

Abstract
Neoadjuvant chemotherapy (NAC) induces a pathological complete response (pCR) in ~30% of patients with breast cancer. However, many patients have residual cancer after chemotherapy, which correlates with a higher risk of metastatic recurrence and poorer outcome than those who achieve a pCR. We hypothesized that molecular profiling of tumors after NAC would identify genes associated with drug resistance. Digital transcript counting was used to profile surgically resected breast cancers after NAC. Low concentrations of dual specificity protein phosphatase 4 (DUSP4), an ERK phosphatase, correlated with high post-NAC tumor cell proliferation and with basal-like breast cancer (BLBC) status. BLBC had higher DUSP4 promoter methylation and gene expression patterns of Ras-ERK pathway activation relative to other breast cancer subtypes. DUSP4 overexpression increased chemotherapy-induced apoptosis, whereas DUSP4 depletion dampened the response to chemotherapy. Reduced DUSP4 expression in primary tumors after NAC was associated with treatment-refractory high Ki-67 scores and shorter recurrence-free survival. Finally, inhibition of mitogen-activated protein kinase kinase (MEK) synergized with docetaxel treatment in BLBC xenografts. Thus, DUSP4 downregulation activates the Ras-ERK pathway in BLBC, resulting in an attenuated response to anti-cancer chemotherapy.
Chemotherapy is the standard treatment for patients with triple negative breast cancers (TNBCs), which are estrogen-receptor protein (ER), progesterone-receptor protein (PR) and human epidermal growth factor receptor 2 (HER2) negative. Although NAC is effective in reducing the size of the primary tumor before surgery, residual disease after NAC is common and is associated with higher risk of metastatic recurrence compared to patients achieving a pCR. A growing amount of evidence shows that chemotherapeutic agents spare cancer-initiating or stem-like cells1–4. Thus, we hypothesized that molecular profiling of treatment-refractory tumor cells may reveal alterations that are associated with drug resistance, metastatic recurrence and disease progression.
Here we used NanoString analyses5 to interrogate gene expression patterns in 49 residual breast tumors after NAC to identify causal effectors of drug resistance. We quantified the levels of 355 transcripts and evaluated them for association with Ki-67 immunohistochemistry (IHC) score in tumors after NAC. From this analysis, we identified DUSP4, a negative regulator of extracellular-regulated kinase (ERK) activity, as a potential mediator of resistance to chemotherapy and a tumor suppressor in BLBC. We further characterized the role of DUSP4 in BLBC through analyses of microarray data and in vitro and in vivo studies. We also provide evidence that loss of DUSP4 may underlie Ras-ERK pathway activation in BLBC, which can be targeted clinically with inhibitors of MEK.
RESULTS
We performed NanoString gene expression profiling on 49 formalin-fixed paraffin-embedded (FFPE) archival tissues from breast cancers resected after NAC (Fig. 1a and Supplementary Table 1). Because high tumor cell proliferation after NAC, as measured by Ki-67 IHC score, correlates with long-term outcome6,7, we used this biomarker as a surrogate endpoint for the effects of therapy. This cohort was enriched with TNBC specimens, where chemotherapy is the standard of care. The Ki-67 score ranged from 2.44–99.03% (Fig. 1b) and was associated with hormone receptors and HER2 status, with the highest positivity present in the TNBC samples (Fig. 1c).
Figure 1.

Ki-67–associated gene expression in chemotherapy-refractory breast cancers. (a) Scheme for the analysis of gene expression patterns in tumor-sparse FFPE tissues. HK genes, housekeeper genes. (b) Representative IHC of breast cancers after NAC with low, intermediate and high Ki-67 scores. Scale bars, 50 μm. (c) Association of pretreatment receptor status with Ki-67 score after chemotherapy. P = 0.0015 by analysis of variance (ANOVA) followed by Bonferroni post-hoc t test correction. **P < 0.01. TN, triple negative. Data are mean ± s.e.m. (d) Heatmap depicting the gene expression patterns in 49 tumors after NAC assayed by NanoString digital RNA transcript counting. Clinical (HER2, ER, PR) and molecular parameters are annotated for the samples (x axis), and gene signature or metagene membership is annotated for the genes (y axis). Red indicates high expression, and blue indicates low expression. NL, normal-like (e) Ki-67 score after NAC is plotted according to molecular subtype. P < 0.0001 by ANOVA followed by Bonferroni post-hoc t test correction, **P < 0.01, ***P < 0.001.
Gene expression profiling in archival tissues after NAC
Because of limitations in the number of genes that can be simultaneously assayed by NanoString, we assembled a priority list of transcripts to quantify. We interrogated the literature to identify gene signatures that are associated with high-grade, chemotherapy-resistant tumors, including the 21-gene Recurrence Score (Oncotype DX) signature8, an 18-gene chemo-resistance signature (CHEMO)9, a 50-gene stromal metagene signature (STROMAL_META)10 and a 13-gene wingless-related MMTV integration site (Wnt) pathway signature that predicts metastatic behavior (WNT/METS)11. We also analyzed other genes known to be involved in breast cancer that were not included in these signatures (Supplementary Table 2). Additionally, we incorporated class discovery approaches into the analysis (see Online Methods). Briefly, we subjected ER− breast tumors from the European Organisation for Research and Treatment of Cancer (EORTC) 10994 study that did not achieve a complete response to NAC10 to hierarchical clustering to identify the two most biologically distinct groups of tumors (Supplementary Fig. 1a). We hypothesized that these tumor and patient groups may correspond to Ki-67hi and Ki-67lo phenotypes after NAC. We tested the 354 microarray probe sets (244 unique genes, hereafter referred as the CLUSTER signature) differentiating these two classes for overlap with the Molecular Signatures Database (http://www.broadinstitute.org/; c2 curated), and we found them to be enriched for genes indicative of Ras activation, breast cancer 1 (BRCA1) deficiency (cluster A) and ER activation (cluster B) (Supplementary Fig. 1b). These genes clustered a panel of 50 breast cancer cell lines according to molecular subtype (Supplementary Fig. 1c), and we combined this panel with the previously published signatures to form a target set comprising 355 unique transcripts.
We profiled the RNA extracted from the 49 tumor blocks (Supplementary Fig. 2a–e) in duplicate using NanoString. The inter-replicate correlations ranged from 0.9985 to ~1.0 (Supplementary Fig. 2f). The seven control genes, selected from existing microdissected tumor microarray data as having low interpatient variability, were well correlated, suggesting that they were appropriate normalization factors (Supplementary Fig. 2g). We excluded 10 of the 355 transcripts because of poor signal-to-noise ratios. The normalized data and sample annotations are shown in Supplementary Tables 3 and 4. As an internal control, digital transcript counts for ESR1 (encoding ER), PGR (encoding PR) and ERBB2 (encoding HER2) were highly associated with clinical ER, PR and HER2 status (Supplementary Fig. 3a–f). Ki-67 score in the tumors after NAC was also highly correlated with expression of MKI67 (Pearson’s r = 0.7, P < 0.0001) (Supplementary Fig. 3g). These findings show the integrity of the acquired gene expression data.
Ki-67 score after NAC varies with molecular subtype
Molecular subtype is a strong prognostic variable in breast cancer and should be considered when performing gene expression profiling of breast tumors6,7,12,13. To determine the molecular subtype of the 49 tumors, we used unsupervised hierarchical clustering of the expression data, which produced four distinct clusters (Fig. 1d). The gene expression patterns of known basal and luminal markers differentiated between the two most prominent clusters (basal-like and luminal, n = 22 and 13, respectively); the third cluster contained the majority of the HER2-overexpressing tumors (n = 10). The fourth cluster contained both basal and luminal markers, and we designated it as the ‘normal-like’ cluster (n = 4).
