

Abstract
Background. Toll-like receptors (TLRs) play a role in the pathophysiology of sepsis and multiple organ failure. This study examined the effect of CpG oligodeoxynucleotide (CpG-ODN), the TLR9 ligand, on polymicrobial sepsis–induced cardiac dysfunction.
Methods. Male C57BL/6 mice were treated with CpG-ODN, control CpG-ODN (control-ODN), or inhibitory CpG-ODN (iCpG-ODN) 1 hour prior to cecal ligation and puncture (CLP)–induced sepsis. Mice that underwent sham surgery served as sham controls. Cardiac function was examined by echocardiography before and 6 hours after CLP.
Results. Cardiac function was significantly decreased 6 hours after CLP. CpG-ODN prevented CLP-induced cardiac dysfunction, as evidenced by maintenance of the ejection fraction and fractional shortening. Control-ODN or iCpG-ODN did not alter CLP-induced cardiac dysfunction. CpG-ODN significantly attenuated CLP-induced myocardial apoptosis and increased myocardial Akt and extracellular-signal-related kinase (ERK) phosphorylation levels following CLP. In vitro experiments demonstrated that CpG-ODN promotes an association between TLR9 and Ras, resulting in Akt and ERK phosphorylation. Inhibition of phosphoinositide 3-kinase (PI3K) by Ly294002 or inhibition of ERK by U0126 in vivo abolished CpG-ODN attenuation of CLP-induced cardiac dysfunction.
Conclusions. CpG-ODN prevents CLP-induced cardiac dysfunction, in part through activation of PI3K/Akt and ERK signaling. Modulation of TLR9 could be an effective approach for treatment of cardiovascular dysfunction in patients with sepsis or septic shock.
Keywords: TLR9, CpG-ODN, Sepsis, cardiac function, PI3K/Akt signaling, ERK
Sepsis is the most important cause of morbidity and mortality in intensive care units (ICUs), and cardiovascular dysfunction contributes to the high morbidity and mortality of this condition [1, 2]. Approximately 60% of patients admitted to the ICU have cardiac dysfunction, with an associated mortality rate of 70%–90%, compared with 20% among septic patients without cardiovascular dysfunction [2, 3].
The innate immune and inflammatory responses mediated by Toll-like receptors (TLRs) [4] are involved in the pathophysiology of sepsis and multiple organ failure [1]. TLRs recognize pathogen-associated molecular patterns [4] and predominately activate nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), which controls the expression of inflammatory cytokine genes [4]. TLR2 and TLR4, which are expressed on the cell surface, have been implicated in cardiac dysfunction in sepsis [5–7]. TLR9 is located intracellularly in endosomes and recognizes unmethylated CpG-DNA from bacteria and viruses, as well as endogenous DNA [8, 9]. Synthetic CpG oligodeoxynucleotide (CpG-ODN) activates TLR9-mediated signaling [8, 10]. Rice et al showed that treatment of rats with CpG-ODN reduced mortality due to polymicrobial sepsis [11]. Weighardt et al reported that challenging mice with CpG-ODN substantially increased the resistance against acute polymicrobial sepsis [12]. Mathur et al showed that pretreatment of mice with CpG-C class 24 hours prior to endotoxin challenge improved the ejection fraction 6 hours after LPS stimulation [13]. However, the effect of CpG-ODN on cardiac function in polymicrobial sepsis has not been investigated.
Activation of the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway has been shown to attenuate cardiac dysfunction [6] and increase survival in polymicrobial sepsis [14]. Recent studies have identified cross talk between the PI3K/Akt pathway and TLR signaling [15–17]. Activation of the PI3K/Akt signaling pathway may serve as a negative feedback regulator for TLR-mediated innate immune inflammatory responses [14, 16, 18, 19]. However, the effect of CpG-ODN on the activation of PI3K/Akt signaling in cardiac function during polymicrobial sepsis has not been studied.
ERK1/2 are related protein-serine/threonine kinases that participate in the Ras-Raf-MEK-ERK signal transduction cascade [20]. Activated ERK plays a protective role in cardiac ischemic injury [21]. However, the role of ERK in sepsis is not clear. Zhang et al [22] reported that lipopolysaccharide (LPS) activates mitogen-activated protein kinase phosphatase 1, which attenuates ERK1/2 and p38 activation, inhibits myocardial tumor necrosis factor α (TNF-α) production, and improves cardiac function in endotoxemia. Kim et al [23] reported that myrrh, a widely used antibacterial and antiinflammatory agent, attenuated CLP-induced liver damage and inhibited LPS-induced production of inflammatory mediators, through inhibition of c-Jun N-terminal kinase (JNK) but not ERK and p38 activation [23]. CpG-ODN increased ERK activity [24, 25]. However, the role of ERK activation induced by CpG-ODN in cardiac function during polymicrobial sepsis has not been examined.
In the present study, we examined the effect of CpG-ODN on polymicrobial sepsis–induced cardiac dysfunction. We observed that CpG-ODN significantly attenuated cardiac dysfunction during polymicrobial sepsis. The mechanisms involve activation of both PI3K/Akt and ERK signaling.
METHODS
Experimental Animals
Male C57BL/6 mice were obtained from Jackson Laboratory (Indianapolis, IN).The mice were maintained in the Division of Laboratory Animal Resources at East Tennessee State University. The experiments outlined in this study conform to National Institutes of Health guidelines for animal experimentation [25a]. All aspects of the animal care and experimental protocols were approved by the East Tennessee State University Committee on Animal Care.
