

Abstract
Expansion of the genetic alphabet with an unnatural base pair is a long-standing goal of synthetic biology. We have developed a class of unnatural base pairs, formed between d5SICS and analogues of dMMO2, that are efficiently and selectively replicated by the Klenow Fragment (Kf) DNA polymerase. In an effort to further characterize and optimize replication, we report the synthesis of five new dMMO2 analogues bearing different substituents designed to be oriented into the developing major groove and an analysis of their insertion opposite d5SICS by Kf and Thermus aquaticus DNA polymerase I (Taq). We also expand the analysis of the previously optimized pair, dNaM-d5SICS, to include replication by Taq. Finally, the efficiency and fidelity of PCR amplification of the base pairs by Taq or Deep Vent polymerases is examined. The resulting structure-activity relationship data suggest that the major determinants of efficient replication are the minimization of desolvation effects and the introduction of favorable hydrophobic packing, and that Taq is more sensitive than Kf to structural changes. In addition, we identify an analogue, dNMO1, that is a better partner for d5SICS than any of the previously identified dMMO2 analogues with the exception of dNaM. We also find that dNaM-d5SICS is replicated by both Kf and Taq with rates approaching those of a natural base pair.
Keywords: DNA, Klenow fragment, Taq, PCR, Genetic Alphabet
Introduction
The four letter genetic alphabet is conserved throughout nature and is based on the complementary shape and hydrogen-bonding (H-bonding) of the natural purines and pyrimidines. For over two decades, efforts have been focused on the development of unnatural base pairs where pairing is mediated by orthogonal H-bonding patterns, and progress along this route continues.[1-4] However, we[5-16] and others,[17-21] have demonstrated that hydrophobic and packing forces are also sufficient to underlie the efficient and selective replication of an unnatural base pair. If sufficiently well replicated, such unnatural base pairs could be used as part of an expanded genetic system and would allow for an increase in the information potential of a genome, but will likely be useful immediately for in vitro applications, such as the site-specific labeling of enzymatically synthesized DNA or RNA with novel functionality (i.e. fluorophores or reactive moieties) for SELEX (Systematic Evolution of Ligands by Exponential Enrichment)[22] or nanomaterial applications.[23]
Two of the most promising unnatural base pairs that we have identified are those formed by d5SICS and either dMMO2 (dMMO2-d5SICS) or dNaM (dNaM-d5SICS) (Figure 1A and B).[5,6,9,13,14]. Previous studies with the Klenow fragment of E. coli DNA polymerase I (Kf) and T7 RNA polymerase have demonstrated that d5SICS-dNaM is replicated and transcribed with greater efficiency than dMMO2-d5SICS.[14] However, the dMMO2 scaffold is simpler and more atom-economical than dNaM. In addition, the dMMO2 scaffold provides more options for linker attachment, which is required for site-specific labeling of the DNA, including attachment at the site analogous to that widely used with the natural pyrimidines[24] and not available with dNaM. Thus, our current efforts to expand the genetic alphabet include the continued evaluation of dNaM-d5SICS, with a particular focus on recognition by other polymerases that enable specific applications such as PCR, as well as to continue to generate structure-activity relationship (SAR) data that will facilitate the further optimization of dMMO2-d5SICS.
Figure 1.

A) dMMO2-d5SICS unnatural base pair. B) dNaM and dDMO. C) dMMO2 analogues synthesized and evaluated in this study. Only nucleobase moieties are shown; sugar and phosphate backbone are omitted for clarity.
A limiting step of replicating DNA containing dMMO2-d5SICS is the incorporation of dMMO2TP opposite d5SICS. Because existing SAR data suggests that this step is more sensitive to modifications of the triphosphate than of the templating nucleobase, we have focused on derivatizing dMMO2TP. The SAR data also clearly reveal that the ortho methoxy group is absolutely required for replication (the methyl group facilitates triphosphate insertion and the oxygen atom facilitates continued primer extension once the unnatural nucleotide is incorporated at the terminus [6]). In addition, the accumulated SAR data also suggest that para derivatization is more promising than meta derivatization.[13,15] Initial efforts to identify beneficial modifications at the para position yielded dDMO (Figure 1B). While the dDMO-d5SICS unnatural base pair is replicated more efficiently than the parental dMMO2-d5SICS pair, it is still replicated less efficiently than dNaM-d5SICS.[8]
To continue our efforts to optimize the dMMO2 scaffold, we now report the synthesis and characterization of five new derivatives bearing different moieties at the para position (Figure 1C). The specific modifications to the dMMO2 scaffold were selected to systematically vary the size, shape, electronic properties, and H-bonding potential of the nucleobase. The unnatural triphosphates were analyzed by determining the steady-state efficiencies with which they are inserted opposite d5SICS in a DNA template by Kf or by Thermus aquaticus DNA polymerase I (Taq). To further explore dNaM-d5SICS, we also characterized its synthesis in both strand contexts (i.e. insertion of dNaMTP opposite d5SICS and d5SICSTP opposite dNaM) by Taq. To facilitate the detailed comparison of all of the analogues in side-by-side experiments, the previously reported efficiencies of dNaM-d5SICS synthesis by Kf were re-determined, as were the insertion efficiencies of dMMO2TP and dDMOTP opposite d5SICS by Kf and Taq. Finally, each triphosphate was also analyzed by characterizing the PCR amplification of the corresponding unnatural base pair with d5SICS by Taq or Deep Vent polymerases. The results reveal important, and in some cases, polymerase-specific SAR data, and we find that dNMO1TP is better optimized than the other new analogues as a partner for d5SICS. The current analysis also reveals that dNaM-d5SICS is synthesized by Kf better than previously appreciated, and synthesized equally well by Taq, solidifying its position as the most promising unnatural base pair identified to date.
Results
Nucleotide design, synthesis, and evaluation
The unnatural nucleotides dAMO1, dAMO2, dAMO3, dPMO1, and dNMO1 (Figure 1C) were designed to position different substituents in the developing major groove during replication. Each substituent is attached via a single bond to the position para to the glycosidic bond because SAR data suggest that rotational flexibility is important for efficient triphosphate incorporation[8] (although dNaM is an interesting and incompletely understood exception), presumably by allowing for the optimization of the developing interactions with d5SICS and/or the DNA polymerase. The most simple derivative of the series, dAMO1, bears only an amino substituent at the para position. In dAMO2 and dAMO3, the amino moiety is modified with acetyl and trifluoroacetyl groups, which increase the size and reduce the ability of the substituent to donate electron density into the aromatic ring of the nucleobase scaffold. The nitro group of dNMO1 is a potent electron withdrawing substituent. In contrast, incorporating the nitrogen atom within the context of the pyrrolo ring of dPMO1 should have more modest electronic effects, but significantly increase the aromatic surface area of the scaffold.
The unnatural nucleotide derivatives were synthesized as shown in Scheme 1. Briefly, the modified nucleoside 3 was obtained in three steps via Heck coupling between the 2′-deoxyribose glycal 1 and the appropriately iodinated N-Cbz-protected anisidine 2, followed by deprotection of the sugar moiety and selective reduction of the resulting 3′ keto group. Acceptable yields of the Heck coupling product required the use of an electron withdrawing group to protect the aromatic amine. The major coupling product was the desired β-anomer, confirmed by NOE experiments, which was separated from the minor α-anomer by silica gel column chromatography. The hydroxyl groups were simultaneously protected with tetraisopropyldisiloxane groups, and the Cbz group was removed by hydrogenation. Compound 4 was used as a common precursor to introduce diversity and access each of the desired nucleotides. We found that even under mildly acidic conditions, compound 4 is prone to epimerize to a mixture of α- and β-anomers, requiring careful control of all reaction conditions.