Similar to hormone receptors (ER and PR) and HER2 status (Fig. 1c), we found a strong association between molecular subtype and Ki-67 score after NAC, which was the highest in the BLBCs (Fig. 1e). However, Ki-67 scores after NAC had high intra-subtype variability, particularly in BLBC, reflecting the heterogeneity of the basal-like subtype.
Gene sets associated with Ki-67 scores in treated tumors
We next applied the CLUSTER signature, described above, to the 49 breast tumors. We hypothesized that the collective expression of these genes may be associated with Ki-67 scores after NAC. Indeed, the CLUSTER signature (Supplementary Table 2) was highly correlated with Ki-67 score after NAC, as well as with the grade, mitotic index and molecular subtype of the tumors (Supplementary Fig. 4a–e). Three of the four preselected signatures (from CHEMO9, WNT/METS11 and Oncotype DX8) were also associated with Ki-67 score after NAC (Supplementary Fig. 5). Both the STROMAL_META signature and the CHEMO signature were previously reported to predict resistance to chemotherapy9,10. Paradoxically, the signature scores for CHEMO and WNT/WETS here were inversely correlated with a high Ki-67 score (we observed the lowest chemotherapy resistance scores for the tumors with the highest expression of Ki-67). This finding suggests two possibilities: (i) many gene signatures predicting sensitivity to chemotherapy are driven primarily by proliferation-related genes, as highly proliferative tumors are often responsive to this type of treatment and/or (ii) gene expression profiling of the tumors after NAC produces different information than profiling before NAC. The Oncotype DX algorithm maintained a strong predictive capacity in tumors after NAC, presumably because it is heavily weighted to proliferation- associated genes (Supplementary Fig. 5d). Although Ki-67 score after NAC prognosticates patient outcome but not necessarily chemoresistance, it is intuitive that those cells that retain high proliferation despite treatment will probably be resistant to chemotherapeutic agents.
Ras-ERK pathway activation in breast tumors after NAC
To identify targetable pathways in chemotherapy-refractory cancer cells, we tested genes that significantly correlated with a high Ki-67 score (P < 0.05) for overlap with the Molecular Signatures Database. To limit the identification of signatures associated with the subtypes of breast cancer (basal or luminal and clinical ER or HER2 status), we restricted the analysis to the 22 BLBC tumors, thereby focusing on pathways and gene sets that may be activated in tumors with high Ki-67 scores specifically within BLBC. Two of the 20 enriched gene sets represented genes downregulated by the activated proto-oncogene KRAS, a member of the Ras protein family (Supplementary Table 5)14,15. The expression of these genes was inversely correlated with Ki-67 score after NAC, suggesting that activation of KRAS is associated with high tumor cell proliferation after treatment. This was surprising to us, as mutations in KRAS are rare in human breast tumors16,17, and sequencing of KRAS codon 12 in the BLBCs from this cohort did not identify any mutations (data not shown). Thus, the gene signatures observed in BLBCs expressing a high Ki-67 score that associate with Ras-ERK pathway activation probably occur through mechanisms other than KRAS mutations.
DUSP4 loss associates with BLBC and Ras-ERK activation
Consistent with activation of the Ras-ERK pathway, low expression of DUSP4, a negative regulator of ERK1 and ERK2, had the strongest correlation with high Ki-67 score (P = 0.001; Fig. 2a). Low DUSP4 expression also correlated with BLBC subtype but retained its inverse association with Ki-67 score when considering only basal-like tumors (Spearman’s r = −0.64, P = 0.001). To confirm this finding, we examined DUSP4 expression as measured by NanoString in a second cohort of 89 TNBCs after NAC (Fig. 2b). Probes specific for each of the known DUSP4 transcript variants (NM_001394 and NM_057158 (ref. 18)) correlated significantly with the Ki-67 score after NAC (P = 0.0348 and P = 0.0008, respectively). In existing microarray datasets, DUSP4 mRNA levels were lowest in BLBC cell lines and tumors (Fig. 2c,d)19–21 despite their high intra-subtype variability. Moreover, reanalysis of two recent methylation studies in cohorts of 138 (ref. 22) and 28 breast cancers23 showed that DUSP4 promoter methylation associates with BLBC (Fig. 2e and Supplementary Fig. 6).
Figure 2.

DUSP4 expression is deficient in BLBC and associates with Ras-ERK pathway activation. (a) Heatmap of the z-score–normalized DUSP4 mRNA transcript counts and Ki-67 scores (shown in Fig. 1c,d) (blue, low transcript counts; red, high transcript counts). The P value shown was calculated by Spearman’s trend test. B, basal-like; L, luminal; H, HER2; MI, mitotic index; NA, not applicable. (b) Association of Ki-67 score and z-score–normalized digital transcript counts for the two known variants of DUSP4, NM_001394 (DUSP4v1) and NM_057158 (DUSP4v2), assayed by NanoString in 89 primary TNBCs treated with NAC. The P values shown were calculated by Pearson’s trend test. (c) Association of molecular subtype (as previously reported20) with DUSP4 gene expression (Affymetrix probe set 204014_at) in the ICBP-50 panel of breast cancer cell lines. P = 0.003 by ANOVA followed by Bonferroni post-hoc t test correction. **P < 0.01. (d) Association of molecular subtype and DUSP4 gene expression (204014 _at) in 230 breast tumors from the MAQC-II study21. P <0.0001 by ANOVA followed by Bonferroni post-hoc t test correction. **P < 0.01, ***P < 0.001. LumA, luminal A; LumB, luminal B. (e) Association of DUSP4 promoter methylation data with reported molecular subtype22 in 138 annotated breast tumors. P = 0.01 by ANOVA followed by Bonferroni post-hoc t test correction. +P < 0.1, *P < 0.05, **P < 0.01. (f) Association of the Ras-ERK pathway score24 (Online Methods) with pERK1/2 signal, as measured by RPPA in 42 of 50 cell lines in the ICBP-50 panel. The P value shown was calculated using Pearson’s trend test. (g) Association of the Ras-ERK pathway score for 230 breast tumors of patients in the MAQC-II study21 with molecular subtype, as determined by PAM50 analysis. P < 0.0001 by ANOVA followed by Bonferroni post-hoc t test correction.***P < 0.001. (h) Kaplan-Meier analysis of 286 breast tumors (Gene Expression Omnibus accession GSE2034)25 dichotomized at the median DUSP4 (204014_at) expression. Data in e and g are mean ± s.e.m.
We next examined the relationship between DUSP4 expression and a Ras-ERK pathway activation gene signature24. First, we confirmed that a published gene expression-based Ras-ERK pathway activation score correlated with the amount of phosphorylation at Thr202 and Tyr204 in ERK1 and Thr185 and Tyr187 of ERK2 respectively (pERK1/2T202/Y204) determined by reverse-phase protein arrays (RPPA) in the Integrative Cancer Biology Program (ICBP)-50 breast cancer cell line panel (Pearson’s r = 0.57, P < 0.0001; Fig. 2f). The signature also correlated with basal-like and HER2+ primary tumors but not with the luminal A and B subtypes (Fig. 2g; n = 230)21. HER2 strongly activates Ras-ERK signaling, potentially explaining the association with this molecular subtype. Moreover, in a dataset of 286 breast cancers from patients who did not receive adjuvant therapy25, low DUSP4 expression predicted shorter recurrence-free survival time (P = 0.0004, hazard ratio = 1.99, Cox proportional hazard univariate P = 0.0005; Fig. 2h). Notably, DUSP4 mRNA expression retained its significance after adjusting for molecular subtype in a multivariate analysis (Cox proportional hazard P = 0.002). These data suggest that low DUSP4 expression in resected tumors is a potential biomarker for a high likelihood of residual micrometastatic disease and tumor recurrence that is independent of molecular subtype.