Cecal Ligation and Puncture (CLP) Polymicrobial Sepsis Model
Cecal ligation and puncture was performed to induce sepsis in mice as described previously [5, 6, 14, 26]. Briefly, after the mice were anesthetized by 5.0% isoflurane, a midline incision was made on the anterior abdomen, and the cecum was exposed and ligated with a size 2-0 suture. Two punctures were made through the cecum with an 18-gauge needle, and feces were extruded from the holes. The abdomen was then closed in 2 layers. Sham surgery served as the sham control. Immediately following surgery, a single dose of resuscitative fluid (lactated Ringer solution; 50 mL per kg of body weight) was administered by subcutaneous injection.
Echocardiography
Echocardiography was performed as described previously [6, 26]. M-mode tracings were used to measure the left ventricular (LV) end-systolic diameter and the LV end-diastolic diameter. The fractional shortening index and ejection fraction were calculated as described previously [6, 26].
Experimental Design
To examine the effect of CpG-ODN on cardiac function during sepsis, mice were treated with CpG-ODN (10 µg per 30 g of body weight), control CpG-ODN (control-ODN; 10 µg per 30 g of body weight), or inhibitory CpG-ODN (iCpG-ODN; 100 µg per 30 g of body weight) by intraperitoneal injection 1 hour prior to CLP (n = 6/group). Untreated mice served as a CLP control (n = 6). Mice that underwent sham surgery served as sham controls (n = 6/group). Cardiac function was measured by echocardiography before and 6 hours after CLP [6, 26]. The dose of CpG-ODN was selected on the basis of our previous study [27], which showed that administration of CpG-ODN at 10 µg per 30 g of body weight also significantly improved cardiac function in mice that underwent traumatic hemorrhagic shock. The CpG-ODN (CpG-ODN 1826), control-ODN (control-ODN 1826), and iCpG-ODN (iCpG-ODN 2088) were purchased from InvivoGen (San Diego, CA).
To evaluate the role of the PI3K/Akt and ERK signaling pathways in CpG-ODN–induced cardioprotection, the PI3K-specific inhibitor, LY 294002 (LY; 1 mg per 25 g of body weight) [6] and the ERK inhibitor (U0126; 300 µg per 30 g of body weight) were administered to mice 15 minutes prior to CpG-ODN administration (n = 6/group). Cardiac function was measured by echocardiography before and 6 hours after CLP.
In Vitro Experiments
H9C2 rat cardiomyoblasts were cultured in Dulbecco's modified Eagle's medium as described previously [18, 28]. The cells were treated with CpG-ODN at a final concentration of 0.3 µM for 0, 5, 15, 30, and 60 minutes, with 4 replicates at each time point. Control-ODN was used at the same dose. The cells were harvested, and cellular proteins were isolated. The levels of phosphorylated Akt (p-Akt) and p-ERK were examined by Western blot. The association between Ras and TLR9 was examined by immunoprecipitation, followed by immunoblotting.
Immunoprecipitation
Approximately 800 µg of cellular protein was immunoprecipitated with 2 µg of antibody against Ras (Upstate Biotechnology) for 1 hour at 4°C on a rotator, followed by addition of 20 µL of protein A/G-agarose beads (Santa Cruz), as described previously [18, 28]. The immunoprecipitates were washed 3 times in the lysis washing buffer, suspended in 25 µL of loading buffer, and boiled for 5 minutes before the immunoprecipitates were subjected to immunoblotting.
Western Blotting
Western blots were performed as described previously [5, 6, 14, 26]. The membranes were incubated with appropriate primary antibody, including anti-Fas (CD95), anti-FasL, anti-TLR9, (Santa Cruz Biotech, Santa Cruz, CA), anti-phospho-Akt, anti-Akt, anti-phospho-ERK, and anti-ERK (Cell Signaling Technology, Danvers, MA), followed by incubation with peroxidase-conjugated second antibodies (Cell Signaling Technology) and examination with the ECL system (Amersham Pharmacia, Piscataway, NJ). The signals were quantified using a G: Box gel imaging system (Syngene, Fredrick, MD).
In Situ Apoptosis Assay
Cardiac myocyte apoptosis was examined by the TUNEL assay (Roche Applied Science, Indianapolis, IN) in the heart sections, according to the instructions provided by the manufacturer, as described previously [6, 14, 26]. Three slides from each block were evaluated for the percentage of cells that were apoptotic. Four fields of each slide were randomly examined using a defined rectangular field area with a magnification of 40×.
Caspase Activity
Caspase-3/7 and -8 activities in heart tissues were measured using a Caspase-Glo assay kit (Promega) according to the manufacturer's protocol.
Statistics
All other data were expressed as mean ± standard error of the mean. Comparisons of data between groups were made using 1-way analysis of variance, and the Tukey procedure for multiple range tests was performed. P < .05 was considered to be significant.
RESULTS
CpG-ODN Attenuated Cardiac Dysfunction in CLP-Induced Sepsis
As shown in Table 1, cardiac function in untreated CLP mice was markedly decreased, as evidenced by a 31.6% decrease in the ejection fraction and a 40.3% decrease in the fractional shortening index, respectively, compared with baseline. In CpG-ODN–treated CLP mice, the ejection fraction and fractional shortening index were significantly greater (by 35.7% and 45.4%, respectively) than values for untreated CLP mice. Cardiac output and stoke volume in CpG-ODN–treated mice also significantly increased (by 1.3-fold and 1.1-fold, respectively), compared with the untreated CLP group. Administration of either control-ODN or iCpG-ODN did not affect CLP-induced cardiac dysfunction. There were no significant differences in the parameters of cardiac function between control-ODN, iCpG-ODN, and untreated CLP-mice.
Table 1.
