-E.
Pharmacodynamic effects are an important consideration in determining the optimal timing of MRI scanning. The timing of MR scans in relation to dosing should be considered for bolus IV administration (i.e. x number of weeks post-dosing) as well as the pharmacokinetic levels. However, duration of dosing (i.e., exposure) should also be considered.
Frequency of surveillance scans in amyloid lowering trials to detect ARIA-E. It is likely not feasible to monitor frequently enough to capture all ARIA-E. It is also not clear that “missing” transient ARIA-E that is clinically asymptomatic will result in any untoward effects. In addition to scheduled surveillance scans, scans should also be prompted by onset of symptom clusters suggestive of ARIA-E. Taken together, surveillance and triggered scans should adequately characterize the incidence and behavior of ARIA. It is important to consider how scans will be read and tracked in these drug development programs.
A short interval rescan provision should be built into each protocol to reassess subjects who develop ARIA during treatment, particularly if they are symptomatic.
3. Reading and Reporting Standards
a) Image interpretation/ascertainment
It should be recognized that variable levels of diagnostic certainty are inherent in any radiological diagnosis. ARIA may be definitely present or absent, but often uncertainty exists and allowance for this should be made in their ratings.
A radiological reading of a “possible” mH (ARIA-H) should not be an exclusionary criterion. Inclusion/exclusion should only be based on readings labeled definite mH. Guidance should also recognize that distinction between “possible” and “definite” may require serial MRI scans – i.e. that changes in diagnostic confidence will inevitably occur over the course of a series of scans, as is true in all diagnostic imaging. Change in categorization of a mH from “possible” to “definite” in the course of a series of scans that “move” a trial participant over an exclusionary threshold should not constitute a “retrospective” protocol violation.
ARIA-E should be interpreted both for severity as well as relevance to clinical symptoms. ARIA-E can be manifest as rather subtle alterations in MR signal, and can be missed. It is not clear, however, that missing very subtle asymptomatic ARIA will have any clinical consequence. Additionally, fulminant ARIA-E can be misidentified as other processes (subarachnoid hemorrhage, venous infarction). Thus it is important to consider how scans will be read and tracked in these drug development programs.
b) Procedures for reading and reporting
There are pros and cons to “central reading” vs. “local” reads. Central reading has the advantage of a small number of individuals with detailed knowledge of ARIA to monitor all scans, increasing both accuracy and inter-rater reliability. Most hospitals require a local read to be performed in any case for safety, and generally this can be accomplished quickly with rapid notification to the investigator.
If monitoring for ARIA is to be done with local reads, it will be important to educate both the local radiologists and the investigators about the MR appearance of ARIA, particularly the various MR manifestations associated with ARIA with multiple case examples. It may be useful to also provide a “central” resource for local radiologists to send possible ARIA cases for immediate consultation.
A detailed reporting form (with checkboxes) covering the spectrum of findings should be provided to the local radiologist that specifically asks about presence or absence of signal abnormalities consistent with ARIA-E (presence of increased signal on FLAIR sequences consistent with parenchymal edema or effusions in the leptomeninges or sulcal space) and/or ARIA-H (presence of mH or hemosiderosis).
It may also be useful to develop a more quantitative scoring or rating scale for ARIA, especially for use by central monitoring, to provide additional information about the potential relationship between ARIA-H and ARIA-E, as well as to clinical symptoms/outcome. This scoring system might include detailed information about anatomic location of ARIA-E and/or ARIA-H, and a severity index.
4. Thresholds for exclusion based on ARIA in clinical trials with amyloid lowering therapies
In response to the original FDA guidance on exclusion of individuals with evidence of ARIA-H at baseline or incidence of ARIA in trials, the Workgroup discussed potential thresholds for number of mH extensively. The Workgroup felt it was obviously important to protect clinical trial participants from potential adverse outcomes related to ARIA, but felt that it was also important not to be unnecessarily stringent in excluding participants with ARIA, based on the current literature and publicly available information. It was felt to be important to elucidate the effects of treatment in the general AD clinic population, where patients with evidence of ARIA-H at baseline would be likely to receive treatment with considerably less monitoring than is possible in a clinical trial. Given the frequency of mH and the incomplete knowledge of their clinical significance, an ascertainment of the risk of meaningful clinical complications during amyloid treatment is a recognized need. Stringent criteria would also limit the acquisition of knowledge needed to optimally manage AD patients with underlying CAA and/or hypertensive small vessel cerebrovascular disease. An additional consideration is that discontinuation of asymptomatic patients from ongoing treatment with incident ARIA would also preclude the evaluation of potential clinical benefits associated with vascular Aβ clearance in the AD population. Thus, given the importance of allowing the field to advance the development of amyloid-modifying treatments for AD, the Workgroup developed recommendations for exclusion criteria based on the recognition that there are limited data available to justify strict exclusions on the basis of baseline ARIA-H, and that current MR methods and clinical reading procedures are imperfect in the detection and tracking of small numbers of mH. It is likely that recommendations will continue to evolve as additional data from large clinical trial databases become available.