DUSP4 loss associates with high ERK activity in BLBC
IHC staining for DUSP4 and pERK1/2T202/Y204 in 17 BLBCs after NAC revealed an inverse correlation between pERK1/2 and DUSP4 (Fig. 3a,b), suggesting that DUSP4 deficiency associates with Ras-ERK pathway activation in chemotherapy-refractory BLBC. Sections from a HER2-gene–amplified BT-474 xenograft stained high for both DUSP4 and pERK1/2 (Fig. 3a), consistent with the observation that HER2-overexpressing tumors have high mRNA expression and an activated Ras-ERK signature (Fig. 2d,g). In a panel of breast cancer cell lines, DUSP4 concentrations were inversely associated with pERK1/2 and pETS-1 (an ERK1/2 substrate) expression and with Ras-ERK gene signature score (Fig. 3c). In addition, DUSP4 mRNA expression was inversely correlated with Ras-ERK pathway score (P = 0.0064) in a series of 230 primary breast cancers (Fig. 3d).
Figure 3.

DUSP4 expression is inversely associated with Ras-ERK pathway activation. (a) Four representative BLBC sections stained by IHC for pERK1/2 and DUSP4. A HER2 gene-amplified BT-474 xenograft (pERK1/2hi, DUSP4hi) was included as a control. Scale bars, 50 μm. (b) Association of DUSP4 and pERK1/2 H scores (nuclear and cytoplasmic; see Online Methods for an explanation of H scores). The P value shown was calculated by Pearson’s trend test. (c) Immunoblot analysis of breast cancer cell lines harvested at subconfluence under normal growth conditions. The Ras-ERK score from Figure 2e is noted below. B, basal-like; L, luminal. Asterisks indicate cell lines with a known activating Ras and/or Raf mutation. (d) Association of the Ras-ERK pathway score and DUSP4 gene expression (204014_at). The P value shown was calculated by Pearson’s trend test.
DUSP4 expression modulates ERK activity in BLBC
We next tested the effect of restoring DUSP4 expression in BLBC cell lines. Reconstitution of DUSP4 inhibited the phosphorylation of ERK1/2 and downstream effectors ETS-1 and ELK-1, members of the ETS oncogene family (Fig. 4a). In contrast, these effectors were activated after transduction with constitutively active MEK1. DUSP4 overexpression reduced the viability of MDA-231 and BT-549 cells but had little effect on MDA-436 cells (Fig. 4b).
Figure 4.

DUSP4 regulates MAPK signaling and cell viability.
(a) Immunoblot analysis for MAPK pathway activation after adenoviral transduction with GFP, constitutively active MEK1 (AdMEK1ca), or DUSP4 (AdDUSP4). HA, hemagglutinin. (b) Sulfarhodamine B (SRB) viability analysis of the cells in a 72 h after transduction. Student’s t test, ***P < 0.001. (c) Immunoblot analysis of breast cancer cell lines transiently transfected for 72 h with one of two sequences of siRNA targeting DUSP4 (siDUSP4, 2 or 3) or a nontargeting siRNA control (siCONTROL, C). (d) Quantitative RT-PCR analysis of DUSP4 mRNA expression, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 48 h after siRNA transfection with the siDUSP4 construct 3 described in c). Data in b and d are mean ± s.d.
To study the effects of loss of DUSP4, we transfected breast cancer cells with siRNA targeting DUSP4 (siDUSP4) (Fig. 4c,d). Treatment with either of two siDUSP4 sequences had variable effects on ERK phosphorylation but consistently induced activation and expression of the ERK effector ETS-1 in three out of three basal-like cell lines (MDA-231, BT-549 and MDA-436). We speculate that ETS-1 activation may be a better marker of ERK activity in the nucleus, where DUSP4 localizes, than total cellular phosphorylation of ERK.
Knockdown of DUSP4 attenuates response to chemotherapy
We next examined whether DUSP4 loss exerts a chemoprotective effect in breast epithelial cells. SiRNA knockdown of DUSP4 increased the half-maximal inhibitory concentration (IC50) of docetaxel, enhanced MEK-dependent cell proliferation and protected against docetaxel-induced apoptosis in MCF-10A immortalized human mammary epithelial cells (Supplementary Fig. 7a–d). We obtained similar results in MDA-231 and MCF-7 breast cancer cells transfected with siDUSP4 (Supplementary Fig. 7e,f). To confirm the specificity of this result to DUSP4, we transduced MDA-231 cells with wild-type DUSP4 or with a synonymous DUSP4 mutant that is resistant to siRNA-mediated degradation (Supplementary Fig. 7g–i). Transduction with the siRNA-resistant DUSP4 mutant but not with wild-type DUSP4 partially abrogated the effect of siDUSP4 on docetaxel-induced caspase 3 and 7 cleavage in MDA-231 cells (Supplementary Fig. 7j). Collectively, these results suggest that DUSP4 loss protects breast cancer cells from chemotherapy-induced apoptosis.
DUSP4 loss after NAC correlates with poor patient outcome
Our in vitro studies suggested a role for DUSP4 in dampening the response to chemotherapy. To further support a chemoprotective effect of DUSP4 loss, at least one of two scenarios must occur: (i) pre-treatment concentrations of DUSP4 must predict clinical response to NAC, or (ii) cells expressing low amounts of DUSP4 must be enriched after NAC. In the EORTC 10994 (ref. 10) and MAQC-II breast cancer studies21, after adjusting for molecular subtype, DUSP4 expression before treatment was not associated with pathological complete response (pCR) (data not shown), thus arguing against the first possibility. We next asked whether DUSP4 expression was reduced in the residual tumors following NAC. We assayed the pretreatment biopsies from 15 patients in our cohort for Ki-67 score and DUSP4 expression and compared these results to those from the residual tumor after NAC. Patients that lost DUSP4 expression also had an increase in the tumor Ki-67 score after NAC. Of note, these patients had a shorter recurrence-free survival (Fig. 5a,b). We confirmed the association of loss of DUSP4 expression with poor clinical response to NAC in two other published cohorts (Fig. 5c). In one of these studies, 32 breast tumors were sampled before and after four cycles of NAC with epirubicin and cyclophosphamide, which was followed by four cycles of docetaxel26. All patients with expression data at both time points had residual disease after therapy. Patients with a substantial volumetric response by ultrasonography showed increased DUSP4 expression after NAC, whereas patients with a poor response to NAC lost or maintained low DUSP4 expression. A similar finding was made by a second study comprised of 15 patients who were treated with four cycles of docetaxel and capecitabine. Data from tumors sampled at baseline and at surgery were analyzed. Patients were categorized as responders or non-responders based on the change in tumor size by clinical exam and pathological response27. Thus, the combined analyses of these three studies suggest a causal association between DUSP4 deficiency and a lower response to anti-cancer chemotherapy.
Figure 5.