The Toll-like Receptor 9 Ligand, CpG Oligodeoxynucleotide (CPG-ODN), Attenuated Cardiac Dysfunction in Mice During Sepsis Induced by Cecal Ligation and Puncture
| Index | Time Relative to Cecal Ligation and Puncture, by Treatment Group |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Untreated |
Control-ODN |
CpG-ODN |
Inhibitory CpG-ODN |
LY294002 |
LY294002 CpG-ODN |
U0126 |
U0126 CpG-ODN |
|||||||||
| Before | After | Before | After | Before | After | Before | After | Before | After | Before | After | Before | After | Before | After | |
| Heart rate, beats/min | 425.6 ± 18.37 | 372.2 ± 22.04a | 435.2 ± 13.26 | 388.1 ± 16.09b | 438.5 ± 25.20 | 416.3 ± 13.94c | 428.3 ± 31.14 | 405.0 ± 7.95 | 441.2 ± 10.15 | 376.1 ± 12.70 | 420.2 ± 13.01 | 400.3 ± 14.61 | 430.9 ± 11.37 | 369.6 ± 14.17 | 422.7 ± 12.33 | 416.4 ± 20.14 |
| Ejection fraction, % | 64.9 ± 7.30 | 44.8 ± 4.25a | 67.6 ± 5.82 | 53.6 ± 3.48b | 67.5 ± 3.88 | 61.32 ± 4.60c | 65.9 ± 1.43 | 49.3 ± 4.78d | 63.2 ± 5.56 | 50.2 ± 4.34 | 64.6 ± 6.51 | 56.7 ± 4.74d | 65.2 ± 4.27 | 51.1 ± 2.78 | 67.2 ± 4.25 | 51.6 ± 7.15d |
| FS, % | 35.3 ± 5.49 | 21.4 ± 2.36a | 37.3 ± 4.48 | 25.2 ± 3.73b | 38.8 ± 5.14 | 33.14 ± 2.19c | 35.8 ± 1.06 | 24.0 ± 4.90d | 33.8 ± 2.74 | 25.4 ± 4.32 | 34.4 ± 5.23 | 27.7 ± 3.73d | 35.3 ± 3.65 | 23.9 ± 3.42 | 40.0 ± 3.24 | 25.9 ± 3.84d |
| LVESD, mm | 2.4 ± 1.01 | 1.7 ± 0.42 | 2.2 ± 0.44 | 1.8 ± 0.66 | 2.3 ± 0.73 | 1.8 ± 0.57 | 2.4 ± 0.33 | 1.7 ± 0.92 | 2.2 ± 0.53 | 1.6 ± 0.41 | 2.2 ± 0.37 | 1.9 ± 0.42 | 2.6 ± 0.69 | 1.7 ± 0.49 | 2.2 ± 0.15 | 1.8 ± 0.33 |
| LVEDD, mm | 6.4 ± 1.04 | 3.1 ± 0.56a | 6.7 ± 0.72 | 3.8 ± 0.64b | 5.9 ± 1.09 | 4.3 ± 1.28 | 6.6 ± 0.92 | 3.2 ± 0.93 | 6.5 ± 0.76 | 3.4 ± 0.29 | 6.9 ± 0.84 | 4.0 ± 0.53 | 6.4 ± 0.72 | 3.5 ± 0.47 | 6.8 ± 1.13 | 4.41 ± 0.71 |
| Stroke volume, µL | 40.1 ± 8.63 | 13.9 ± 1.90a | 44.6 ± 5.98 | 19.7 ± 4.63b | 43.3 ± 8.66 | 33.7 ± 3.76c | 46.7 ± 6.11 | 14.3 ± 1.93d | 43.2 ± 2.37 | 18.1 ± 1.96 | 47.8 ± 3.15 | 23.3 ± 3.05d | 38.0 ± 5.08 | 17.9 ± 4.12 | 45.9 ± 2.97 | 19.5 ± 4.81d |
| Cardiac output, µL/min | 18 816.5 ± 2771.08 | 5068.5 ± 593.89a | 19 514.8 ± 2803.23 | 7738.1 ± 1266.92b | 18 981.6 ± 4029.69 | 14 051.5 ± 1941.27c | 20 001.3 ± 3085.75 | 5799.9 ± 805.56d | 19 065.1 ± 1202.34 | 6818.5 ± 623.89 | 20 064.1 ± 1127.15 | 9326.1 ± 516.24d | 16 391.8 ± 1346.19 | 6604.2 ± 712.36 | 19 405.2 ± 957.31 | 6866.1 ± 579.21d |
Data are mean ± standard error of the mean. Mice were treated with CpG-ODN, control-ODN, or inhibitory CpG-ODN 1 hour before cecal ligation and puncture. In separate experiments, mice were injected with the phosphoinositide 3-kinase inhibitor, LY294002, or the extracellular-signal-related kinase inhibitor, U0126, 15 minutes before CpG-ODN administration. There were 6 mice in each group. Cardiac function was measured by echocardiography before and 6 hours after cecal ligation and puncture.
Abbreviations: FS, fractional shortening index; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter.
a *P < .05, compared with before CLP in the untreated group.
b *P < .05, compared with before CLP in the control-ODN group.
c *P < .05, compared with after CLP in the control-ODN group.
d *P < .05, compared with after CLP in the CpG-ODN group.
CpG-ODN Attenuated CLP-Induced Cardiac Myocyte Apoptosis
Figure 1A shows that CLP significantly increased myocardial apoptosis by 23.8-fold, caspase-3/7 by 30.2%, and caspase-8 by 45.8%, compared with sham control (Figure 1B). In contrast, CpG-ODN significantly attenuated CLP-increased myocardial apoptosis and caspase-8/caspase-3/7 activities in the myocardium. Neither control-ODN nor iCpG-ODN affected CLP-induced increases in myocardial apoptosis.