Toward dNMO1, the aromatic amine of 4 was first oxidized using a potassium iodide-tert-butyl hydroperoxide catalyst, which cleanly converted the amine to the nitro group, while approaches based on in situ generation of the dimethyldioxirane or the use of methyltrioxorhenium/O2 resulted in incomplete oxidation to the nitroso and hydroxylamine compounds, as well as the production of azoxy self coupled products. dNMO1(5a) was finally obtained by removal of the silyl protecting groups. Protected dPMO1 was obtained via acid-free condensation[25] in aqueous 2,5-dimethoxytetrahydrofuran with microwave irradiation at 140 °C, which afforded the pure β-anomer, and the free nucleoside 5b was obtained by removal of the silyl protecting group. Compounds 5c and 5d were obtained by acylation with acetic anhydride or trifluoroacetic anhydride respectively, followed by TBAF-mediated sugar deprotection.
Free nucleosides 5a - d were converted to the corresponding triphosphates 6a - d using Ludwig conditions[26] and purified by anion exchange chromatography, followed by HPLC. dAMO1TP (6e) was obtained from dAMO3TP (6d) by ammonia-mediated deprotection of the amino group at room temperature. The purity of each triphosphate was confirmed by 31P NMR, HPLC, and MALDI-TOF (Supporting Information). The triphosphates of dMMO2, dDMO, dNaM and d5SICS were prepared as described previously,[6,8,13] and the phosphoramidites of dNaM and d5SICS were prepared and incorporated in to DNA as described previously.[13]
Each triphosphate was analyzed by examining its insertion opposite a correct or incorrect nucleotide in a DNA template by Kf and Taq under steady-state conditions, which provides a convenient assay to measure the overall rate at which product is formed.[27] The most interpretable data is the second order rate constant (or efficiency, kcat/KM) relating the duplex-bound polymerase and free triphosphate to the rate limiting transition state for the multiple turnover reaction. Assays using Kf were performed at 25 °C, which is close to the enzyme's optimal temperature of 37 °C. While Taq is a thermophilic polymerases, assays using Taq were performed at 50 °C to allow for the use of the same primer-template substrates used with Kf and in previous studies. Although it is approximately 20 °C below optimal, Taq does retain a significant level of activity at this temperature.[28] Each analogue was also examined as a partner for d5SICS by PCR.
Efficiency of insertion of dMMO2TP, dDMOTP, and each natural dNTP opposite d5SICS
To gauge the efficiency of unnatural base pair synthesis, we first analyzed the insertion of dATP opposite dT in sequence context I (Table 1). In good agreement with previous data, we found that dATP was inserted opposite dT in the template with a kcat/KM of 7.7 × 108 M-1min-1. We then examined the insertion of dMMO2TP and dDMOTP opposite d5SICS under identical conditions. We found that that dMMO2TP and dDMOTP are inserted with efficiencies of 3.8 × 105 and 2.2 × 106 M-1min-1, respectively. To determine fidelity, we measured the rate of insertion of each natural triphosphate opposite d5SICS in the same sequence context. We found that dATP, dGTP, and dTTP are inserted with efficiencies between 9 × 103 and 1.5 × 105 M-1min-1, while dCTP is not inserted at a detectable level (kcat/KM < 1 × 103 M-1min-1). All of the data are in good agreement with previously reported data.[6,8] We also determined the efficiency with which Taq inserts dATP opposite dT and dMMO2TP opposite d5SICS in sequence context I and observed values of 8.2 × 107 M-1min-1 and 9.7 × 104 M-1min-1, respectively, also in good agreement with previously reported data.[5]
Table 1.
Kinetic data for Kf-mediated insertion of triphosphates (dYTP) opposite d5SICS or dNaM in the template in sequence context I; insertion of dATP opposite dT is provided for comparison.
| 5′–d(TAATACGACTCACTATAGGGAGA) | ||||
|---|---|---|---|---|
| 3′–d(ATTATGCTGAGTGATATCCCTCTXGCTAGGTTACGGCAGGATCGC) | ||||
|
| ||||
| X | Y | kcat (min-1) | KM (μM) | kcat/KM (× 105 M-1min-1) |
| T | A | 4.1 ± 0.3 | 0.0053 ± 0.0004 | 7700 |
| 5SICS | NaM | 14.6 ± 0.8 | 0.25 ± 0.04 | 580 |
| DMO | 27.0 ± 1.5 | 12.2 ± 1.5 | 22 | |
| MMO2 | 13.7 ± 2.0 | 35.8 ± 0.6 | 3.8 | |
| NMO1 | 50.1 ± 8.8 | 22.0 ± 3.5 | 23 | |
| PMO1 | 41.7 ± 6.7 | 20.7 ± 2.6 | 20 | |
| AMO1 | 15.3 ± 3.5 | 178 ± 62 | 0.86 | |
| AMO3 | 13.4 ± 2.8 | 117 ± 30 | 1.1 | |
| AMO2 | 7.0 ± 1.0 | 464 ± 100 | 0.15 | |
| 5SICS | 12.6 ± 0.7 | 44.2 ± 11.7 | 2.9 | |
| A | 2.1 ± 0.3 | 52.4 ± 7.2 | 0.40 | |
| G | 11.7 ± 1.3 | 75.5 ± 3.7 | 1.5 | |
| C | n.d.[a] | n.d.[a] | < 0.01 | |
| T | 2.1 ±0.4 | 230 ± 11 | 0.091 | |
| NaM | 5SICS | 8.3 ± 1.1 | 0.039 ± 0.004 | 2100 |
| NaM | 51.5 ± 3.4 | 5.4 ± 0.3 | 95 | |
| A | 24.7 ± 4.7 | 14.0 ± 0.8 | 18 | |
| G | n.d.[a] | n.d.[a] | < 0.01 | |
| C | n.d.[a] | n.d.[a] | < 0.01 | |
| T | 1.6 ± 0.2 | 129 ± 15 | 0.12 | |
Below limits of detection.
Insertion efficiencies of dMMO2TP derivatives opposite d5SICS
The insertion by Kf of each dMMO2 triphosphate derivative opposite d5SICS was characterized in sequence context I (Table 1). We found that the most simple derivative, dAMO1TP, is inserted opposite d5SICS with a second order rate constant of 8.6 × 104 M-1min-1. dAMO3TP is inserted with a similar efficiency (1.1 × 105 M-1min-1), while dAMO2TP is inserted 6-fold less efficiently (1.5 × 104 M-1min-1). However, dPMO1TP and dNMO1TP are inserted with higher efficiencies of 2.0 × 106 and 2.3 × 106 M-1min-1, respectively. The increased efficiency of dPMO1TP and dNMO1TP insertion opposite d5SICS by Kf results from both an increase in the apparent kcat and a decrease in the apparent KM.
We next characterized the ability of Taq to insert each dMMO2TP derivative opposite d5SICS in the same sequence context (Table 2). We found that Taq inserts dDMOTP with an efficiency of 1.6 × 105 M-1min-1, which is virtually identical to the efficiency of dMMO2TP insertion. We found that Taq inserts dAMO1TP, dAMO2TP, and dAMO3TP opposite d5SICS much less efficiently, with second order rate constants of 4.9 × 103 M-1min-1, 1.2 × 103 M-1min-1, and 1.6 × 103 M-1min-1, respectively. While insertion of dPMO1TP opposite d5SICS by Taq is also not very efficient (3.2 × 104 M-1min-1), insertion of dNMO1TP is significantly more efficient (4.3 × 105 M-1min-1).
Table 2.