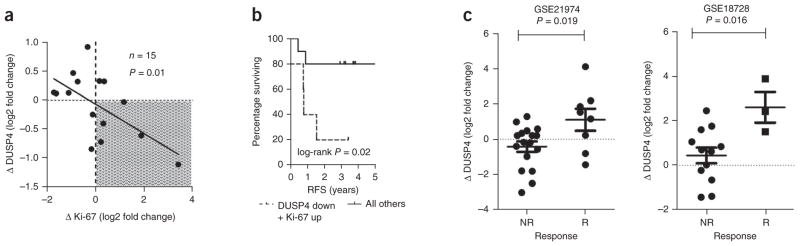
Reduction of DUSP4 expression after NAC correlates with enhanced tumor Ki-67 score and shorter RFS. (a) Association of DUSP4 gene expression and Ki-67 score in 15 breast tumors (paired diagnostic biopsies and surgical specimens after NAC). The P value shown was calculated by Pearson’s trend test. (b) Kaplan-Meier survival analysis of the population from the thatched area in a (loss of DUSP4 (DUSP down) and gain of Ki-67 score (Ki-67 up)) compared to all other tumors. (c) Association of the change in DUSP4 gene expression from paired breast tumors before and after NAC with response to NAC in two published studies26,27. A two-tailed t test was used to test for differences between patients who had a substantial tumor response (R) to NAC and those that did not (NR).
Inhibition of MEK improves chemotherapy-induced apoptosis
Given the role of DUSP4 in negative regulation of ERK1 and ERK2, we hypothesized that low expression of DUSP4 may be partly responsible for Ras-ERK pathway dependency and may be a potential biomarker for response to MEK inhibition. In support of this notion, we found that DUSP4 mRNA expression correlated inversely with sensitivity to the MEK inhibitors CI-1040 and U0126 across 17 BLBC cell lines, particularly when we excluded cell lines with loss of phosphatase and tensin homolog (PTEN) (Supplementary Fig. 8). Thus, we next determined whether MEK inhibition with AZD6244 (selumetinib) enhances docetaxel-induced apoptosis in breast cancer cells. Selumetinib was anti-proliferative in BLBC cells and antagonized the effects of docetaxel (data not shown), as has been observed by others28. However, three out of four BLBC cell lines treated with sequenced selumetinib after docetaxel treatment showed higher levels of cleaved caspase 3 and cleaved poly-(ADP-ribose) polymerase (PARP) compared to lines treated with docetaxel alone (Fig. 6a). Similar to selumetinib, transduction of DUSP4 also enhanced docetaxel-induced apoptosis in MDA-231 and MDA-436 cells (Fig. 6b,c).
Figure 6.

Pharmacological or genetic inhibition of MEK improves chemotherapy-induced apoptosis in vitro and in vivo. (a) Immunoblot analysis of breast cancer cell lines treated for 6 h with docetaxel or DMSO in normal growth medium, followed by drug washout and treatment with DMSO or selumetinib for 72 h. (b) Immunofluorescence for cleaved caspase 3 and DUSP4 in MDA-231 and MDA-436 cells transduced with adenovirus encoding DUSP4 (AdDUSP4) or LacZ (AdLacZ, control) followed by 24 h of docetaxel (Doc) or DMSO treatment. DUSP4 is shown in red, cleaved caspase 3 (cCASP3) is shown in green, and DAPI is shown in blue. The overlay (yellow) is shown in the lower right quadrant of each group. Scale bar, 50 μm. (c) Quantification of the immunofluorescence in b. FITC (cleaved caspase 3) signals were normalized to DAPI and are shown as mean ± s.d. (n = 5). The Kruskal-Wallis (nonparametric ANOVA equivalent; KW) test was used to test for differences among the groups, with a Dunn’s post-test to compare individual groups. (d) Tumor growth curves of MDA-231 xenografts randomized to treatment with selumetinib, docetaxel, the combination of both (combo) or vehicle control (n = 9 per arm). The results are shown on the linear scale (left) and the log2 scale (center) to visualize the differences in smaller tumors. Boxplot of final tumor volumes (day 24, right). The treatment interaction was tested by two-way ANOVA. Data are mean ± range. (e) Immunoblot analysis of xenografts harvested after 3 d of treatment, 1 h after the last dose of selumetinib (Sel).
To confirm these findings in vivo, we established MDA-231 xenografts in athymic mice and randomized them to treatment with docetaxel or vehicle, each with or without selumetinib. Treatment with selumetinib alone arrested the growth of MDA-231 xenografts, whereas treatment with docetaxel alone transiently delayed tumor growth. In contrast, we found marked tumor regression in the combination treatment arm (Fig. 6d). There was significant interaction effect (P = 0.003) between selumetinib and docetaxel in the effect on tumor size, suggesting therapeutic synergy. Immunoblot analyses of tumor lysates harvested after 3 days of treatment confirmed downregulation of pERK1/2 and ETS-1 in xenografts treated with selumetinib alone or in combination with docetaxel (Fig. 6e). These data provide further evidence that activation of the Ras-MAPK pathway can dampen the response to anti-cancer chemotherapy in vivo and support clinical trials combining these agents in DUSP4-deficient BLBC.
DISCUSSION
Archival FFPE tissues are a large source of primary tumor material that can be used for the identification of molecular markers that are associated with cancer behavior and patient outcome. Recent advances in cancer stem cell biology and tumor cell drug resistance have suggested the importance of molecularly profiling tumors after in vivo drug selection of treatment-refractory cancer cell subpopulations6,29–31. However, tumors after treatment are often paucicellular and include substantial necrotic and non-tumor cell contamination. Thus, accurate profiling in these specimens can be challenging. Using macrodissection coupled with digital expression analyses in treatment-refractory breast tumors after NAC, we report here a discovery-based methodology that may be useful in identifying mechanisms of drug resistance. Because patients with residual tumors after NAC, particularly those with high residual cell proliferation, have a higher probability of early metastatic recurrence and death than those who achieve a pCR, we hypothesize that the gene expression profile in these refractory cancers should mirror the expression profile of the residual micrometastases6,7. Exploring these profiles may enable the discovery of molecular alterations driving tumor recurrence, paving the way for rational adjuvant therapies targeting micrometastases.
Gene expression profiling of specimens after NAC resulted in several key findings. First, the Ki-67 score after NAC was highly related to the molecular and clinical subtype of breast cancer. To our knowledge, this association has not been previously shown. Second, in line with the hypothesis that residual tumors with high Ki-67 scores after chemotherapy harbor biomarkers and causal effectors of drug resistance, we identified loss of the phosphatase DUSP4 as a targetable mediator of drug resistance.
DUSP4 deregulation has been observed in lung cancer cell lines and primary tumors, and epigenetic silencing of DUSP4 has been noted in glioblastomas32–34. A previous study showed DUSP4 copy number loss by fluorescence in situ hybridization in breast cancers35. Thus, copy number loss may also influence DUSP4 expression, resulting in a loss of negative feedback mechanisms to Ras-ERK pathway activation and a dependence on this pathway for tumor cell proliferation and survival. Supporting this notion, we found that DUSP4 mRNA expression correlated inversely with sensitivity to the MEK inhibitors CI-1040 and U0126 in BLBC cell lines, particularly when excluding PTEN-null cell lines. PTEN loss increases phosphoinositide-3-kinase (PI3K) pathway activation, potentially providing MEK-independent growth and survival signals. Thus, we predict that DUSP4 expression (coupled with PTEN status) is a testable biomarker for sensitivity to MEK inhibitors in patients with BLBC. Furthermore, as DUSP4 expression was reduced in breast tumors that responded poorly to NAC, we propose that patients with residual disease after NAC may benefit from adjuvant therapy with an inhibitor of MEK.