Figure 1.

CpG oligodeoxynucleotide (CpG-ODN) attenuated cecal ligation and puncture (CLP)–induced myocardial apoptosis. Mice were treated with CpG-ODN, control CpG-ODN (control-ODN), and inhibitory CpG-ODN (iCpG-ODN) by intraperitoneal injection 1 hour prior to CLP (n = 6/group). Sham surgery served as a sham control (n = 6/group). Hearts were harvested, and cellular proteins were prepared. A, Myocardial apoptosis was examined by the TUNEL assay. Red arrows indicate cardiac myocyte apoptosis. Quantitative data are shown at right. B, Caspase-3 and -8 activities in the myocardium were measured by enzyme-linked immunosorbent assay kits. C, Fas and FasL levels in the myocardium were measured by Western blotting. Representative blots are shown above in graphs. *P < .05. Abbreviation: RLU, relative light units.
CLP-Increased Fas and FasL Expression in the Myocardium Was Prevented by CpG-ODN Administration
Fas/FasL-mediated apoptotic signaling plays a role in the induction of apoptosis [29]. We examined the effect of CpG-ODN on Fas/FasL-mediated apoptotic signaling in the myocardium of CLP mice. Figure 1C shows that CLP-associated sepsis markedly increased the levels of Fas (65.1%) and FasL (30.4%), compared with sham control. CpG-ODN prevented CLP-increased myocardial Fas and FasL levels. Neither control-ODN nor iCpG-ODN altered CLP-increased Fas/FasL levels in the myocardium.
CpG-ODN Increased Akt Phosphorylation in the Myocardium Following CLP
Activation of the PI3K/Akt signaling pathway has been reported to protect against sepsis-induced cardiac dysfunction and myocardial ischemic injury [6, 28]. Figure 2 shows that the levels of phospho-Akt and phospho-GSK-3β in CLP mice were markedly decreased (by 53.1% and 61.6%, respectively), compared with sham control. In contrast, CpG-ODN significantly attenuated CLP-decreased levels of myocardial p-Akt and p–GSK-3β, compared with the untreated CLP group. Neither control-ODN nor iCpG-ODN markedly affected CLP-decreased myocardial p-Akt and p-GSK-3β levels, compared with the untreated CLP group.
Figure 2.
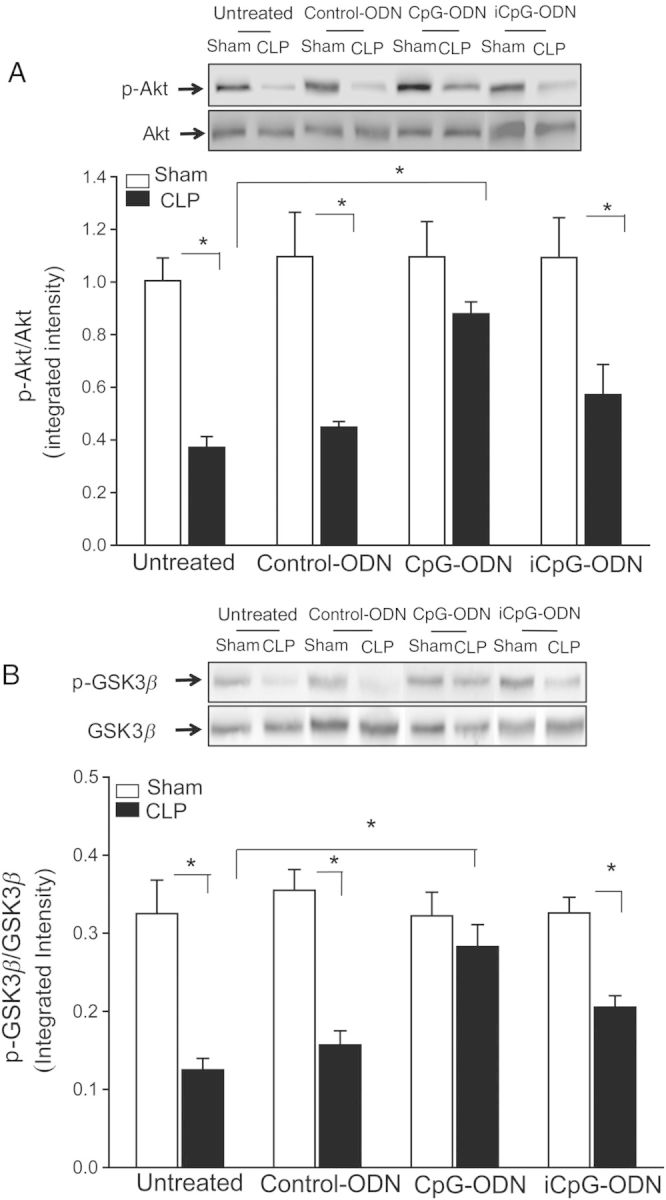
Cecal ligation and puncture (CLP) decreased the levels of Akt and glycogen synthase kinase G β (GSK3β) phosphorylation was attenuated by CpG oligodeoxynucleotide (CpG-ODN). Mice were treated with CpG-ODN, control CpG-ODN (control-ODN), and inhibitory CpG-ODN (iCpG-ODN) by intraperitoneal injection 1 hour prior to CLP (n = 6/group). Sham surgery served as a sham control (n = 6/group). Hearts were harvested, and cellular proteins were prepared. The levels of phosphorylation of Akt (p-Akt; A) and GSK-3β (p-GSK3β; B) were examined by Western blotting. Representative blots are shown above the graph. Abbreviations: S, Sham; C, CLP; C-ODN, Control-ODN; CpG, CpG-ODN; iCpG, inhibitory CpG-ODN. *P < .05.