Kinetic data for Taq-mediated insertion of triphosphates (dYTP) opposite d5SICS or dNaM in the template in sequence context I; insertion of dATP opposite dT is provided for comparison.
| 5′–d(TAATACGACTCACTATAGGGAGA) | ||||
|---|---|---|---|---|
| 3′–d(ATTATGCTGAGTGATATCCCTCTXGCTAGGTTACGGCAGGATCGC) | ||||
|
| ||||
| X | Y | kcat (min-1) | KM (μM) | kcat/KM (× 105 M-1min-1) |
| T | A | 2.6 ± 0.2 | 0.032 ± 0.002 | 820 |
| 5SICS | NaM | 2.7 ± 0.4 | 0.35 ± 0.04 | 76 |
| DMO | 3.3 ± 1.7 | 21.0 ± 0.5 | 1.6 | |
| MMO2 | 3.4 ± 0.3 | 35.0 ± 6.0 | 0.97 | |
| NMO1 | 5.5 ± 0.2 | 12.8 ± 3.4 | 4.3 | |
| PMO1 | 3.6 ± 1.4 | 113 ± 25 | 0.32 | |
| AMO1 | 0.83 ± 0.13 | 170 ± 31 | 0.049 | |
| AMO3 | 0.35 ± 0.06 | 214 ± 28 | 0.016 | |
| AMO2 | 0.45± 0.06 | 370 ± 36 | 0.012 | |
| 5SICS | 3.4 ± 0.5 | 189 ± 16 | 0.18 | |
| A | 0.5 ± 0.05 | 147 ± 7 | 0.035 | |
| G | 4.5 ± 0.1 | 216 ± 42 | 0.21 | |
| C | n.d.[a] | n.d.[a] | < 0.01 | |
| T | 0.8 ± 0.1 | 303 ± 19 | 0.026 | |
| NaM | 5SICS | 2.4 ± 0.6 | 0.34 ± 0.03 | 71 |
| NaM | 7.9± 1.4 | 151± 9 | 0.52 | |
| A | 4.0 ± 0.8 | 84.0 ± 4.0 | 0.48 | |
| G | n.d.[a] | n.d.[a] | < 0.01 | |
| C | n.d.[a] | n.d.[a] | < 0.01 | |
| T | 1.0 ± 0.2 | 421± 71 | 0.024 | |
Below limits of detection.
Of the new dMMO2 analogues examined, the most promising in sequence context I are dPMO1TP, and especially dNMO1TP, thus we next determined whether their insertion efficiency by Kf opposite d5SICS is dependent on sequence context, using sequence context II (Table 3). Again, for comparison, we re-determined the efficiencies of dATP, dMMO2TP or dDMOTP insertion opposite their cognate base in the same sequence context. The natural base pair is synthesized with an efficiency of 5.8 × 108 M-1min-1, while dMMO2TP is inserted opposite d5SICS with an efficiency of 1.9 × 105 M-1min-1; both values are in good agreement with previously reported data.[13] Kf inserts dDMOTP opposite d5SICS with an efficiency of 3.9 × 105 M-1min-1. Interestingly, we found that insertion of both dPMO1TP and dNMO1TP opposite d5SICS by Kf is approximately 4- and 8-fold more efficient than insertion of dDMOTP and dMMO2TP, respectively.
Table 3.
Kinetic data for Kf-mediated insertion of triphosphates (dYTP) opposite d5SICS or dNaM in the template in sequence context II; insertion of dATP opposite dT is provided for comparison.
| 5′–d(TAATACGACTCACTATAGGGAGC) | ||||
|---|---|---|---|---|
| 3′–d(ATTATGCTGAGTGATATCCCTCGXTCTAGGTTACGGCAGGATCGC) | ||||
|
| ||||
| X | Y | kcat (min-1) | KM (μM) | kcat/KM (× 105 M-1min-1) |
| T | A | 0.95 ± 0.10 | 0.0017 ± 0.0001 | 5800 |
| 5SICS | NaM | 11.4 ± 2.3 | 0.41 ± 0.01 | 280 |
| DMO | 14.2 ± 1.1 | 35.8 ± 1.5 | 3.9 | |
| MMO2 | 8.6 ± 0.2 | 46.6 ± 7.1 | 1.9 | |
| NMO1 | 33.6 ± 7.0 | 23.0 ± 0.1 | 15 | |
| PMO1 | 19.0 ± 1.6 | 13.5 ± 1.5 | 14 | |
| NaM | 5SICS | 5.8 ± 1.1 | 0.049 ± 0.003 | 1200 |
Efficiency and fidelity of dNaM-d5SICS synthesis
To provide a reference for how efficiently and selectively Taq synthesizes dNaM-d5SICS, we first measured the efficiency of Kf-mediated synthesis of dNaM-d5SICS and all possible mispairs in both strand contexts of sequence context I (Table 1). We found that d5SICSTP is inserted opposite dNaM with an efficiency of 2.1 × 108 M-1min-1, in good agreement with previously reported data.[13] However, we found that dNaMTP is inserted opposite d5SICS with an efficiency of 5.8 × 107 M-1min-1. While significantly greater than reported previously for this sequence context, it is similar to that reported for the same insertion in sequence context II.[13] To confirm the rates for insertion in sequence context II, we reanalyzed the synthesis of dNaM-d5SICS in both strand contexts of sequence context II (Table 3). We found that d5SICSTP is inserted opposite dNaM and that dNaMTP is inserted opposite d5SICS with efficiencies of 1.2 × 108 M-1min-1, and 2.8 × 107 M-1min-1, respectively, both in good agreement with the previously published data.
As already mentioned, the most efficiently inserted dNTP opposite d5SICS by Kf is dGTP, which we found to proceed with an efficiency of 1.5 × 105 M-1min-1, while dATP and dTTP are inserted less efficiently, dCTP is not inserted at detectable level, and d5SICSTP is inserted with an efficiency of 2.9 × 105 M-1min-1 (Table 1). With dNaM in the template, the natural triphosphate most efficiently inserted by Kf is dATP, which proceeds with an efficiency of 1.8 × 106 M-1min-1 (Table 1). dTTP is also inserted, but with a reduced efficiency of 1.2 × 104 M-1min-1, while dGTP and dCTP are not inserted at detectable levels (kcat/KM < 103 M-1min-1). dNaMTP is inserted opposite dNaM with an efficiency of 9.5 × 106 M-1min-1. The rates for the synthesis of these mispairs are all in good agreement with previously published data.[13]
We next determined the efficiency and fidelity of dNaM-d5SICS synthesis by Taq in both strand contexts of sequence context I. Again, for comparison we first measured the efficiency with which Taq inserts dATP opposite dT in the same sequence context, which we found to be 8.2 × 107 M-1min-1. We found that Taq inserts dNaMTP opposite d5SICS and d5SICSTP opposite dNaM with efficiencies of 7.6 × 106 M-1min-1 and 7.1 × 106 M-1min-1, respectively. With d5SICS in the template, Taq inserts dGTP more efficiently than the other natural triphosphates, with an efficiency of 2.1 × 104 M-1min-1, followed by dATP and dTTP, with efficiencies of 3.5 × 103 M-1min-1 and 2.6 × 103 M-1min-1. Taq does not insert dCTP at a detectable rate (<103 M-1min-1). The most competitive mispair synthesized results from the insertion of d5SICSTP, which proceeds with an efficiency of only 1.8 × 104 M-1min-1. With dNaM in the template, Taq inserts dATP most efficiently, with a second order rate constant of 4.8 × 104 M-1min-1. dTTP is inserted with an efficiency of 2.4 × 103 M-1min-1, while dGTP and dCTP are inserted with undetectable rates (< 103 M-1min-1). dNaMTP is inserted opposite dNaM by Taq with an efficiency of 5.2 × 104 M-1min-1.