a) Exclusions for presence of baseline ARIA-H (mH or hemosiderosis)
There are only very limited data about the risks of amyloid-modifying treatment with amyloid-lowering therapy in patients with evidence of ARIA-H at baseline. It is recognized that substantial numbers of lobar mH likely reflect the presence and severity of CAA, raising diagnostic and therapeutic considerations. Current prevalence estimates in mild to moderate AD are that 80% of patients with mH will have less than or equal to 2 mH. Given the uncertainty of risk and concerns about CAA severity, the Workgroup supports the recommendation that the cutoff value of 4 mH be used for exclusion in trials of amyloid-modifying therapies for AD. This threshold would allow the potential for imaging measurement variability to be taken into account and reflect the uncertainty regarding the clinical relevance of small numbers of mH.
b) Exclusionary criteria for incident ARIA-H (based, in part, on using the recommended technical MR characteristics noted above)
Development of asymptomatic ARIA-H may result from Aβ clearance associated with amyloid lowering therapy. As well because ARIA-H may also occur within the natural history of AD, we recommend that the appearance of incident mH or hemosiderosis not automatically disqualify a patient from further treatment. Rather, we suggest that discontinuation of trial participants with incident ARIA-H be reserved for those in whom these MRI findings are associated with significant clinical symptoms or evidence of precipitous clinical decline. Until there are more available data, clinical consideration may also be given to discontinuing specific patients with a large number of incident mHs who are still asymptomatic. The Workgroup recommends that as more data becomes available, this recommendation would merit continued review.
Recommendations for further research into the mechanisms underlying ARIA
In parallel with close monitoring of patients in ongoing clinical trials of amyloid-modifying treatments, further research is clearly needed to elucidate the mechanisms underlying the phenomena observed in ARIA. In particular, it would be valuable to utilize animal models to determine whether amyloid-lowering therapies indeed is associated with increased vascular permeability, perhaps through fluorescent labeling plasma proteins of different sizes. Additional understanding of the genetic and age factors that favor vascular clearance of amyloid might also be achieved through transgenic breeding, perhaps crossing transgenics containing human isoforms of ApoE with APP/PS-1 mutants.
In human studies, the combination of PET amyloid imaging with frequent MRI monitoring in ApoE ε4 carriers receiving amyloid-lowering therapies may serve to clarify whether ARIA will occur preferentially in regions with high amyloid burden, and to demonstrate evidence of amyloid clearance proximate to ARIA. Most importantly, longitudinal natural history studies are needed to better understand the spontaneous occurrence of ARIA and the associated clinical course, as well as detailed analyses of the cognitive, behavioral, and functional outcomes of individuals who develop ARIA in the setting of amyloid treatment trials.
Table 1.