CpG-ODN Increased ERK1/2 Phosphorylation in the Myocardium Following CLP
Figure 3 shows that CLP did not markedly alter the levels of phosphorylated ERK in the myocardium, compared with sham control. However, CpG-ODN significantly increased the levels of ERK phosphorylation (by 2.4-fold), compared with levels in untreated CLP mice. The levels of ERK phosphorylation in either control-ODN or iCpG-ODN–treated mice were significantly lower than that in CpG-ODN–treated CLP mice.
Figure 3.

CpG oligodeoxynucleotide (CpG-ODN) increased the levels of extracellular-signal-related kinase (ERK) phosphorylation in the myocardium following cecal ligation and puncture (CLP). Mice were treated with CpG-ODN, control CpG-ODN (control-ODN), and inhibitory CpG-ODN (iCpG-ODN) by intraperitoneal injection 1 hour prior to CLP (n = 6/group). Sham surgery served as a sham control (n = 6/group). Hearts were harvested, and cellular proteins were prepared. The levels of phosphorylated ERK (p-ERK) were examined by Western blotting. Representative blots are shown above in the graph. *P < .05.
CpG-ODN Treatment Induced an Association Between TLR9 and Ras in H9C2 Cardiomyoblasts
To investigate the mechanisms by which CpG-ODN increased both Akt and ERK phosphorylation in the myocardium following CLP, we performed in vitro experiments using the H9C2 cell line. Figure 4 shows that CpG-ODN increased both Akt and ERK phosphorylation in a time-dependent manner. Akt phosphorylation was increased at 5 minutes and was highest at 60 minutes following CpG-ODN treatment. ERK phosphorylation was increased at 5 minutes and peaked at 15 minutes after CpG-ODN stimulation.
Figure 4.

CpG oligodeoxynucleotide (CpG-ODN) increased both Akt and extracellular-signal-related kinase (ERK) phosphorylation and induced an association between Ras and Toll-like receptor 9 (TLR9) in H9C2 cells. H9C2 cells were treated with CpG-ODN or control CpG-ODN (control-ODN) at indicated times. Untreated cells served as a control. Cells were harvested, and cellular proteins were isolated for analysis of phosphorylated Akt (p-Akt; A) and phosphorylated ERK (B; p-ERK) by Western blotting. Immunoprecipitation (IP) was performed with a specific anti-Ras antibody, followed by immunoblotting (IB) with a specific anti-TLR9 antibody (C). Representative blots are shown above in the graph. There were 4 replicates for each time point. *P < .05.
Ras is involved in activation of the both Raf1/MEK/ERK signaling and the PI3K/NF-κB pathways [30, 31]. To investigate whether CpG-ODN induces an association between Ras and TLR9 that results in activation of PI3K and ERK, we performed immunoprecipitation with anti-Ras followed by immunoblotting with anti-TLR9. As shown in Figure 4C, TLR9 appeared in the anti-Ras immunoprecipitates at 15 minutes, reached peak level at 30 minutes, and decreased at 60 minutes after CpG-ODN stimulation. Control-ODN did not induce an association between TLR9 and Ras.
Pharmacological Inhibition of PI3K/Akt and ERK1/2 Abrogates CpG-ODN–Attenuated Cardiac Dysfunction in CLP-Induced Sepsis
To determine whether activation of PI3K/Akt signaling contributes to CpG-ODN–associated protection against CLP-induced cardiac dysfunction, we treated mice with a PI3K inhibitor, LY294002, 15 minutes prior to CpG-ODN administration. Cardiac function was measured by echocardiography before and 6 hours after CLP. As shown in Table 1, LY294002 partially abolished CpG-ODN attenuation of CLP-induced cardiac dysfunction. The ejection fraction and fractional shortening index in CpG-ODN + LY294002–treated CLP mice were significantly lower than in CpG-ODN–treated CLP mice but still higher than in untreated CLP mice, suggesting that PI3K inhibition partially abolished CpG-ODN–induced attenuation of cardiac dysfunction in CLP mice. Figure 5A shows that LY294002 significantly prevented CpG-ODN–increased levels of phosphorylated Akt in the myocardium following CLP.
Figure 5.