In HEK293 cells, overexpression of DUSP4 also results in chemo-resistance through repression of apoptosis mediated by mitogen-activated protein kinase 8 (JNK)36. However, these data contrast with other data supporting a breast-cancer–promoting role for JNK37–39. JNK and ERK1/2 pathway crosstalk mediates an epithelial- mesenchymal transition phenotype in breast cancer39, which is a hallmark of chemotherapy-resistant, tumor-initiating cancer cells3,40. Future studies will be needed to elucidate the contributions of JNK and ERK in promoting stem-like behavior in DUSP4-deficient breast cancer cells.
In summary, molecular profiling of chemotherapy-resistant breast cancers after NAC identified loss of the MAPK phosphatase DUSP4 and activation of the Ras-ERK pathway in BLBC. We propose that further molecular profiling of tumor samples after NAC will yield other actionable molecular targets that can be used to eliminate drug-resistant subpopulations of breast cancer cells.
ONLINE METHODS
Tumor samples and patients
Breast tumor blocks were from a cohort of patients who had consented to the use of any deidentified tissues for research purposes under the auspices of an institutional review board–approved protocol (Vanderbilt IRB number 030747). Selection was based on the following factors: (i) the patient received NAC as treatment and had residual disease at surgery; (ii) sufficient FFPE tissue was available for nucleic acid purification; and (iii) presurgical clinical parameters (grade, mitotic index and ER, PR and HER2 status) were available. Samples were included only if they were deemed by an expert pathologist to contain ≥20% tumor cells in the entire section or if they contained a region with ≥20% tumor cells that could be macrodissected. In total, 49 samples met these criteria. Fifteen of 49 patients had pretreatment tumor biopsies available. This methodology was repeated for a cohort of 89 TNBC FFPE specimens resected after NAC. Patients in this second cohort were identified retrospectively at the Instituto Nacional de Enfermedades Neoplásicas, Lima, Perú and were collected under an institutionally approved protocol (INEN 10-018).
Macrodissection
To perform macrodissection, 3–5 serial 10-μm sections of tumor were adhered to uncharged slides using nuclease-free water. One additional 5-μm adjacent section was stained for H&E. The tumor hotspot region was outlined by an expert breast histopathologist. Each slide from the block was then overlaid on the H&E-stained slide and oriented according to the features of the section. The area surrounding the tumor-dense target region was scraped away using a sterile razor blade; the remaining tumor region was scraped into a 1.7-ml tube using a fresh blade. This process was repeated for all of the sections for each macrodissected sample.
Nucleic acid purification
Nucleic acid purification was performed using the Agencourt FormaPure Kit (Beckman Coulter, Beverly, MA) according to the manufacturer’s instructions. Total nucleic acid was extracted from the samples. DNAse treatment was performed on 1 μg total nucleic acid before nCounter analysis.
NanoString nCounter analysis
RNA samples were provided to NanoString (Seattle, WA) for analysis. Samples were assayed on a Bioanalyzer (Agilent, Santa Clara, CA) to determine the concentration of intact RNA (Supplementary Fig. 2). Code sets were synthesized targeting 355 genes and 14 controls (369-plex code set). The raw transcript counts were normalized by dividing by the geometric mean of the seven preselected normalization housekeeper genes (WAS, CD40, B2M, NAGA, TUBB, NPAS2 and POLR1B), which cover a range of levels of constitutive expression.
Molecular subtyping
Intrinsic subtyping was performed on the NanoString data by hierarchical clustering. Four clusters were identified. On the basis of the gene expressions of known basal (KIT, EGFR, KRT5 and KRT14), luminal (ESR1, PGR and GATA3) and HER2-enriched (ERBB2) markers, these clusters were annotated accordingly. For external microarray data sets, PAM50 molecular subtyping was performed using the genefu package in R on the scaled log2-normalized microarray data13,41.
Immunohistochemistry
Immunohistochemistry was performed for Ki-67 (m7240; Dako, Denmark), pERK1/2 (Cell Signaling, 9101) and DUSP4 (Santa Cruz, sc-10797). FFPE tumor sections were scanned at 100× magnification, and the area containing the highest number of positive cells in each case was selected. Positive and negative tumor cells were manually counted at 400×; the percentage of positive cells was calculated with at least 700 viable cells. Antigen retrieval for Ki-67 was performed using HpH Buffer (pH 8.0) in a decloaking chamber (Biocare Medical, Concord, MA). The antibody to Ki-67 (m7240; Dako, Denmark) was used at a 1:75 dilution overnight. Visualization was performed using the 4plus Detection System (Biocare) and 3,3′-diaminobenzidine (DAB) (Dako) as the chromogen. IHC was performed for DUSP4 (Santa Cruz, sc-10797) according to the following parameters: antigen retrieval using citrate buffer, pH 6.0 (decloaking chamber); dilution of 1:100; overnight incubation at 4 °C; and the Envision Visualization System from Dako. IHC was performed for pERK1/2 (Cell Signaling, 9101) according to the following parameters: antigen retrieval using citrate buffer, pH 6.0 (decloaking chamber); dilution of 1:80; overnight incubation at 4 °C; and the Envision Visualization System from Dako using DAB (Dako). DUSP4- and pERK1/2-stained tumor regions were scored independently for cytoplasmic and nuclear staining by an expert histopathologist by calculating the product of the percentage of cells staining at each intensity level and the intensity level (1+ to 3+ intensity, as estimated by an expert pathologist). An H score was then calculated by summing the individual intensity level scores. A composite H score was calculated by summing the H scores for the nuclear and cytoplasmic regions.
Statistical analyses
Hierarchical clustering, linear regression, Kaplan-Meier analyses, ANOVA and Student’s t tests were performed in R (http://cran.r-project.org/) or GraphPad Prism (GraphPad Software, La Jolla, CA). Receiver operator characteristic curves were generated using JMP 7 (SAS Institute, Cary, NC). Bonferroni-corrected post-hoc t tests were used to make selected comparisons in multigroup analyses after a significant result was obtained using ANOVA.
Cell lines
All cell lines were obtained from American Type Culture Collection (Rockland, MD). MDA-231, MCF-7, MDA-436 and T47D cells were cultured in DMEM with 10% FBS (Gibco). SUM159PT cells were cultured in DMEM with 5% FBS and 0.5 μg/ml hydrocortisone. BT-549 cells were cultured in RPMI with 10% FBS. MCF-10A cells were grown in DMEM and F12 at a 1:1 ratio with 5% horse serum, 10 μg/ml insulin, 100 ng/ml cholera toxin, 0.5 μg/ml hydrocortisone and 20 ng/ml epidermal growth factor.
Inhibitors
Docetaxel was obtained from the Vanderbilt University Hospital Outpatient Pharmacy and diluted in DMSO to a stock concentration of 1.25 mM. Selumetinib was provided by AstraZeneca and reconstituted in DMSO at a stock concentration of 10 μM. Selumetinib was tested in cells at a final concentration of 1 μM.