Microhemorrhage prevalence and incidence rates in referred clinic samples, epidemiology surveys, clinical trials
| Citation | Description of the population | Sample size | Baseline prevalence | Incidence | Technical features |
|---|---|---|---|---|---|
| Goos et al Neurol 2010 [30] | Memory clinic – includes demented and non-demented individuals | 254 | Overall 19% | Controls 11% MCI 21% AD 12% Overall 12% over average 1.9 yrs |
2D T2*GRE; 1.0T; 5mm skip 1mm slices; TE=22ms |
| Goos et al 2011 [38] | Memory clinic Comparing GRE versus SWI on the same 1.5 T MRI | 141 | Overall: 23% GRE 40% SWI AD 20% GRE 39% SWI |
2D T2*GRE; 1.5T; 5mm skip 1.5mm slices TE=25ms | 3D SWI; 1.5T; 2mm, skip 2mm slices TE=40ms |
| Cordonnier Neurol 2006 [39] | Memory clinic | 772 | VasD 65% AD 18% MCI 20% SubMm 10% Overall 17% |
2D T2*GRE; 1.0T; 5mm skip 1.5mm slices; TE=22ms | |
| Vernooij Neurol 2008 [19] | Population based sample | 1062 | 60–69, 17.8% 70–79, 31.3% >80yo, 38.3% |
3D T2*GRE; 1.5T; 1.6mm; Skip 0mm slices TE=31ms | |
| Jeerakathil et al Stroke 2004 [27] | Healthy community based sample (Framingham) | 472 | 4.7% | 2D T2*GRE; 1.0T; 5mm skip 0.5 slices; TE=26ms | |
| Pettersen et al Arch Neurol 2008 {Pettersen, 2008 #2342 | Memory Clinic | 105 | AD 29%; Healthy controls 12% | 2D T2*GRE; 1.5T; 6mm skip 2mm slices; TE=33ms | |
| Sveinbjornsdottir et al JNNP 2008 [28] | Healthy community based sample (AGES) | 1962 | 11.1% | 2D GRE-EPI; 1.5T; 3mm skip 0 slices; TE=50ms | |
| Werring et al Brain 2004 [32] | Suspected stroke or transient ischaemic attack | 214 | 13.6 | T2*; 1.5T; TE=40ms | |
| Lee SH, et al J Neurol Neurosurg Psych[40] | Hypertensive population | 129 | 55.8 | T2 GRE; 1.5T; 5 mm skip 2 mm slices; TE=15ms | |
| Yakushiji et al Stroke 2008 [41] | No history of neurological disorder | 518 | 6.8% | T2*GRE; 1.5T; 7mm skip 1.4 mm slices; TE=20ms | |
| Igase, et al Circ J 2009 [42] | Neurologically Healthy population | 377 | 5.6% | T2*GRE; 3.0 T mH definition < 5 mm. NOTE: 20/21 pts with mH had one mH with one pt having 3 mH | |
| Hanyu et al J Neurol 2003[43] | AD (mean MMSE 17.9); Controls without neurological deficits | 59 (AD) 55 (no neurolog. Deficit |
32.2% (AD) 7.3% (no neurolog. Deficit) |
T2*GRE; 1.5T; 5mm skip 1.5 mm slices; TE=26 ms | |
| Roob et al Neurol 1999 [44] | Population based sample | 280 | 6.4% | 2D T2*GRE; 1.5T; 5mm With 10% gap; TE=16–20ms | |
| Tsushima et al Neuroradiology 2002 [45] | Population based sample | 450 | 3.1% | 2D T2*GRE; 1.0T; 5 mm skip 2.5 mm slices; TE=30ms | |
| Bednar et al 2010 [46] | Mild-moderate AD (MMSE 16–26); Screen Data for Therapeutic trial | 231 | 21.6% | 2D T2*GRE; 1.5T; 5 mm skip 1 mm slices; TE=20ms | |
| Novartis Data on file | Mild AD (MMSE 20–26) patients recruited for a multinational active immunotherapy trial | 137 | Any mb: 38% >1 mb: 21% >2 mb: 12% |
N/A | 2D T2*GRE, 1.5T 2, TE=23, 5 mm slices, no gap |
| Poels et al 2010 [47]; Follow-up to Vernooij, et al Neurol 2008 [19] | Community- dwelling people aged 45 years and older | 3979 | Range from 6.5% in 45–50 yo to 35.7% in ≥ 80 yo (15.3% overall had at least 1 mH) | N/A | 3D T2*GRE; 1.5T; 1.6mm; Skip 0mm slices TE=31ms |
Footnotes
Disclosures:
Dr. Reisa A. Sperling has served as a site investigator for Avid, Bristol-Myers-Squibb, Elan, Janssen, Pfizer, and Wyeth, and a consultant to Avid (unpaid), Bayer, Bristol-Myers-Squibb, Elan, Eisai, Janssen, Pfizer, and Wyeth. Dr. Clifford R. Jack Jr. serves as a consultant for Janssen, Lilly, GE, Johnson and Johnson, Eisai, and Élan, and is an investigator in clinical trials sponsored by Pfizer, Allon and Baxter, Inc. Dr. Sandra E. Black has received contract research funds to the Cognitive Neurology and Stroke Research Units from Roche, GlaxoSmithKline, Novartis Pharmaceuticals, Myriad Pharmaceuticals, Pfizer, Sanofi-Aventis, Boehringer Ingelheim, Novo