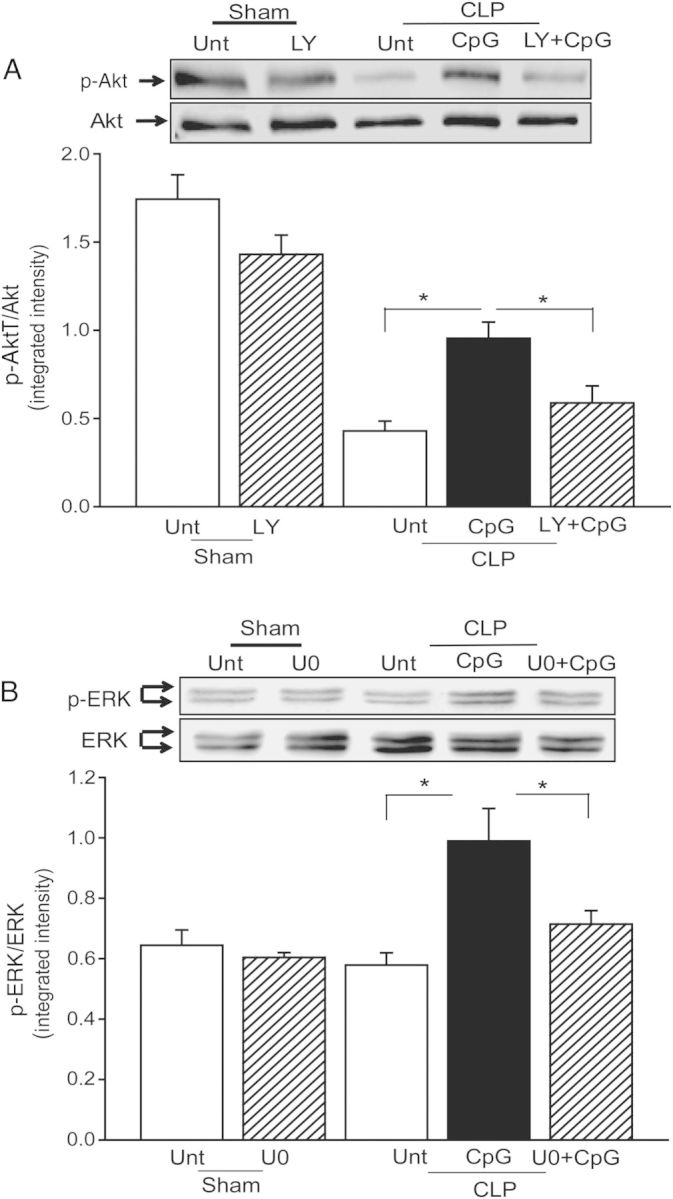
Phosphoinositide 3-kinase (PI3K) or extracellular-signal-related kinase (ERK) inhibition abrogated CpG oligodeoxynucleotide (CpG-ODN)–induced attenuation of cardiac dysfunction in polymicrobial sepsis. Mice were treated with the PI3K-specific inhibitor Ly294002 (LY) or the ERK-specific inhibitor U0126 (U0) 15 minutes prior to CpG-ODN administration. Cardiac function was measured by echocardiography before and 6 hours after cecal ligation and puncture. Hearts were harvested, and cellular proteins were isolated for analysis of phosphorylation of Akt (p-Akt; A) and phosphorylation of ERK (p-ERK; B) by Western blotting. Representative blots are shown above in the graph. There were 6 mice in each group. *P < .05. Abbreviations: LY+CpG, CpG-ODN+LY294002; Unt, untreated; U + CpG: CpG-ODN+U0126.
We also examined the role of activation of ERK in CpG-ODN–attenuated cardiac dysfunction in CLP-septic mice. Mice were treated with a selective inhibitor of ERK1/2, U0126, 15 minutes prior to CpG-ODN administration. Cardiac function was measured before and 6 hours after CLP. The ejection fraction and fractional shortening index of U0126 + CpG-ODN–treated CLP mice were significantly reduced (by 18.7% and by 25.6%), compared with CpG-ODN–treated CLP mice (Table 1), indicating that ERK inhibition partially abolished CpG-ODN–induced protection against CLP-induced cardiac dysfunction. Figure 5B shows that U0126 prevented CpG-ODN–increased ERK1/2 phosphorylation in the myocardium following CLP.
DISCUSSION
The important finding in the present study is that the TLR9 ligand, CpG-ODN, significantly attenuated CLP-induced cardiac dysfunction. CpG-ODN activated the PI3K/Akt and ERK signaling pathways through a complex of TLR9 with Ras, a small GPT-binding protein. Inhibition of either PI3K or ERK significantly reduced the cardioprotection of CpG-ODN in polymicrobial septic mice. The data indicate that modulation of TLR9 by its ligand, CpG-ODN, could be an effective approach for treating patients with sepsis or septic shock.
Cardiovascular dysfunction is an early and fatal complication of sepsis and septic shock and contributes to the high mortality rate among septic patients in the ICU [1, 2]. Rossi et al reported that the foci of myocytolysis and cardiomyocyte tumefaction were present in human heart tissues obtained from long-term severe sepsis and septic shock [32] and in murine hearts 24 hours after CLP [33]. In addition, there were higher numbers of infiltrated macrophages and increased expression of TNF-α, as well as oxidative stress in the myocardium after sepsis and septic shock [32, 33]. We have observed infiltrated macrophages and neutrophils in heart tissues (data not shown). It is possible that the innate immune and inflammatory responses play a role in cardiac dysfunction during early stage of sepsis and septic shock, while cardiac structural changes contribute to cardiac dysfunction in the later phase of sepsis and septic shock [34]. We have observed that pretreatment of mice with CpG-ODN significantly attenuated CLP-induced cardiac dysfunction. CpG-ODN is a synthetic ODN that is recognized by TLR9 [8, 10]. Rice et al [11] and Weighardt et al [12] have reported that CpG-ODN increased survival among rats and mice with acute polymicrobial sepsis [11, 12]. Therefore, attenuation of cardiac dysfunction could be an important mechanism by which CpG-ODN increases survival during sepsis and septic shock [11].
Cardiac myocyte apoptosis plays an important role in cardiac dysfunction during sepsis and septic shock [35]. The present study showed that CpG-ODN attenuated CLP–associated increases in myocardial apoptosis, suggesting that reduction in myocardial apoptosis by CpG-ODN may be a mechanism responsible for the attenuation of cardiac dysfunction in CLP mice. Several studies have shown that CpG-ODN decreased apoptosis in macrophages [36] and dendritic cells [37] by upregulation of cellular inhibitor of apoptosis proteins, Bcl-2, and Bcl-x [37]. We have observed that CpG-ODN prevented increased levels of Fas and FasL in the myocardium after CLP. The data indicate that CpG-ODN attenuation of CLP-associated increases in myocardial apoptosis is mediated, in part, through inhibition of the Fas-mediated apoptotic signaling pathway. Consistently, CLP-associated increases in caspase-8 and caspase-3 activities were markedly attenuated by CpG-ODN.