Xenograft experiments
MDA-231 cells (1 × 106) were injected into the left inguinal mammary fatpads of female BALB/c athymic mice (Harlan Laboratories) in 100 μl 1:1 DMEM and growth-factor–reduced Matrigel (BD Biosciences). All tumors were palpable within 10 d. Tumor volume in mm3 was measured three times weekly using the formula: volume = width2 × length/2. When tumors were ≥100 mm3, mice were randomized to receive saline (control) or docetaxel (15 mg per kg body weight weekly intraperitoneally), each with or without selumetinib (25 mg per kg body weight emulsified in 50 μl 0.1% methylcellulose and 0.1% Tween-80, twice daily orally; n = 9 per group) or vehicle gavage. Mice were treated through day 24, at which time they were killed for tumor collection 1 h after the last administration of selumetinib. In some cases, tumors were collected after 3 d of therapy to assess the inhibition of drug targets in situ.
Immunoblotting
Immunoblotting was carried out as described42. Antibodies to the following were used for immunoblotting: pERK1/2 (phosphorylated at Thr202 or Tyr204; 9101; 1:5,000), cleaved caspase 3 (9664; 1:500), PARP (9542; 1:1,000), calnexin (2433; 1:5,000), pELK-1 (9181; 1:1,000), ELK-1 (9182; 1:1,000), pcJun (2361; 1:1,000), cJun (9165; 1:1,000) (all from Cell Signaling), pETS-1 (phosphorylated at Thr38, Invitrogen, 44-1104G; 1:500), ETS-1 (Santa Cruz, sc-350; 1:1,000), DUSP4 (Abcam, ab7259; 1:500) and actin (Sigma, A2066; 1:10,000).
Gene set and metagene selection
The STROMAL_META metagene group comprised a set of 50 genes shown to be correlated with the seed stromal gene DCN (decorin) fitted on an external dataset of patients with breast cancer10. The high expression of this metagene group predicts intrinsic resistance to chemotherapy and is reported elsewhere10. The CHEMO gene set is a set of 31 genes; high expression of five of these genes is associated with pCR to NAC, and high expression of the other 26 genes in the set is associated with lack of pCR after NAC. These genes were identified on a training set of 82 breast tumors to which clinical response to NAC was known, and they were then validated on a set of 51 additional breast tumors9. The WNT/METS gene set is a 13-gene component of a larger signature that was identified in an orthotopic human breast cancer xenograft that was metastatic to lung11. These 13 genes were identified by gene ontology to be linked to the Wnt pathway and were associated with reduced time to metastasis, poor prognosis and reduced overall survival in patients with breast cancer11.
The CLUSTER gene set was identified by unsupervised techniques to select genes potentially correlated with Ki-67 score after therapy and, thus, with poor patient outcome. To accomplish this, 102 tumor samples that were previously published as part of the EORTC 10994 study10 were queried using hierarchical clustering to identify subclasses within the study population that did not undergo a pCR after NAC. Expression of ER protein and its associated expression signature can easily mask other gene expression patterns of biological relevance43. Therefore, to ensure that an ER-driven signature was not directly selected for interrogation, we further reduced the class discovery sample set to only the 37 ER− tumors that did not have a pCR in response to NAC. These 37 tumors were subjected to hierarchical clustering (genes with >100% coefficient of variation; CV and an average signal intensity of at least 100) to identify two transcriptionally distinct classes of ER− tumors. The 354 probe sets (P < 0.01, t-sample t test), which differentiated these two classes (called here the CLUSTER signature), mapped to 244 unique Entrez identification numbers.
Selection of normalization genes
To identify candidate normalization genes a priori, we used previously published laser-capture microdissection data from 50 breast tumors (GSE5847)44. Genes were selected based on a low coefficient of variation among all samples (<10%) and no significant differences in expression between the tumor epithelium and the surrounding stroma. Genes with varying average expressions across all tissue samples were selected to generate a diverse series of normalization genes.
NanoString nCounter analysis
All samples were assayed in duplicate using a 5-μl aliquot for a total of 98 assays. Hybridizations were carried at 65 °C for 18 h after mixing 5 μl of sample with 20 μl NanoString nCounter Reporter Probe and 5 μl Capture Probe. To account for slight differences in assay efficiency (hybridization, purification, binding and so on), the data were normalized to the sum of six positive control RNA spikes. The concentrations of the control RNA spikes ranged from 0.125 to 128 fM. All but one assay (of 98 total) passed quality control metrics for control spike linearity (R2 > 0.95) and sensitivity (control spike detection at 0.5 fM).
Expression data normalization, technical reproducibility and robustness
The correlation of expression across the 348 assayed genes was calculated for each pair of technical replicates. The interreplicate correlation was extremely high for 48 of the 49 samples (replicate r range of 0.998869–0.999991; (Supplementary Fig. 2f)). One sample was not assayed in duplicate because of a technical problem with a replicate that did not produce a signal.
Raw nCounter data from seven preselected normalization genes were first plotted as a correlation matrix to test their similarity of expression before using them to normalize the remaining data. Plotting the expression data in this manner permits the visual identification of individual genes that vary markedly in their pattern of expression relative to other genes across all of the samples. In this case, all seven selected genes had generally high positive correlation to one another, suggesting that they were all appropriate for normalization and that their expression varied according to the amount of input RNA (Supplementary Fig. 2g). One of the normalization genes, NPAS1, was not well correlated with several of the other normalization genes. Thus, to protect against the potential contribution of this gene as a normalization outlier, we used a geometric mean as opposed to an arithmetic mean. Therefore, the geometric mean of the seven transcript counts was calculated to serve as a normalization factor for the remaining data.
Gene set scoring
The CHEMO, WNT/METS and STROMA_META gene sets were scored by first summing the upregulated genes and downregulated genes from each respective gene set. The gene set score was then calculated as: gene set score = (upregulated gene component score) − (downregulated gene component score).
The Ras-ERK activation signature was comprised of 57 upregulated probe sets, as reported previously24. The 57 probe sets were extracted from the target RMA-normalized log2-transformed Affymetrix U133plus2 dataset. The resulting signal intensities were summed for each sample to generate the Ras-ERK pathway score.
The CLUSTER gene set was scored by summing the normalized log2 transcript counts for genes upregulated in the cluster A and cluster B components. The CLUSTER score was then generated as: CLUSTER score = (cluster A component score) − (cluster B component score).
Recurrence score quantification
The recurrence score (RS) was reproduced based on the methods reported by Paik et al.8. The log2 housekeeper gene– normalized data were used to approximate the ΔCT values used in the Oncotype DX algorithm. The recurrence score was calculated in R as follows (using the normalized transcript counts for each gene where indicated):
GRB7 group score = (0.9 × GRB7) + (0.1 × ERBB2)
ER group score = ((0.8 × ESR1) + (1.2 × PGR) + (1 × BCL2) + (1 × SCUBE2))/4
Proliferation group score = ((1 × survivin) + (1 × MKI67) + (1 × CCNB1))/3
Two genes from the proliferation group were not available; thus, we divided the gene sum by a factor of 3 instead of by a factor of 5, as is reported in the original algorithm8.
Invasion group score = ((1 × CTSL2) + (1 × MMP11))/2
The unscaled recurrence score (RSU) was then calculated as:
The RSU for the 49 patients was then scaled from 0–100 using the rescale function of the genefu package in R41. A scaled RS (RSS) <18 was considered ‘low’; an RSS of 18–31 was considered ‘intermediate’; and RSS >31 was considered ‘high’.
No threshold cutoffs were used in the approximation of the RS as is performed in the Oncotype DX algorithm.