Nordisk, and AstraZeneca. In addition Dr. Black has received Speaker’s honoraria for CME from Janssen-Ortho, Novartis Pharmaceutical, Lundbeck, Pfizer, Eisai and Myriad Pharmaceuticals, and Honoraria for ad hoc consulting from Pfizer, Janssen-Ortho, Novartis Pharmaceuticals, Lundbeck, Myriad Pharmaceuticals, GlaxoSmithKline, Schering-Plough, Elan and Wyeth Pharmaceuticals, Bristol-Myers Squibb and Eisai. Dr Steven M. Greenberg serves as a consultant for Hoffman-La Roche, Janssen Alzheimer Immunotherapy, and Bristol-Myers Squibb Company, and has received honorarium from Medtronic and Pfizer. Dr. Bradley T. Hyman has consulted with several pharmaceutical and biotechnology companies: EMD Serrano, Janssen, Takeda, BMS, Neurophage, Pfizer, Quanterix, foldrx, Elan and Link. Dr. Philip Scheltens serves as a consultant to Roche AG, Novartis AG, Genentech, Danonen Research, Lundbeck, GE Healthcare, Avid. Drs. Martin M. Bednar and Rachel J. Schindler are employed by Pfizer. Dr. Ronald S. Black is a full time employee of Pfizer and owns stock in the company. Dr. H. Robert Brashear is employed by Johnson and Johnson. Dr. Michael Grundman has consulted for Janssen Alzheimer Immunotherapy, Johnson and Johnson, Elan Pharmaceuticals and Avid Radiopharmaceuticals. Dr. Eric R. Siemers is a full time employee and stockholder at Eli Lilly. Dr. Howard H. Feldman is a full time employee and holds stock with Bristol Myers Squibb. Drs. Maria C. Carrillo and William Thies are employees of the Alzheimer’s Association and report no conflicts.
References
- 1.Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010 Apr-Jun;24(2):198–203. doi: 10.1097/WAD.0b013e3181c53b00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009 Nov 18; doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling R, Salloway S, Fox N, Barackos J, Morris K, Francis G, et al., editors. Risk Factors and Clinical Course Associated with Vasogenic Edema in a Phase II Trial of Bapineuzumab. American Academy of Neurology; Seattle, Washington: 2009. [Google Scholar]
- 4.#XX1 RA.
- 5.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002 Nov 15;298(5597):1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 6.Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004 Feb;55(2):250–6. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- 7.Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, et al. Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain. 2005 Mar;128(Pt 3):500–15. doi: 10.1093/brain/awh379. [DOI] [PubMed] [Google Scholar]
- 8.Oh U, Gupta R, Krakauer JW, Khandji AG, Chin SS, Elkind MS. Reversible leukoencephalopathy associated with cerebral amyloid angiopathy. Neurology. 2004 Feb 10;62(3):494–7. doi: 10.1212/01.wnl.0000106951.94624.df. [DOI] [PubMed] [Google Scholar]
- 9.Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007 Apr 24;68(17):1411–6. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg SM, Parisi JE, Keegan BM. A 63-year-old man with headaches and behavioral deterioration. Neurology. 2007 Mar 6;68(10):782–7. doi: 10.1212/01.wnl.0000258985.31455.13. [DOI] [PubMed] [Google Scholar]
- 11.DiFrancesco JC, Brioschi M, Brighina L, Ruffmann C, Saracchi E, Costantino G, et al. Anti-Abeta autoantibodies in the CSF of a patient with CAA-related inflammation: a case report. Neurology. 2011 Mar 1;76(9):842–4. doi: 10.1212/WNL.0b013e31820e773c. [DOI] [PubMed] [Google Scholar]
- 12.Stott VL, Hurrell MA, Anderson TJ. Reversible posterior leukoencephalopathy syndrome: a misnomer reviewed. Intern Med J. 2005 Feb;35(2):83–90. doi: 10.1111/j.1445-5994.2004.00750.x. [DOI] [PubMed] [Google Scholar]
- 13.Tungkasaereerak C, Phanthumchinda K. Reversible posterior leukoencephalopathy syndrome: a retrospective study in King Chulalongkorn Memorial Hospital. J Med Assoc Thai. 2008 Mar;91(3):427–32. [PubMed] [Google Scholar]
- 14.XX2 R.