Activation of the PI3K/Akt signaling pathway attenuates cardiac myocyte apoptosis in polymicrobial sepsis [6, 28]. CpG-ODN significantly attenuated CLP-decreased myocardial Akt and GSK-3β phosphorylation. The data suggests that CpG-ODN can activate the PI3K/Akt signaling pathway. Recent evidence demonstrated that there is cross talk between TLRs and the PI3K/Akt signaling pathway [15]. We have previously reported that modulation of TLR2 or TLR4 with their ligands activated the PI3K/Akt signaling pathway [18, 28]. To determine whether activation of the PI3K/Akt signaling pathway is responsible for CpG-ODN–attenuated cardiac dysfunction in CLP mice, we treated mice with a PI3K-specific inhibitor, LY294002, prior to CpG-ODN administration and observed that PI3K inhibition partially abolished CpG-ODN–induced cardioprotection in CLP mice, indicating that CpG-ODN–induced protection occurs, in part, through activation of the PI3K/Akt signaling pathway. The data also suggest that an additional mechanism could be involved in CpG-ODN–induced protection against CLP-induced cardiac dysfunction.
Activation of ERK signaling has been reported to protect cells from injury [38]. CpG-ODN can activate the MAPK pathway [39]. We observed that CLP did not induce ERK phosphorylation in the myocardium. However, CpG-ODN significantly induced ERK phosphorylation in the myocardium of CLP mice. CpG-ODN did not induce phosphorylation of either JNK or p38 MAPK in the myocardium following CLP (data not shown), suggesting that CpG-ODN may specifically activate myocardial ERK following CLP. ERK inhibition with the selective inhibitor U0126 partially abolished CpG-ODN–induced protection against CLP-induced cardiac dysfunction, indicating that activation of ERK contributed to attenuation of cardiac dysfunction by CpG-ODN in CLP-septic mice.
Ras is a small GTP-binding protein that plays a role in the activation of both PI3K/Akt and ERK signaling pathways [30]. To determine the role of Ras in CpG-ODN–activated PI3K/Akt and ERK signaling pathways, we performed in vitro experiments that used H9C2 cardiomyoblasts. CpG-ODN induced an association of TLR9 with Ras, as evidenced by TLR9 present in the anti-Ras immunoprecipitates. The data suggest that CpG-ODN–associated activation of both PI3K/Akt and ERK signaling pathways is mediated by an interaction between TLR9 and Ras. Our observation is consistent with the study by Xu et al [39], which showed that CpG-ODN induced an association between Ras and TLR9 in macrophages [39]. It is well-known that Ras interacts with Raf-1, resulting in activation of both PI3K/Akt and ERK signaling pathways [40]. At present, we do not understand how TLR9 associates with Ras following CpG-ODN administration. Collectively, these data suggest that CpG-ODN–associated attenuation of cardiac dysfunction in CLP mice is mediated, in part, via an association of TLR9 with Ras, resulting in activation of both PI3K/Akt and ERK signaling pathways.
Notes
Financial support. This work was supported by the National Institutes of Health (grants HL071837 [to C. L.], GM083016 [to C. L. and D. W.], and GM53522 [to D. W.]), HL091405, HL092459 and a Merit Review Grant from the Department of Veterans Affairs [to KS].
Potential conflict of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Bulmer BJ. Cardiovascular dysfunction in sepsis and critical illness. Vet Clin North Am Small Anim Pract. 2011;41:717–26. doi: 10.1016/j.cvsm.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Parrillo JE, Parker MM, Natanson C, et al. Septic shock in humans: Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–42. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 3.Vieillard-Baron A, Caille V, Charron C, Belliard G, Page G, Jardin BF. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med. 2008;36:1701–6. doi: 10.1097/CCM.0b013e318174db05. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R. Toll-like Receptors and Innate Immunity. Nature Reviews Immunology. 2001;1:135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 5.Williams DL, Ha T, Li C, et al. Modulation of tissue toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–18. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- 6.Ha T, Lu C, Liu L, et al. TLR2 ligands attenuate cardiac dysfunction in polymicrobial sepsis via a phosphoinositide-3-kinase dependent mechanism. Am J Physiol Heart Circ Physiol. 2010;298:H984–91. doi: 10.1152/ajpheart.01109.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou L, Feng Y, Chen YJ, et al. Toll-like receptor 2 plays a critical role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2010;38:1335–42. doi: 10.1097/CCM.0b013e3181d99e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Advanced Drug Delivery Reviews. 2008;60:795–804. doi: 10.1016/j.addr.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Gupta GK, Agrawal DK. CpG oligodeoxynucleotides as TLR9 agonists: therapeutic application in allergy and asthma. BioDrugs. 2010;24:225–35. doi: 10.2165/11536140-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nature Reviews Drug Discovery. 2006;5:471–84. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 11.Rice L, Orlow D, Ceonzo K, et al. CpG oligodeoxynucleotide protection in polymicrobial sepsis is dependent on interleukin-17. J Infect Dis. 2005;191:1368–76. doi: 10.1086/428452. [DOI] [PubMed] [Google Scholar]
- 12.Weighardt H, Feterowski C, Veit M, Rump M, Wagner H, Holzmann B. Increased resistance against acute polymicrobial sepsis in mice challenged with immunostimulatory CpG oligodeoxynucleotides is related to an enhanced innate effector cell response. J Immunol. 2000;165:4537–43. doi: 10.4049/jimmunol.165.8.4537. [DOI] [PubMed] [Google Scholar]