Microarray data sets
Raw microarray data from 230 unique breast tumors from the MAQC-II project were downloaded from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession GSE20194 (ref. 21). Microarray data were generated from pretreatment fine-needle aspirates of the primary tumor and from the surgical specimen after NAC. The patients were treated with 6 months of NAC. Raw microarray data from the study published by Wang et al.25 and clinical follow-up data were extracted from GEO under accession GSE2034.
Raw microarray data for the ICBP-50 were extracted from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession E-TABM-157 (ref. 20). All data were log2 transformed and RMA normalized in R before analysis. Normalized probe-level methylation data from 189 annotated breast tumors were downloaded from GEO under accession GSE22210 (ref. 22). DUSP4 promoter methylation levels were plotted according to molecular subtype as reported by the authors of ref 22. Only tumors classified as either HER2-enriched, basal like or luminal (A or B) were included in the analysis (n = 138). Normalized probe-level methylation data from 28 annotated breast tumors were downloaded from GEO under accession GSE22135 (ref. 23). DUSP4 promoter methylation levels were plotted according to molecular subtype as reported by the authors of ref. 23. Only tumors classified as either HER2-enriched, basal-like or luminal (A or B) were included in the analysis.
Microarray data from 32 tumors sampled before and after four cycles of NAC with epirubicine and cyclophosphamide every 3 weeks followed by four cycles of docetaxel were downloaded from GEO under accession GSE21974 (ref. 45). All patients with gene expression data at both time points had residual tumor in the breast after therapy. Patients were classified as responders or non-responders according to substantial tumor volume response by ultrasonigraph (as defined by the investigators). Additionally, microarray data from 15 patients treated with four cycles of NAC (docetaxel and capecitabine) were downloaded from GEO under accession GSE18728. Only those patients with pretreatment and surgical specimens were included. Patients were categorized as responders or non-responders based on change in tumor size by clinical exam and pathologic response as assessed and reported by the investigators27.
Reverse-phase protein array analysis
Breast cancer cell lines in the ICBP-50 panel were maintained in culture as described20. RPPA of lysates from 42 cell lines in the ICBP-50 panel was performed as described46–49. In brief, cell lysates were normalized to 1 μg/μl concentration as assessed by bicinchoninic acid assay and boiled with 1% SDS. Supernatants were manually diluted fivefold with lysis buffer. An Aushon BioSystems 2470 arrayer (Burlington, MA) created 1,056-sample arrays on nitrocellulose-coated FAST slides (Schleicher and Schuell BioScience, Inc.). Slides were probed with validated primary antibodies to pERK1/2 (Cell Signaling, 4377; 1:1,000) and total ERK2 (Santa Cruz, SC-154; 1:250) and signal amplified using a DakoCytomation-catalyzed system. Secondary antibodies were used as a starting point for amplification. Slides were scanned, analyzed and quantified using Microvigene software (VigeneTech Inc., Carlisle, MA) to generate spot signal intensities, which were processed by the R package SuperCurve (version 1.01, http://bioinformatics.mdanderson.org/OOMPA)47. A fitted curve (‘supercurve’) was plotted, with the signal intensities on the y axis and the relative log2 concentration of each protein on the x axis, using the nonparametric, monotone increasing B-spline model47. Protein concentrations were derived from the supercurve for each lysate by curve fitting and were normalized by median polish. Protein measurements were corrected for loading as previously described46,49,50.
Adenovirus transduction
The transduction procedure and the documentation of GFP-expressing (AdGFP) and the constitutively active MEK1 (AdMEK1ca) adenoviruses were conducted as previously reported42. The AdMEK1ca construct was kindly provided by E.P. Black (University of Kentucky, Lexington KY). Adenovirus expressing DUSP4 (AdDUSP4) was purchased from Vector Biolabs (Philadelphia, PA).
Lentiviral transduction of wild-type and siRNA-resistant DUSP4
The destination vector pLX301 was purchased from Addgene51. The Gateway entry vector for the wild-type DUSP4 open reading frame (pENTR221) was purchased from Open Biosystems (100066579). Six synonymous point mutations were introduced in the target coding sequence of siDUSP4 construct 3 to render the resulting transcript resistant to the siRNA (services provided by GENEWIZ, South Plainfield, NJ). The resulting sequences are shown below (siRNA target in bold).
wild type: GACTGCCCAAACCACTTTGAAGGACACTATCAGTACA AGTGCATCCCAGTGGAAGATAAC;
mutated: GACTGCCCAAACCACTTTGAGGGTCATTACCAATATA AGTGCATCCCAGTGGAAGATAAC
Gateway recombination was performed using the Invitrogen Clonase II LR Kit, resulting in the wild-type pLX301-DUSP4 and pLX301-DUSP4 mutant constructs. Lentivirus was packaged in 293T cells by cotransfection with the plasmids psPAX2 and pMD2G. MDA-231 cells were transduced with viral supernatant and selected with puromycin for over 1 week before plating for experiments.
SiRNA knockdown of DUSP4
Cells were reverse transfected by plating 2 × 105 cells in 5 ml of growth medium in 60-mm dishes containing precomplexed Lipofectamine RNAiMAX (Invitrogen) and siRNA in Optimem (Gibco) medium. After 24 h, cells were transferred to 6-well or 96-well plates for drug treatment. SiRNA duplexes were obtained from QIAGEN (siCONTROL: 5′-GGAAGCAGACTCACTCTTATA-3′) or Dharmacon (ON-TARGETplus, siDUSP4 construct 1: 5′-GUACAUCGAUGCCGUGAAG-3′; siDUSP4 construct 2: 5′-CAUCACGGCUCUGUUGAAU-3′; and siDUSP4 construct 3: 5′-GAAG GACACUAUCAGUACA-3′). All siRNAs were used at a final concentration of 20 nM.
Immunofluorescence
MDA-231 and MDA-436 cells grown in 4-well chamber slides were fixed in 10% neutral buffered formalin, washed twice with PBS and then blocked for 30 min in 3% cold fish gelatin (Sigma-Aldrich) in PBST (0.1% Tween-20 in PBS, pH 7.4). Slides were incubated overnight at 4 °C with a rabbit antibody to DUSP4 (Abcam, ab7259, 1:200) and a goat antibody to cleaved caspase 3 (Santa Cruz Biotechnologies, sc-22171, 1:100), washed five times with PBS (2 min per wash) and incubated with fluorochrome-conjugated donkey antibody for rabbit or goat IgG. (Santa Cruz Biotechnologies, 1:100 in PBS) for 2 h at 4 °C, washed five times with PBS (2 min per wash) and mounted using Vectashield with DAPI (Vector Laboratories, Burlingame, VT). Images were captured using ProgRes software and a Jenoptik ProgRes digital camera mounted on a Motic AE31 microscope. To quantify activated caspase 3, four or five random images (FITC for cleaved caspase 3 and DAPI) were taken from each well. The images were converted to a binary signal and quantified by densitometry using ImageJ. Each caspase 3 measurement was normalized to its paired DAPI measurement to control for cell number.
Cell viability and apoptosis assays
Sulforhodamine B assays were performed as previously described42. Apoptosis assays (Caspase-Glo) were performed in 96-well black-walled plates according to the manufacturer’s protocol (Promega, Madison, WI). SRB and Caspase-Glo assay data were normalized by subtracting the signal in a blank well and then dividing by the signal from untreated cells where indicated (percentage of control viability).