- 15.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995 Aug;38(2):254–9. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 16.Sperling RA, et al., editors. International Conference on Alzheimer’s Diseae. 2011. [Google Scholar]
- 17.Cordonnier C, Potter GM, Jackson CA, Doubal F, Keir S, Sudlow CL, et al. improving interrater agreement about brain microbleeds: development of the Brain Observer MicroBleed Scale (BOMBS) Stroke. 2009 Jan;40(1):94–9. doi: 10.1161/STROKEAHA.108.526996. [DOI] [PubMed] [Google Scholar]
- 18.Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010 Apr 27;74(17):1346–50. doi: 10.1212/WNL.0b013e3181dad605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008 Apr 1;70(14):1208–14. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 20.Haacke EM, Xu Y, Cheng YC, Reichenbach JR. Susceptibility weighted imaging (SWI) Magn Reson Med. 2004 Sep;52(3):612–8. doi: 10.1002/mrm.20198. [DOI] [PubMed] [Google Scholar]
- 21.Kirsch W, McAuley G, Holshouser B, Petersen F, Ayaz M, Vinters HV, et al. Serial susceptibility weighted MRI measures brain iron and microbleeds in dementia. J Alzheimers Dis. 2009;17(3):599–609. doi: 10.3233/JAD-2009-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stehling C, Wersching H, Kloska SP, Kirchhof P, Ring J, Nassenstein I, et al. Detection of asymptomatic cerebral microbleeds: a comparative study at 1.5 and 3.0 T. Acad Radiol. 2008 Jul;15(7):895–900. doi: 10.1016/j.acra.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, Schmidt R, et al. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol. 1999 Apr;20(4):637–42. [PMC free article] [PubMed] [Google Scholar]
- 24.Moody DM, Brown WR, Challa VR, Stump DA, Reboussin DM, Legault C. Brain microemboli associated with cardiopulmonary bypass: a histologic and magnetic resonance imaging study. Ann Thorac Surg. 1995 May;59(5):1304–7. doi: 10.1016/0003-4975(95)00057-r. [DOI] [PubMed] [Google Scholar]
- 25.Nakata-Kudo Y, Mizuno T, Yamada K, Shiga K, Yoshikawa K, Mori S, et al. Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord. 2006;22(1):8–14. doi: 10.1159/000092958. [DOI] [PubMed] [Google Scholar]
- 26.Lee SH, Kim SM, Kim N, Yoon BW, Roh JK. Cortico-subcortical distribution of microbleeds is different between hypertension and cerebral amyloid angiopathy. J Neurol Sci. 2007 Jul 15;258(1–2):111–4. doi: 10.1016/j.jns.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Jeerakathil T, Wolf PA, Beiser A, Hald JK, Au R, Kase CS, et al. Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke. 2004 Aug;35(8):1831–5. doi: 10.1161/01.STR.0000131809.35202.1b. [DOI] [PubMed] [Google Scholar]
- 28.Sveinbjornsdottir S, Sigurdsson S, Aspelund T, Kjartansson O, Eiriksdottir G, Valtysdottir B, et al. Cerebral microbleeds in the population based AGES-Reykjavik study: prevalence and location. J Neurol Neurosurg Psychiatry. 2008 Sep;79(9):1002–6. doi: 10.1136/jnnp.2007.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregoire SM, Brown MM, Kallis C, Jager HR, Yousry TA, Werring DJ. MRI detection of new microbleeds in patients with ischemic stroke: five-year cohort follow-up study. Stroke. 2010 Jan;41(1):184–6. doi: 10.1161/STROKEAHA.109.568469. [DOI] [PubMed] [Google Scholar]
- 30.Goos JD, Henneman WJ, Sluimer JD, Vrenken H, Sluimer IC, Barkhof F, et al. Incidence of cerebral microbleeds: a longitudinal study in a memory clinic population. Neurology. 2010 Jun 15;74(24):1954–60. doi: 10.1212/WNL.0b013e3181e396ea. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009 Feb;8(2):165–74. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werring DJ, Frazer DW, Coward LJ, Losseff NA, Watt H, Cipolotti L, et al. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain. 2004 Oct;127(Pt 10):2265–75. doi: 10.1093/brain/awh253. [DOI] [PubMed] [Google Scholar]
- 33.Fiehler J. Cerebral microbleeds: old leaks and new haemorrhages. Int J Stroke. 2006 Aug;1(3):122–30. doi: 10.1111/j.1747-4949.2006.00042.x. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008 Jun;65(6):790–5. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 35.Jeon SB, Kang DW, Cho AH, Lee EM, Choi CG, Kwon SU, et al. Initial microbleeds at MR imaging can predict recurrent intracerebral hemorrhage. J Neurol. 2007 Apr;254(4):508–12. doi: 10.1007/s00415-006-0406-6. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke. 2004 Jun;35(6):1415–20. doi: 10.1161/01.STR.0000126807.69758.0e. [DOI] [PubMed] [Google Scholar]