Efficiency and fidelity of PCR amplification
To begin to examine unnatural base pair replication, which includes synthesis and extension in both strand contexts, we first explored the Taq-mediated PCR amplification of DNA containing d5SICS paired opposite either dNaM, dMMO2, dDMO, dNMO1, or dPMO1. Taq was employed despite its low fidelity for replication of the unnatural base pair, because it lacks exonuclease proofreading activity, to facilitate comparison with the steady-state kinetic data. In addition, the unnatural base pairs were incorporated into 134 nt DNA template, D6, in the middle of a six nucleotide randomized region.[9] The randomized template was selected to provide the strictest possible measure of fidelity as sequences with inherently low fidelity are expected to lose the unnatural base pair and then efficiently amplify. While it is not practical to characterize fidelity in these reactions due to the significant read through, sequencing traces clearly indicate that while d5SICS-dNaM is best replicated, amongst the other analogues, dNMO1 is optimized for pairing opposite d5SICS, followed by dDMO, dMMO2, and dPMO1 (Supporting Information, Fig. S28).
To examine amplification under more practical and high fidelity conditions we explored PCR using the exonuclease proficient Deep Vent polymerase of the same unnatural base pairs positioned in the middle of the 149 nt duplex referred to as D1, which was used previously to characterize the amplification of dNaM-d5SICS and dMMO2-d5SICS[9] (Table 4 and Fig. S29). In this case fidelity was characterized as described previously.[9] In agreement with previous results, dNaM-d5SICS is replicated with a remarkable fidelity of 99.7. Interestingly, dNMO1-d5SICS, dDMO-d5SICS, and dMMO2-d5SICS were also amplified with a similar fidelity, while as predicted by the steady-state kinetic data, dPMO1-d5SICS is amplified with a significantly lower fidelity.
Table 4.
Fidelities of Deep Vent-mediated PCR amplification of unnatural base pairs. [a]
| Base pair | Fidelity[b] |
|---|---|
| dNaM-d5SICS | 99.7 |
| dDMO-d5SICS | 99.7 |
| dMMO2-d5SICS | 99.7 |
| dPMO1-d5SICS | 92.4 |
| dNMO1-d5SICS | 99.5 |
See text for experimental details.
Fidelity is defined as % unnatural base pair retention per doubling, see Ref. [9].
Discussion
The identification dMMO2-d5SICS was a landmark in our efforts to identify an unnatural base pair, and early efforts directed toward its optimization yielded dDMO-d5SICS and in particular dNaM-d5SICS. While the former is replicated slightly better than dMMO2-d5SICS, dNaM-d5SICS is replicated significantly better, although whether this was unique to Kf, or a general property of the unnatural base pair was not known. In addition, the strategy of using nucleobases with little or no structural homology to their natural counterparts makes possible many different substituents, and it is unclear whether dNaM-d5SICS represents the best route to optimize dMMO2-d5SICS, especially considering the increased potential for linker modification of the more simple scaffolds.
In an effort to optimize the dMMO2 scaffold for pairing with d5SICS, we examined derivatives with different substituents in place of the para methyl group. Specifically, five different substituents expected to alter the physicochemical properties of the nucleobases were examined, including amine, amide, trifluoroamide, nitro, and pyrrolo groups. The amine substituent is electron donating and expected to introduce a dipole along the C-N bond directed toward the aromatic ring. The amine group is also expected to form H-bonds with water molecules. While the amide groups are less electron donating, along with increased steric demands, they introduce increased H-bonding relative to the amine. The pyrrolo substituent reduces the electron donating ability of the nitrogen and adds steric bulk, but within the context of decreased H-bonding and increased stacking potential. In contrast, the nitro group is electron withdrawing and expected to introduce a significant dipole oriented along the carbon-nitrogen bond away from the aromatic ring. However, like the pyrrolo substituent, the nitro group is expected to decrease H-bonding, being only a moderate H-bond acceptor, and increase the ability of the nucleobase analog to stack with flanking nucleobases within the developing duplex.
With both Kf and Taq, the analogues examined clearly separate into two groups: dAMO1TP, dAMO2TP, and dAMO3TP are inserted less efficiently opposite d5SICS than dMMO2TP; and dNMO1TP and dPMO1TP are inserted more efficiently. For dAMO1TP, the decrease is 4-fold with Kf, while with Taq it is 20-fold, with the difference between the polymerases largely due to changes in the apparent kcat; the modification increases kcat with Kf and decreases it with Taq while the KM is similarly increased (∼5-fold) with both enzymes. Because, the amino group is expected to increase the electron density of the nucleobase ring, the observed decrease in insertion efficiencies suggests that any favorable increase in packing due to increased polarizability is offset by other deleterious factors, such as forced desolvation of the amino group, which is consistent with a similar effect on KM observed with both enzymes. Modification of the amine with the acetyl group of dAMO2TP decreases insertion efficiency by both enzymes, relative to dMMO2TP, by a factor of 26 with Kf and more than a factor of 83 with Taq. The large reduction in efficiency with Taq results from a significant decrease in apparent binding, and an even larger decrease in turnover. Modification of the amino group with the trifluoroacetyl group of dAMO3TP also reduced the efficiency of insertion, but very differently with the two enzymes. With Kf, the decrease is only 4-fold relative to dMMO2TP, but it is more than 60-fold with Taq. The large decrease in recognition by Taq again results from both reduced binding and reduced turnover. Thus, insertion of these analogues appears to be limited by desolvation and steric or electrostatic clashes that are polymerase specific and generally more severe with Taq.
Relative to dMMO2TP, the behavior of dPMO1TP and dNMO1TP are very different from that of the other analogues with both Kf and Taq. With Kf, in both sequence contexts I and II, dPMO1TP and dNMO1TP are each inserted approximately 5-fold more efficiently, due to increased binding and increased turnover. In contrast, with in sequence context I, dPMO1TP is inserted by Taq 3-fold less efficiently then dMMO2TP, due to reduced binding, but dNMO1TP is inserted 4-fold more efficiently, due to a small increase in turnover and a slightly larger increase in apparent binding affinity. The data suggest that with Kf the presence of the nitro or the pyrrole group likely reduces the cost of desolvation relative to the amine or amide and also mediates favorable packing interactions with d5SICS or the polymerase in the developing transition state. As with the other analogues, Taq appears to be less accommodating to alterations in the size of the substituent, tolerating the nitro substituent, but not the pyrrole group.
The detailed kinetic analysis focused on the insertion of the different triphosphate analogue opposite d5SICS. However, replication requires base pair synthesis in the other strand context, the insertion of d5SICSTP opposite the analogue in template DNA, as well as the continued primer elongation in both strand contexts. The results of the PCR analysis suggest that the nitro and pyrrolo substituents of dNMO1 and dPMO1, respectively, do not significantly interfere with any of these other steps of replication. Moreover, the fidelities observed during PCR with Taq indicate that the improvement in steady-state triphosphate insertion characterized for dNMO1TP is also manifest as improved replication.
Generally, the data obtained with the five new analogues examined suggest that optimizing hydrophobicity, including reducing the cost of desolvation, and improving packing in the developing major groove are the most efficacious routes to optimization of the dMMO2 scaffold, although the interactions must be more carefully manipulated with Taq than with Kf, apparently due to a more discriminating active site. The importance of hydrophobicity and packing is also consistent with the remarkable insertion efficiency of dNaM opposite d5SICS. If supported by further study, these arguments suggest that, at least from the perspective of the para position of the dMMO2 scaffold, the developing major groove of the duplex within the polymerase active site is able to accommodate planar aromatic groups with favorable packing interactions, but the waters and metal ions found within the major groove of free duplex DNA are not yet available. Regardless of the underlying mechanism, the steady-state kinetic and PCR data indicate that dNMO1 is the most promising dMMO2 analogue yet identified.