Supplementary Material
Acknowledgments
We would like to thank E. Penni Black (University of Kentucky), who provided the AdMEK1ca adenoviral construct. The authors would also like to thank P.D. Smith and AstraZeneca for supplying selumetinib utilized in the in vivo experiments. This work was supported by Breast Cancer Specialized Program of Research Excellence (SPORE) grant P50CA98131, Vanderbilt-Ingram Cancer Center Support grant P30CA68485, a grant from the Breast Cancer Research Foundation, the American Cancer Society Clinical Research Professorship grant CRP-07-234 and the Lee Jeans Translational Breast Cancer Research Program (to C.L.A.). G.B.M. was supported by the SU2C Dream Team award, PO1CA099031 and the Komen Promise grant KG 081694. J.S. and M.D. were funded by the Breakthrough Breast Cancer and the Royal Marsden National Institutes of Health Research Biomedical Research Centre.
Footnotes
Note: Supplementary information is available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
J.M.B. designed and performed the experiments, and authored the manuscript. R.S.C. provided expert guidance on in vivo models and performed immuno-fluorescence. M.G.K., N.M.G.-I. and M.E.S. provided pathology support and performed IHC staining and scoring. D.B.V., T.W.M. and N.E.B. provided scientific advice. K.S.-H., A.M.G.-A. and G.B.M. performed RPPA analysis. J.S., M.D., J.A.P. and H.L.G. provided post-NAC patient tumor samples. J.J.S. and I.M.M. identified the Vanderbilt cohort and managed the clinical database, including performing retrospective chart review. M.E.S. and I.M.M. functioned as honest brokers to maintain patient privacy. C.L.A. provided scientific direction, established collaborations, prepared the manuscript with J.M.B. and allocated funding for the work. All the authors participated in the preparation of the manuscript.
References
- 1.Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS ONE. 2008;3:e3077. doi: 10.1371/journal.pone.0003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morel AP, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Creighton CJ, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calcagno AM, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiss GK, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 6.Guarneri V, et al. A prognostic model based on nodal status and Ki-67 predicts the risk of recurrence and death in breast cancer patients with residual disease after preoperative chemotherapy. Ann Oncol. 2009;20:1193–1198. doi: 10.1093/annonc/mdn761. [DOI] [PubMed] [Google Scholar]
- 7.Jones RL, et al. The prognostic significance of Ki67 before and after neoadjuvant chemotherapy in breast cancer. Breast Cancer Res Treat. 2009;116:53–68. doi: 10.1007/s10549-008-0081-7. [DOI] [PubMed] [Google Scholar]
- 8.Paik S, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 9.Hess KR, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- 10.Farmer P, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 11.DiMeo TA, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheang MC, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweet-Cordero A, et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37:48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 15.Chiaradonna F, et al. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene. 2006;25:5391–5404. doi: 10.1038/sj.onc.1209528. [DOI] [PubMed] [Google Scholar]
- 16.Sánchez-Munoz A, et al. Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer. 2010;10:136. doi: 10.1186/1471-2407-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forbes SA, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr Protoc Hum Genet. 2008;Chapter 10(Unit 10.11) doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cadalbert LC, et al. Differential regulation of MAP kinase activation by a novel splice variant of human MAP kinase phosphatase-2. Cell Signal. 2010;22:357–365. doi: 10.1016/j.cellsig.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Mirzoeva OK, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popovici V, et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010;12:R5. doi: 10.1186/bcr2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm K, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bediaga NG, et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010;12:R77. doi: 10.1186/bcr2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pratilas CA, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 26.Stickeler E, et al. Basal-like molecular subtype and HER4 up-regulation and response to neoadjuvant chemotherapy in breast cancer. Oncol Rep. 2011;26:1037–1045. doi: 10.3892/or.2011.1392. [DOI] [PubMed] [Google Scholar]
- 27.Korde LA, et al. Gene expression pathway analysis to predict response to neoadjuvant docetaxel and capecitabine for breast cancer. Breast Cancer Res Treat. 2010;119:685–699. doi: 10.1007/s10549-009-0651-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yacoub A, et al. Sequence dependent exposure of mammary carcinoma cells to Taxotere and the MEK1/2 inhibitor U0126 causes enhanced cell killing in vitro. Cancer Biol Ther. 2003;2:670–676. [PubMed] [Google Scholar]
- 29.Dowsett M, et al. Short-term changes in Ki-67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence-free survival. Clin Cancer Res. 2005;11:951s–958s. [PubMed] [Google Scholar]
- 30.Dowsett M, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. 2007;99:167–170. doi: 10.1093/jnci/djk020. [DOI] [PubMed] [Google Scholar]
- 31.Miller WR, et al. Gene expression profiles differentiating between breast cancers clinically responsive or resistant to letrozole. J Clin Oncol. 2009;27:1382–1387. doi: 10.1200/JCO.2008.16.8849. [DOI] [PubMed] [Google Scholar]
- 32.Britson JS, Barton F, Balko JM, Black EP. Deregulation of DUSP activity in EGFR-mutant lung cancer cell lines contributes to sustained ERK1/2 signaling. Biochem Biophys Res Commun. 2009;390:849–854. doi: 10.1016/j.bbrc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 33.Chitale D, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28:2773–2783. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waha A, et al. Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010;70:1689–1699. doi: 10.1158/0008-5472.CAN-09-3218. [DOI] [PubMed] [Google Scholar]
- 35.Armes JE, et al. Candidate tumor-suppressor genes on chromosome arm 8p in early-onset and high-grade breast cancers. Oncogene. 2004;23:5697–5702. doi: 10.1038/sj.onc.1207740. [DOI] [PubMed] [Google Scholar]
- 36.Cadalbert L, Sloss CM, Cameron P, Plevin R. Conditional expression of MAP kinase phosphatase-2 protects against genotoxic stress-induced apoptosis by binding and selective dephosphorylation of nuclear activated c-jun N-terminal kinase. Cell Signal. 2005;17:1254–1264. doi: 10.1016/j.cellsig.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Brumby AM, et al. Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics. 2011:105–125. doi: 10.1534/genetics.111.127910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, et al. Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol Cancer Res. 2010;8:266–277. doi: 10.1158/1541-7786.MCR-09-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:253–260. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- 41.Haibe-Kains B. genefu R package: Relevant Functions for Gene Expression Analysis. Especially in Breast Cancer. 2009 < http://www.bioconductor.org/packages/release/bioc/html/genefu.html>.
- 42.Balko JM, Jones BR, Coakley VL, Black EP. Combined MEK and EGFR inhibition demonstrates synergistic activity in EGFR-dependent NSCLC. Cancer Biol Ther. 2009;8:522–530. doi: 10.4161/cbt.8.6.7690. [DOI] [PubMed] [Google Scholar]
- 43.Pusztai L. Gene expression profiling of breast cancer. Breast Cancer Res. 2009;11 (suppl 3):S11. doi: 10.1186/bcr2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boersma BJ, et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer. 2008;122:1324–1332. doi: 10.1002/ijc.23237. [DOI] [PubMed] [Google Scholar]
- 45.Stickeler E, et al. Basal-like molecular subtype and HER4 up-regulation and response to neoadjuvant chemotherapy in breast cancer. Oncol Rep. 2011;26:1037–1045. doi: 10.3892/or.2011.1392. [DOI] [PubMed] [Google Scholar]
- 46.Hennessy BT, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. 2007;13:7421–7431. doi: 10.1158/1078-0432.CCR-07-0760. [DOI] [PubMed] [Google Scholar]
- 47.Hu J, et al. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 48.Liang J, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 49.Stemke-Hale K, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tibes R, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.