- 37.Vernooij MW, Ikram MA, Hofman A, Krestin GP, Breteler MM, van der Lugt A. Superficial siderosis in the general population. Neurology. 2009 Jul 21;73(3):202–5. doi: 10.1212/WNL.0b013e3181ae7c5e. [DOI] [PubMed] [Google Scholar]
- 38.Goos JD. Stroke. In Press. [Google Scholar]
- 39.Cordonnier C, van der Flier WM, Sluimer JD, Leys D, Barkhof F, Scheltens P. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006 May 9;66(9):1356–60. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 40.Lee SH, Bae HJ, Ko SB, Kim H, Yoon BW, Roh JK. Comparative analysis of the spatial distribution and severity of cerebral microbleeds and old lacunes. J Neurol Neurosurg Psychiatry. 2004 Mar;75(3):423–7. doi: 10.1136/jnnp.2003.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yakushiji Y, Nishiyama M, Yakushiji S, Hirotsu T, Uchino A, Nakajima J, et al. Brain microbleeds and global cognitive function in adults without neurological disorder. Stroke. 2008 Dec;39(12):3323–8. doi: 10.1161/STROKEAHA.108.516112. [DOI] [PubMed] [Google Scholar]
- 42.Igase M, Tabara Y, Igase K, Nagai T, Ochi N, Kido T, et al. Asymptomatic cerebral microbleeds seen in healthy subjects have a strong association with asymptomatic lacunar infarction. Circ J. 2009 Mar;73(3):530–3. doi: 10.1253/circj.cj-08-0764. [DOI] [PubMed] [Google Scholar]
- 43.Hanyu H, Tanaka Y, Shimizu S, Takasaki M, Abe K. Cerebral microbleeds in Alzheimer’s disease. J Neurol. 2003 Dec;250(12):1496–7. doi: 10.1007/s00415-003-0245-7. [DOI] [PubMed] [Google Scholar]
- 44.Roob G, Schmidt R, Kapeller P, Lechner A, Hartung HP, Fazekas F. MRI evidence of past cerebral microbleeds in a healthy elderly population. Neurology. 1999 Mar 23;52(5):991–4. doi: 10.1212/wnl.52.5.991. [DOI] [PubMed] [Google Scholar]
- 45.Tsushima Y, Tanizaki Y, Aoki J, Endo K. MR detection of microhemorrhages in neurologically healthy adults. Neuroradiology. 2002 Jan;44(1):31–6. doi: 10.1007/s002340100649. [DOI] [PubMed] [Google Scholar]
- 46.Bednar M, Kantarci K, Kupiec J, Burstein A, Landen J, Reyes D, et al. The prevalence of brain microhemorrhages and infarcts in a cohort of patients with mild–moderate Alzheimer’s disease. ICAD. 2010:P2–372. [Google Scholar]
- 47.Poels MM, Vernooij MW, Ikram MA, Hofman A, Krestin GP, van der Lugt A, et al. Prevalence and risk factors of cerebral microbleeds: an update of the Rotterdam scan study. Stroke. 2010 Oct;41(10 Suppl):S103–6. doi: 10.1161/STROKEAHA.110.595181. [DOI] [PubMed] [Google Scholar]
- 48.Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, Sjogren M, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. 2001 Jun;32(6):1318–22. doi: 10.1161/01.str.32.6.1318. [DOI] [PubMed] [Google Scholar]
- 49.Black S, Gao F, Bilbao J. Understanding white matter disease: imaging-pathological correlations in vascular cognitive impairment. Stroke. 2009 Mar;40(3 Suppl):S48–52. doi: 10.1161/STROKEAHA.108.537704. [DOI] [PubMed] [Google Scholar]
- 50.Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009 Jul;118(1):87–102. doi: 10.1007/s00401-009-0498-z. [DOI] [PubMed] [Google Scholar]
- 51.Chen YW, Gurol ME, Rosand J, Viswanathan A, Rakich SM, Groover TR, et al. Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology. 2006 Jul 11;67(1):83–7. doi: 10.1212/01.wnl.0000223613.57229.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003 Apr;9(4):448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 53.Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain. 2008 Dec;131(Pt 12):3299–310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- 54.Lim SY, Wesley Thevathasan A, Gonzales M, Mitchell PJ, Evans A. Vasogenic oedema with no mass lesion. J Clin Neurosci. 2008 Sep;15(9):1048, 75–6. doi: 10.1016/j.jocn.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 55.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998 Apr;57(4):353–9. doi: 10.1097/00005072-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 56.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007 Sep;62(3):229–34. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 57.Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA, et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol. 2010 Oct;68(4):545–8. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003 Jul 8;61(1):46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 59.Siemers E, Friedrich S, Dean R, Sethuraman G, Demattos R, Jennings D, et al. Safety, tolerability and biomarker effects of an Aβ monoclonal antibody administered to patients with Alzheimer’s disease. Alzheimer’s and Dementia. 2008;4(Suppl 2):T774. [Google Scholar]