Previous data collected for the Kf-mediated synthesis of dNaM-d5SICS suggested that its efficiency is strand and sequence context specific. However, this was based on a the efficiency of a single step in a single sequence context, the insertion of dNaMTP opposite d5SICS in sequence context I, which appeared to be significantly less efficient than that in sequence context II, or than the insertion of d5SICSTP opposite dNaM in either sequence context. The efficiency of this reaction was re-evaluated in the current work and we found that the previous data underestimated the rate approximately 10-fold, which we attribute to the presence of an impurity. The corrected data places the efficiency of dNaMTP insertion on par with that of the others. Thus, the replication of dNaM-d5SICS does not appear to be strongly strand or sequence context dependent. The efficiency of both steps of unnatural base pair synthesis are within 4- to 13-fold that of a natural base pair. Moreover, relative to the most competitive mispair, the overall fidelity when both unnatural base pair synthesis and extension[13] are combined is at least 104. To our knowledge, this represents the most efficient and high fidelity replication reported to date for an unnatural base pair.
The synthesis of dNaM-d5SICS by Kf is remarkably efficient and selective, and it appears to be just as efficiently synthesized by Taq. Taq inserts both dNaMTP opposite d5SICS and d5SICSTP opposite dNaM only 10-fold less efficiently than a natural base pair in the same sequence context. Moreover, none of the natural dNTPs are inserted efficiently opposite either d5SICS and dNaM in the template, resulting in 150-fold or greater fidelities for this step alone. The efficient and selective recognition of dNaM-d5SICS by both Kf and Taq now allow us to conclude that the determinants of efficient replication are inherent to the nucleotides themselves.
Conclusion
The data reveal that reducing the cost of nucleotide desolvation and optimizing packing interactions within the developing major groove are promising routes to optimize the efficiency of polymerase-mediated insertion of the dMMO2TP analogues opposite d5SICS, and that other than dNaM, dNMO1 is the most promising analogue identified to date. While continued efforts toward the optimization of the dNMO1 scaffold are justified by its potential for accommodating linkers, the data reported herein for dNaM-d5SICS demonstrate just how challenging the identification of a more optimized base pair is likely to be. Thus, the data strongly suggest that efforts toward developing an unnatural base pair for in vitro applications should focus on the dNaM scaffold, including efforts to identify suitable sites for linker attachment to facilitate applications based on the site-specific modification of DNA or RNA. Such efforts are currently underway.
Experimental Section
General Methods
All reactions were carried out in oven-dried glassware under inert atmosphere, and all solvents were dried over 4 Å molecular sieves with the exception of tetrahydrofuran, which was distilled from sodium metal. All other reagents were purchased from Fisher or Aldrich. 1H, 13C and 31P NMR spectra were recorded on Bruker DRX-600, DRX-500 or Varian Inova-300 spectrometers. High-resolution mass spectroscopic data were obtained on an ESI-TOF mass spectrometer (Agilent 6200 Series) at the TSRI Open Access Mass Spectrometry Lab and MALDI-TOF mass spectrometry (Applied Biosystems Voyager DE-PRO System 6008) from the TSRI Center for Protein and Nucleic Acid Research.
Synthetic Procedures and Characterizations
Compound 1
Compound 1 was synthesized according to the literature.[29]
Compound 2
To a solution of m-anisidine (2 mL, 16.2 mmol, 1 equiv) in tetrahydrofuran (50 mL) at 0 °C was added NaHCO3 (1.65 g, 19.4 mmol, 1.2 equiv). Benzyl chloroformate (2.8 mL, 19.4 mmol, 1.2 equiv) was then added dropwise under strong stirring. The mixture was allowed to warm to room temperature over 20 min, stirred for an additional hour and then diluted with CH2Cl2. The organic layer was quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to a short silica gel column chromatography with a step gradient of CH2Cl2 (20 – 100%) in hexane affording pure NH-Cbz m-anisidine. To a solution of the Cbz-protected m-anisidine in MeOH (120 mL) at -20 °C was added silver nitrate (4.48 g, 14.37 mmol) and iodine (3.65 g, 14.37 mmol). The mixture was stirred at -20 °C for 1 h, quenched by saturated aqueous Na2S2O3 (80 mL) and filtered through a Celite pad. The filtrate was concentrated to 10% of its original volume and diluted with CH2Cl2 (200 mL). The organic layer was quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of CH2Cl2 (20 – 80%) in hexane. The desired compound 2 was obtained as white foam after evaporation of the solvent (4.04 g, 10.54 mmol, 65% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H, NH), 7.60 (d, J = 8.5 Hz, 1H), 7.40 (m, 5H), 7.27 (d, J = 2.3 Hz, 1H), 6.90 (m, 1H), 5.16 (s, 2H), 3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 158.66, 153.08, 139.53, 139.13, 135.79, 128.67, 128.49, 128.31, 112.37, 102.07, 78.04, 67.22, 56.33. HRMS (ESI+) m/z calcd for C15H15INO3 (M+H)+ 384.0091, found 384.0097.
Compound 3
A mixture of palladium acetate (880 mg, 3.92 mmol, 0.15 equiv) and triphenylarsine (2 g, 3.92, 0.15 equiv) in dry dimethylformamide (150 mL) was stirred under argon atmosphere at room temperature for 20 min. To this mixture was added 2 (15 g, 39.2 mmol, 1.5 eq), 1 (9 g, 26.1 mmol, 1 eq) and tri-n-butylamine (9.3 mL, 39.2 mmol, 1.5 eq) in dimethylformamide (5 mL), and the resulting reaction mixture was stirred under nitrogen at 70 °C for 15 h. The mixture was cooled to 0 °C and 1 m tetrabutylammonium fluoride (44 mL, 44 mmol) in tetrahydrofuran was added and the mixture was stirred for 2 h while warming the reaction to room temperature. The reaction mixture was filtered through Celite and extracted with ethyl acetate and saturated aqueous NaHCO3. The combined organic layers were dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of ethyl acetate (5 – 30%) in CH2Cl2. The eluted product was dissolved in acetic acid (50 mL) and acetonitrile (50 mL). The solution was cooled to 0 °C, sodium triacetoxyborohydride (4.5 g, 21.23 mmol) was added, and the mixture was stirred for 1 h. The reaction mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of methanol (0 – 4%) in ethyl acetate. The desired compound 3 was obtained as white foam after evaporation of the solvent (6.58 g, 17.62 mmol, 45% yield). 1H NMR (400 MHz, MeOD) δ 7.49 – 7.27 (m, 6H, H-ar, H-6), 7.26 – 7.17 (m, 1H, H-3), 6.95 (dd, J = 8.2, 2.0 Hz, 1H, H-5), 5.39 (dd, J = 10.2, 5.5 Hz, 1H, H-1′), 5.17 (s, 2H, OCH2Ar (Cbz)), 4.35 – 4.24 (m, 1H, H-3′), 3.93 (td, J = 5.2, 2.7 Hz, 1H, H-4′), 3.81 (s, 3H, OCH3 ), 3.74 – 3.59 (m, 2H, H-5′,H-5″), 2.27 (ddd, J = 13.1, 5.6, 1.9 Hz, 1H, H-2′), 1.82 (ddd, J = 13.1, 10.3, 6.1 Hz, 1H, H-2″). 13C NMR (100 MHz, MeOD) δ 156.7 (C2 (C-OMe)), 154.4 (NHC=O), 139.1 (C4(C-NHCbz)), 136.6 (Cq, Car), 128.1-127.6 (CH, Ar), 126.0 (C6), 124.6 (C1(c-sugar)), 110.2 (C5), 101.2 (C3), 87.1 (C4′), 74.8 (C1′), 73.0 (C3′), 66.1 (OCH2Ph), 62.7 (C5′), 54.3 (OCH3), 41.7 (C2′). HRMS (ESI+) m/z calcd for C20H24NO6 (M+H)+ 374.1598, found 374.1615.