- 13.Mathur S, Walley KR, Boyd JH. The TLR9 ligand CpG-C attenuates acute inflammatory cardiac dysfunction. Shock. 2011;36:478–83. doi: 10.1097/SHK.0b013e31822d6442. [DOI] [PubMed] [Google Scholar]
- 14.Williams DL, Li C, Ha T, et al. Modulation of the phosphoinositide 3-Kinase pathway alters innate resistance to polymicrobial sepsis. J Immunol. 2004;172:449–56. doi: 10.4049/jimmunol.172.1.449. [DOI] [PubMed] [Google Scholar]
- 15.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 16.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends in Immunology. 2003;24:358–63. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 17.Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide-3-kinase signaling pathway alters host resistance to sepsis, inflammation, and ischemia/reperfusion injury. Shock. 2006;25:432–9. doi: 10.1097/01.shk.0000209542.76305.55. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Ha T, Kelley J, et al. Modulating Toll-like receptor mediated signaling by (1→3)-b-D-glucan rapidly induces cardioprotection. Cardiovasc Res. 2003;61:538–47. doi: 10.1016/j.cardiores.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Guha M, Mackman N. The PI3K-Akt pathway limits LPS activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 20.Roskoski R., Jr ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol Res. 2012;66:105–43. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–46. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, Lu X, Arnold P, et al. Mitogen-activated protein kinase phosphatase-1 inhibits myocardial TNF-a expression and improves cardiac function during endotoxemia. Cardiovasc Res. 2012;93:471–9. doi: 10.1093/cvr/cvr346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MS, Bae GS, Park KC, et al. Myrrh inhibits LPS-induced inflammatory respnse and protects from cecal ligation and puncture-induced sepsis. Evid Based Complement Altern Med. 2012;2012:278718. doi: 10.1155/2012/278718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Nardo D, De Nardo CM, Nquyen T, Hamilton JA, Scholz GM. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol. 2009;183:8110–8. doi: 10.4049/jimmunol.0901031. [DOI] [PubMed] [Google Scholar]
- 25.Lim EJ, Lee SH, Lee JG, et al. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp Mol Med. 2007;39:239–45. doi: 10.1038/emm.2007.27. [DOI] [PubMed] [Google Scholar]
- 25a.National Institutes of Health (NIH) 8th ed. Bethesda, MD: NIH; 2011. Guide for the care and use of laboratory animals. [Google Scholar]
- 26.Ha T, Hua F, Grant D, et al. Glucan phosphate attenuates cardiac dysfunction and inhibits cardiac MIF expression and apoptosis in septic mice. Am J Physiol Heart Circ Physiol. 2006;291:H1910–18. doi: 10.1152/ajpheart.01264.2005. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Gao M, Ha T, et al. The TLR9 agonist, CpG-ODN 1826, ameliorates cardiac dysfunction after trauma-hemorrhage. Shock. 2012;38:146–52. doi: 10.1097/SHK.0b013e31825ce0de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha T, Hu Y, Liu L, et al. TLR2 ligands induce cardioprotection against ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2010;87:694–703. doi: 10.1093/cvr/cvq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehrenschwender M, Wajant H. The role of FasL and Fas in health and disease. Adv Exp Med Biol. 2009;647:64–93. doi: 10.1007/978-0-387-89520-8_5. [DOI] [PubMed] [Google Scholar]
- 30.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002;277:31099–106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- 31.McCubrey JA, Steelman LS, Kempf CR, et al. Therapeutic Resistance Resulting from Mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Signaling Pathways. J Cell Physiol. 2011;226:2762–81. doi: 10.1002/jcp.22647. [DOI] [PubMed] [Google Scholar]
- 32.Rossi MA, Celes MRN, Prado CM, Saggioro FP. Myocardial Structural Changes in Long-Term Human Severe Sepsis/Septic Shock May be Responsible for Cardiac Dysfunction. Shock. 2007;27:10–8. doi: 10.1097/01.shk.0000235141.05528.47. [DOI] [PubMed] [Google Scholar]
- 33.Celes MR, Torres-Duenas D, Prado CM, et al. Increased sarcolemmal permeability as an early event in experimental septic cardiomyopathy: a potential role for oxidative damage to lipids and proteins. Shock. 2010;33:322–31. doi: 10.1097/SHK.0b013e3181b38ef6. [DOI] [PubMed] [Google Scholar]
- 34.Celes MRN, Prado CM, Rossi MA. Sepsis: Going to the Heart of the Matter. Pathobiology. 2013;80:70–86. doi: 10.1159/000341640. [DOI] [PubMed] [Google Scholar]
- 35.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 36.Sester DP, Brion K, Trieu A, et al. CpG DNA Activates Survival in Murine Macrophages through TLR9 and the Phosphatidylinositol 3-Kinase-Akt Pathway. J Immunol. 2006;177:4473–80. doi: 10.4049/jimmunol.177.7.4473. [DOI] [PubMed] [Google Scholar]
- 37.Park Y, Lee SW, Sung YC. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J Immunol. 2002;168:5–8. doi: 10.4049/jimmunol.168.1.5. [DOI] [PubMed] [Google Scholar]
- 38.Kehat I, Davis J, Triburcy M, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–83. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu H, An H, Yu Y, Zhang M, Qi R, Cao X. Ras participates in CpG oligodeoxynucleotide signaling through association with toll-like receptor 9 and promotion of interleukin-1 receptor-associated kinase/tumor necrosis factor receptor-associated factor 6 complex formation in macrophages. J Biol Chem. 2003;278:36334–40. doi: 10.1074/jbc.M305698200. [DOI] [PubMed] [Google Scholar]
- 40.Meier F, Schittek B, Busch S, et al. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]