- 60.XX3. ICAD.
- 61.Hinchey J, Chaves C, Appignani B, Breen J, Pao L, Wang A, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996 Feb 22;334(8):494–500. doi: 10.1056/NEJM199602223340803. [DOI] [PubMed] [Google Scholar]
- 62.Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008 Jun;29(6):1043–9. doi: 10.3174/ajnr.A0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schiff D, Lopes MB. Neuropathological correlates of reversible posterior leukoencephalopathy. Neurocrit Care. 2005;2(3):303–5. doi: 10.1385/NCC:2:3:303. [DOI] [PubMed] [Google Scholar]
- 64.Okeda R, Kawamoto T, Tanaka E, Shimizu H. An autopsy case of drug-induced diffuse cerebral axonopathic leukoencephalopathy: the pathogenesis in relation to reversible posterior leukoencephalopathy syndrome. Neuropathology. 2007 Aug;27(4):364–70. doi: 10.1111/j.1440-1789.2007.00771.x. [DOI] [PubMed] [Google Scholar]
- 65.Greenberg SM, Rapalino O, Frosch MP. Case records of the Massachusetts General Hospital. Case 22–2010. An 87-year-old woman with dementia and a seizure. N Engl J Med. 2010 Jul 22;363(4):373–81. doi: 10.1056/NEJMcpc1004364. [DOI] [PubMed] [Google Scholar]
- 66.Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004 Jan;14(1):11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanaka A, Ueno Y, Nakayama Y, Takano K, Takebayashi S. Small chronic hemorrhages and ischemic lesions in association with spontaneous intracerebral hematomas. Stroke. 1999 Aug;30(8):1637–42. doi: 10.1161/01.str.30.8.1637. [DOI] [PubMed] [Google Scholar]
- 68.Tatsumi S, Shinohara M, Yamamoto T. Direct comparison of histology of microbleeds with postmortem MR images: a case report. Cerebrovasc Dis. 2008;26(2):142–6. doi: 10.1159/000139661. [DOI] [PubMed] [Google Scholar]
- 69.Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, et al. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol. 2009 Nov 25; doi: 10.1007/s00401-009-0615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesbery W, Davis D, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005 Jan;57(1):98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 71.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009 Aug;66(2):200–8. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weiss L, Harlos JP. The validity of negative necropsy reports for metastases in solid organs. J Pathol. 1986 Mar;148(3):203–6. doi: 10.1002/path.1711480302. [DOI] [PubMed] [Google Scholar]
- 73.Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007 Feb 9;144(3):950–60. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prada CM, Garcia-Alloza M, Betensky RA, Zhang-Nunes SX, Greenberg SM, Bacskai BJ, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci. 2007 Feb 21;27(8):1973–80. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schroeter S, Khan K, Barbour R, Doan M, Chen M, Guido T, et al. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci. 2008 Jul 2;28(27):6787–93. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995 Feb 9;373(6514):523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 77.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998 Jan;4(1):97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 78.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996 Oct 4;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 79.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004 Jan 15;13(2):159–70. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 80.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129–40. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003 Jul 31;39(3):409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 82.