Compound 4
To a solution of 3 (2.5 g, 6.7 mmol, 1 equiv) in dry pyridine (100 mL) was added 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (2.6 mL, 8.1 mmol, 1.2 equiv) dropwise over 15 min. The reaction mixture was stirred for 2 h at room temperature under nitrogen atmosphere, concentrated two-fold and diluted with CH2Cl2. The organic layer was quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was dissolved in ethyl acetate (75 mL) and Et3N (250 μL, 1.76 mmol) was added. The resulting solution was treated with 10% Pd/C (150 mg) under H2 atmosphere and allowed to stir until the presence of the starting material could no longer be detected by TLC (∼1 h). The reaction mixture was filtered through Celite and the filtrate extracted with ethyl acetate and saturated aqueous NaHCO3. The combined organic layers were dried (Na2SO4), filtered and evaporated. The residue was subjected to a silica gel column chromatography with a step gradient with ethyl acetate (0 – 5%) in CH2Cl2 containing 0.5% triethylamine. The desired compound 4 was obtained as a colorless oil after evaporation of the solvent (21 g, 4.28 mmol, 64% yield). 1H NMR (500 MHz, CD3CN) δ 7.10 (d, J = 8.1 Hz, 1H, H-6), 6.26 (d, J = 2.1 Hz, 1H, H-3), 6.18 (dd, J = 8.1, 2.1 Hz, 1H, H-5), 5.15 (m, 1H, H-1′), 4.47 (dt, J = 7.7, 5.6 Hz, 1H, H-3′), 4.11 (s, 2H, NH2), 4.05 – 3.84 (m, 2H, H-5′,H-5″), 3.75 – 3.68 (m, 4H, OCH3, H-4′), 2.20 (ddd, J = 12.7, 7.3, 5.4 Hz, 1H, H-2′), 2.00 (m, 1H, H-2″), 1.13 – 1.00 (m, 28H, iPr). 13C NMR (125 MHz, CD3CN) δ 158.3 (C2), 149.6 (C4), 128.0 (C6), 120.2 (C1), 107.1 (C5), 98.6 (C3), 86.3 (C4′), 74.3 (C1′), 74.0 (C3′), 64.2 (C5′), 55.8 (OCH3), 42.5 (C2′), 17.9-13.4 (iPr). HRMS (ESI+) m/z calcd for C24H44NO5Si2 (M+H)+ 482.2752, found 482.2748.
Compound 5a
To a solution of 4 (150 mg, 0.31 mmol, 1 equiv) and potassium iodide (5.1 mg, 0.031 mmol, 0.1 eq.) in CH3CN (1.1 mL) was added a solution of 70% aqueous tert-Butyl hydroperoxide (160 μL, 1.18 mmol, 3.8 equiv) dropwise over a period of 15 min and the mixture was stirred at 75 °C in the absence of light for 2 h. The mixture was quenched with saturated aqueous Na2S2O3, washed with brine, extracted with ethyl acetate, dried (Na2SO4), filtered and evaporated. The resulting residue was dissolved in tetrahydrofuran (1.5 mL), 1 m tetrabutylammonium fluoride (0.5 mL, 0.5 mmol) in tetrahydrofuran was added and the mixture was stirred for 1 h at room temperature. The reaction mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of methanol (0 – 4%) in ethyl acetate. The desired compound 5a was obtained as white foam after evaporation of the solvent (32 mg, 0.12 mmol, 38 %). 1H NMR (500 MHz, CD3CN) δ 7.82 (dd, J = 8.4, 2.2 Hz, 1H, H-5), 7.77 – 7.69 (m, 2H, H-6, H-3), 5.32 (dd, J = 10.1, 5.8 Hz, 1H, H-1′), 4.24 (dt, J = 5.2, 2.1 Hz, 1H, H-3′), 3.91 (s, 3H, OCH3), 3.88 (td, J = 5.0, 2.6 Hz, 1H, H-4′), 3.63 – 3.59 (dd, J = 5.0, 0.9 Hz, 2H, H-5′, H-5″), 2.32 (ddd, J = 13.0, 5.7, 2.0 Hz, 1H), 1.68 (ddd, J = 13.1, 10.1, 5.9 Hz, 1H). 13C NMR (125 MHz, CD3CN) δ 157.4 (C2), 149.0 (C4), 140.3 (C1), 127.1 (C6), 116.6 (C5), 106.1 (C3), 88.5 (C4′), 75.5 (C1′), 73.9 (C3′), 63.7 (C5′), 56.8 (OCH3), 42.9 (C2′). HRMS (ESI+) m/z calcd for C12H16NO6 (M+H)+ 270.0972, found 270.0984.
Compound 5b
A solution of 4 (250 mg, 0.52 mmol, 1 equiv) and 2,5-dimethoxytetrahydrofuran (170 mg, 1.3 mmol, 2.5 equiv) in H2O (0.8 mL) was heated to 140 °C for 30 min in a microwave synthesizer (Biotage AB, Sweden). The reaction was allowed to cool and the resulting mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The resulting residue was dissolved in tetrahydrofuran (3 mL), 1 m tetrabutylammonium fluoride (1 mL, 1 mmol) in tetrahydrofuran was added and the mixture was stirred for 1 h at room temperature. The reaction mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of methanol (0 – 8%) in CH2Cl2. The desired compound 5b was obtained as white foam after evaporation of the solvent (95 mg, 0.33 mmol, 63 %). 1H NMR (600 MHz, CD3CN) δ 7.52 (d, J = 8.0 Hz, 1H, H-6), 7.17 (m, J = 2.1 Hz, 2H, H-7), 7.02 (m, 2H, H-5, H-3), 6.29 (t, J = 2.2 Hz, 2H, H-8), 5.30 (dd, J = 10.2, 5.6 Hz, 1H, H-1′), 4.23 (m, 1H, H-3′), 3.88 (s, 2H, OCH3), 3.83 (td, J = 5.0, 2.6 Hz, 1H, H-4′), 3.60 (t, J = 5.1 Hz, 2H), 3.21 (d, J = 3.7 Hz, 1H, OH-3′), 2.92 (t, J = 5.7 Hz, 1H, OH-5′), 2.22 (m, 1H, H-2′), 1.76 (ddd, J = 13.0, 10.3, 6.0 Hz, 1H, H-2″). 13C NMR (150 MHz, CD3CN) δ 158.1 (C2), 141.6 (C4), 129.1 (C6), 128.0 (C1), 120.2 (C7), 112.6 (C5), 111.1 (C8), 104.0 (C3), 88.2 (C4′), 75.4 (C1′), 74.1 (C3′), 63.8 (C5′), 56.4 (OCH3), 43.1 (C2′). HRMS (ESI+) m/z calcd for C16H20NO4 (M+H)+ 290.1387, found 290.1397.
Compound 5c
To a solution of 4 (250 mg, 0.52 mmol, 1 equiv) in CH2Cl2 (600 μl) was added triethylamine (125 μL, 0.89 mmol, 1.7 equiv) and then acetic anhydride dropwise (60 μL, 0.62 mmol, 1.2 equiv). The reaction mixture was stirred at room temperature for 20 min and then quenched with saturated aqueous NaHCO3. The organic layer was diluted with CH2Cl2, washed with brine, dried (Na2SO4), filtered and evaporated. The resulting residue was dissolved in tetrhydrofuran (2 mL), 1 m tetrabutylammonium fluoride (1 mL, 1 mmol) in tetrahydrofuran was added and the mixture was stirred for 1 h at room temperature. The reaction mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of methanol (0 – 12%) in CH2Cl2. The desired compound 5c was obtained as white foam after evaporation of the solvent (75 mg, 0.27 mmol, 51% yield). 1H NMR (600 MHz, CD3OD) δ 7.40 (d, J = 8.3 Hz, 1H, H-6), 7.33 (d, J = 1.8 Hz, 1H, H-3), 7.02 (dd, J = 8.3, 1.8 Hz, 1H, H-5), 5.38 (dd, J = 10.2, 5.6 Hz, 1H, H-1′), 4.27 (m, 1H, H-3′), 3.90 (td, J = 5.2, 2.7 Hz, 1H, H-4′), 3.80 (s, 3H, OCH3), 3.65 (m, 2H, H-5′, H-5″), 2.27 (ddd, J = 13.1, 5.5, 1.7 Hz, 1H, H-2′), 2.10 (s, 3H, COCH3), 1.80 (ddd, J = 13.2, 10.3, 6.0 Hz, 1H, H-2″). 13C NMR (150 MHz, CD3OD) δ 171.5 (NHC=O), 157.8 (C2), 140.1 (C4), 127.2 (C6), 127.2 (C1), 112.7 (C5), 103.7 (C3), 88.5 (C4′), 76.2 (C1′), 74.4 (C3′), 64.0 (C5′), 55.7 (OCH3), 43.1 (C2′), 23.9 (COCH3). HRMS (ESI+) m/z calcd for C14H20NO5 (M+H)+ 282.1341, found 290.1351.
Compound 5d
To a solution of 4 (350 mg, 0.73 mmol, 1 equiv) in CH2Cl2 (2 mL) at 10 °C was added triethylamine (170 μL, 1.24 mmol, 1.7 equiv) and then trifluoroacetic anhydride dropwise (170 μL, 0.88 mmol, 1.2 equiv). The reaction mixture was stirred at room temperature for 20 min at 10 °C and then quenched with saturated aqueous NaHCO3. The organic layer was diluted with CH2Cl2, washed with brine, dried (Na2SO4), filtered and evaporated. The resulting residue was dissolved in tetrahydrofuran (5 mL), 1 m tetrabutylammonium fluoride (1.9 mL, 1.9 mmol) in tetrahydrofuran was added and the mixture was stirred for 1 h at room temperature. The reaction mixture was diluted with ethyl acetate, quenched with saturated aqueous NaHCO3, washed with brine, dried (Na2SO4), filtered and evaporated. The residue was subjected to silica gel column chromatography with a step gradient of methanol (0 – 10%) in CH2Cl2. The desired compound 5c was obtained as white foam after evaporation of the solvent (200 mg, 0.60 mmol, 81% yield). 1H NMR (600 MHz, CD3CN) δ 9.25 (s, 1H, NH), 7.50 (d, J = 8.2 Hz, 1H, H-6), 7.27 (d, J = 1.9 Hz, 1H, H-3), 7.19 (m, 1H, H-5), 5.27 (dd, J = 10.2, 5.6 Hz, 1H, H-1′), 4.22 (m, 1H, H-3′), 3.82 (d, J = 27.6 Hz, 4H, H-4′, OCH3), 3.59 (d, J = 4.9 Hz, 2H, H-5′, H-5″), 2.22 (m, 1H, H-2′), 1.72 (m, 1H, H-2″). 13C NMR (150 MHz, CD3CN) δ 157.5 (C2), 156.2-155.4 (NHC=O), 136.9 (C4), 129.7 (C6), 127.3 (C1), 119.8-114.1 (CF3), 113.7 (C5), 104.7 (C3), 88.2 (C4′), 75.4 (C1′), 74.1 (C3′), 63.9 (C5′), 56.2 (OCH3), 43.1 (C2′). HRMS (ESI+) m/z calcd for C14H17F3NO5 (M+H)+ 336.1053, found 336.1066.
General Procedure for Triphosphate Synthesis
Proton sponge (1.3 equiv) and the free nucleoside derivative (1.0 equiv) were dissolved in dry trimethyl phosphate (40 equiv) and cooled to -15 °C under nitrogen atmosphere. Freshly distilled POCl3 (1.3 equiv) was added dropwise and the resulting mixture was stirred at -10 °C for 2 h. Tributylamine (6.0 equiv) and a solution of tributylammonium pyrophosphate (5.0 eq.) in dimethylformamide (0.5 M) were added. Over 30 min, the reaction was allowed to warm slowly to 0 °C and then was quenched by addition of 0.5 M aqueous Et3NH2CO3 (TEAB) pH 7.5 (2 vol-equiv). The mixture was diluted two-fold with H2O and the product was isolated on a DEAE Sephadex column (GE Healthcare) with an elution gradient of 0 to 1.2 m TEAB, evaporated, co-distilled with H2O (3×). Additional purification by reverse-phase (C18) HPLC (0 – 35% CH3CN in 0.1 m TEAB, pH 7.5) was performed.
Compound 6a
31P NMR (162 MHz, D2O) δ -10.52 (d, J = 19.8 Hz, γ-P), -10.84 (d, J = 20.2 Hz, α-P), -22.92 (t, J = 20.1 Hz, β-P). MS (MALDI-TOF-, matrix : 9-aminoacridine) (m/z): [M-H]- calcd for C12H17NO15P3, 508.2; found, 508.3. ε(λ = 330 nm) = 2200 M-1 cm-1; ε(λ = 285 nm) = 4800 M-1 cm-1
Compound 6b
31P NMR (162 MHz, D2O) δ -10.22 (d, J = 19.8 Hz, γ-P), -10.75 (d, J = 20.1 Hz, α-P), -22.82 (t, J = 20.0 Hz, β-P). MS (MALDI-TOF, matrix: 9-aminoacridine) (m/z) : [M-H]- calcd for C16H21NO13P3, 528.3; found 527.8. ε (λ = 283 nm) = 5080 M-1 cm-1; ε (λ = 256 nm) = 10850 M-1 cm-1
Compound 6c
31P NMR (202 MHz, D2O) δ -6.35 (d, J = 16.9 Hz, γ-P), -10.74 (d, J = 19.7 Hz, α-P), -22.38 (m, β-P). MS (MALDI-TOF-, matrix : 9-aminoacridine) (m/z) : [M-H] - calcd for C14H21NO14P3, 520.2; found 519.3. ε (λ = 282 nm) = 2700 M-1 cm-1; ε (λ = 248 nm) = 7500 M-1 cm-1
Compound 6d
31P NMR (162 MHz, D2O) δ -10.38 (d, J = 19.8 Hz, γ-P), -10.87 (d, J = 20.2 Hz, α-P), -22.82 (t, J = 20.0 Hz, β-P). MS (MALDI-TOF-, matrix : 9-aminoacridine) (m/z): [M-H]- calcd for C14H18F3NO14P3, 574.2; found 573.9. ε(λ = 285 nm)= 3780 M-1 cm-1; ε (λ = 251 nm)= 7450 M-1 cm-1
Compound 6e