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997 Nov 25;94(24):13287–92. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004 May 7;279(19):20296–306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 84.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999 Jul 8;400(6740):173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 85.Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006 May 17;26(20):5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004 Dec 8;1(1):24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karlnoski RA, Rosenthal A, Alamed J, Ronan V, Gordon MN, Gottschall PE, et al. Deglycosylated anti-Abeta antibody dose-response effects on pathology and memory in APP transgenic mice. J Neuroimmune Pharmacol. 2008 Sep;3(3):187–97. doi: 10.1007/s11481-008-9114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005 Jan 19;25(3):629–36. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burbach GJ, Vlachos A, Ghebremedhin E, Del Turco D, Coomaraswamy J, Staufenbiel M, et al. Vessel ultrastructure in APP23 transgenic mice after passive anti-Abeta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007 Feb;28(2):202–12. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 90.Petrushina I, Ghochikyan A, Mkrtichyan M, Mamikonyan G, Movsesyan N, Ajdari R, et al. Mannan-Abeta28 conjugate prevents Abeta-plaque deposition, but increases microhemorrhages in the brains of vaccinated Tg2576 (APPsw) mice. J Neuroinflammation. 2008;5:42. doi: 10.1186/1742-2094-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets. 2009 Mar;8(1):50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wisniewski T, Boutajangout A. Immunotherapeutic approaches for Alzheimer’s disease in transgenic mouse models. Brain Struct Funct. 2010 Mar;214(2–3):201–18. doi: 10.1007/s00429-009-0236-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220(2):95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- 94.Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, et al. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009 Mar 17;106(11):4501–6. doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009 Feb 22; doi: 10.1007/s00401-009-0498-z. [DOI] [PubMed] [Google Scholar]
- 96.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008 Apr;18(2):253–66. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008 Apr;34(2):131–44. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 98.Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol. 2006 Feb 21;238(4):962–74. doi: 10.1016/j.jtbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 99.Weller RO, Massey A, Kuo YM, Roher AE. Cerebral amyloid angiopathy: accumulation of A beta in interstitial fluid drainage pathways in Alzheimer’s disease. Ann N Y Acad Sci. 2000 Apr;903:110–7. doi: 10.1111/j.1749-6632.2000.tb06356.x. [DOI] [PubMed] [Google Scholar]
- 100.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998 Sep;153(3):725–33. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo F, Rustay NR, Seifert T, Roesner B, Hradil V, Hillen H, et al. Magnetic resonance imaging detection and time course of cerebral microhemorrhages during passive immunotherapy in living amyloid precursor protein transgenic mice. J Pharmacol Exp Ther. 2010 Dec;335(3):580–8. doi: 10.1124/jpet.110.172932. [DOI] [PubMed] [Google Scholar]
- 102.Meyer-Luehmann M, Mora JR, Mielke M, Spires-Jones TL, de Calignon A, von Andrian UH, et al. T cell mediated cerebral hemorrhages and microhemorrhages during passive Abeta immunization in APPPS1 transgenic mice. Mol Neurodegener. 2011;6:22. doi: 10.1186/1750-1326-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Domnitz SB, Robbins EM, Hoang AW, Garcia-Alloza M, Hyman BT, Rebeck GW, et al. Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J Neuropathol Exp Neurol. 2005 Jul;64(7):588–94. doi: 10.1097/01.jnen.0000171644.00180.fc. [DOI] [PubMed] [Google Scholar]
- 104.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006 Dec;24(3):516–24. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 105.Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, et al. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006 Jan 11;26(2):365–71. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]