A solution of 6d in aqueous ammonia (28% NH3 w/v) (1 mL) was stirred for 1 h. Ammonia was removed by vacuum concentration (SpeedVac, 20 min) and the resulting solution was separated by reverse-phase (C18) HPLC (0 – 35% CH3CN in 0.1 m TEAB, pH 7.5) providing pure compound 6e. 31P NMR (162 MHz, D2O) δ -9.21 (d, J = 20.0 Hz, γ-P), -10.85 (d, J = 20.4 Hz, α-P), -22.62 (t, J = 20.6 Hz, β-P). MS (MALDI-TOF, matrix: 9-aminoacridine) (m/z): [M-H]- calcd for C12H19NO13P3, 478.2; found 477.8. ε(λ = 284 nm) = 2450 M-1 cm-1; ε(λ = 240 nm) = 9250 M-1 cm-1
dNaMTP
dNaMTP was synthesized using the General Procedure described above starting from dNaM nucleoside (Berry & Associates Inc.). 31P NMR (162 MHz, D2O) δ -8.96 (d, J = 19.4 Hz, γ-P), -10.81 (d, J = 20.1 Hz, α-P), -22.66 (t, J = 20.2 Hz, β-P). MS (MALDI-TOF-, matrix : 9-aminoacridine) (m/z): [M-H]- calcd for C16H20O13P3, 513.2; found 513.4. ε(λ = 230 nm) = 75,000 M-1 cm-1
d5SICSTP
d5SICSTP was synthesized using the General Procedure described above starting from the d5SICS nucleoside (Berry & Associates Inc.). 31P NMR (162 MHz, D2O) δ -10.11 (d, J = 20.0 Hz, γ-P), -11.05 (d, J = 20.2 Hz, α-P), -22.75 (t, J = 20.0 Hz, β-P). MS (MALDI-TOF-, matrix : 9-aminoacridine) (m/z) : [M-H]- calcd for C15H19NO12P3S, 530.3; found 529.3. ε(λ = 365 nm) = 3,950 M-1 cm-1
Gel-Based Kinetic Assays
Primer oligonucleotides (Integrated DNA Technologies) were 5′-radiolabeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]-ATP (GE Biosciences) and annealed to template oligonucleotides[13] by heating to 95 °C followed by slow cooling to room temperature. Reactions were initiated by adding a solution of 2× dNTP solution (5 μL) to a solution containing polymerase (0.10 – 1.23 nM) and primer template (40 nM) in reaction buffer (5 μL); Klenow reaction buffer (50 mm Tris-HCl, pH 7.5, 10 mm DTT and 50 μg/mL acetylated BSA) for Kf polymerase, ThermoPol reaction buffer (20 mM Tris-HCl, 10 mm (NH4)2SO4, 10 mm KCl, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) for Taq polymerase. After incubation at 25 °C (Kf) or 50 °C (Taq) for 3-10 min the reactions were quenched with 20 μL of loading dye (95% formamide, 20 mm EDTA, and sufficient amounts of bromophenol blue and xylene cyanole). Reaction products were resolved by 15% polyacrylamide gel electrophoresis, and gel band intensities corresponding to the extended and unextended primers were quantified by phosphorimaging (Storm Imager, Molecular Dynamics) and Quantity One (BioRad) software. Plots of kobs versus triphosphate concentration were fit to the Michaelis-Menten equation using the program Origin (Microcal Software) to determine Vmax and KM. kcat was determined from Vmax by normalizing by the total enzyme concentration. Each reaction was run in triplicate and standard deviations for both kinetic parameters were determined (see Tables 1 – 3). An example of the raw kinetic data is shown in Figure S27 in the Supporting Information.
PCR amplification
DNA templates D1 and D6 were synthesized as described previously.[9] PCR amplification (see SI for details and sequences) was carried out starting with 0.1 ng of D6 or 1 ng of D1 (Taq or DeepVent, respectively) in 1× ThermoPol reaction buffer with the following modifications: MgSO4 adjusted to 6.0 mm, 0.6 mm or 0.7 mm each natural dNTP (Taq or DeepVent, respectively), 0.1 mm each unnatural triphosphate, 1 μm each primer (see SI for sequences), and 0.03 unit/μL of Taq or 0.02 unit/μL of DeepVent (exo+) in an iCycler Thermal Cycler (Bio-Rad) under following thermal cycling conditions: 94 °C, 30 s; 48 °C, 30 s; 65 °C, 4 min, 18 or 13 cycles (Taq or DeepVent, respectively). Upon completion, PCR products were purified utilizing the PureLink™ PCR purification kit (Invitrogen), quantified by fluorescent dye binding (Quant-iT dsDNA HS Assay kit, Invitrogen) and sequenced on 3730 DNA Analyzer (Applied Biosystems) to determine fidelity of unnatural base pair replication (see SI and Ref. [9 ] for details).
Supplementary Material
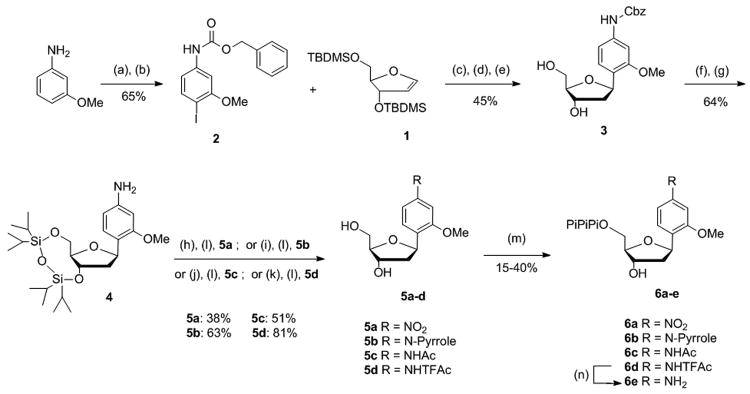
Scheme 1. Conditions: (a) CBz-Cl, NaHCO3, THF, rt, 20 min; (b) I2, Ag2SO4, MeOH, - 20 °C, 1 h; (c) Pd(OAc)2, AsPh3, nBu3N, DMF, 70 °C, 15 h; (d) TBAF 1 m in THF, 0 °C → rt, 2 h; (e) NaBH(OAc)3, AcOH, CH3CN, 0 °C, 1 h; (f) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane, pyridine, rt, 2 h; (g) 10% Pd/C, H2, EtOAc, rt, 1 h; (h) KI, tbutyl hydroperoxide aq 70%, CH3CN, dark, 75 °C, 2 h; (i) 2,5-dimethoxytetrahydrofuran, H2O, microwave-140 °C, 30 min; (j) Ac2O, Et3N, CH2Cl2, rt, 20 min; (k) trifluoroacetic anhydride, Et3N, CH2Cl2, 10 °C, 20 min; (l) TBAF 1 m in THF, 1 h; (m) proton sponge, POCl3, PO(OMe)3, -15 °C → -10 °C, 3 h then Bu3N, (Bu3NH)2H2P2O7 in DMF, -10 °C → 0 °C, 30 min then TEAB buffer (0.5 m), rt, 10 min; (n) NH4OH 30%, rt, 1 h.
Acknowledgments
Funding for this work was provided by the National Institute for Health (GM060005). We wish to thank Dr. Phillip Ordoukhanian, director of The Center for Protein and Nucleic Acids Research at The Scripps Research Institute, for analytical support.
References
- 1.Horlacher J, Hottiger M, Podust VN, Hübscher U, Benner SA. Proc Natl Acad Sci USA. 1995;92:6329–6333. doi: 10.1073/pnas.92.14.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutz MJ, Held HA, Hottiger M, Hübscher U, Benner SA. Nucleic Acids Res. 1996;24:1308–1313. doi: 10.1093/nar/24.7.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Chen F, Chamberlin SG, Benner SA. Angew Chem Int Ed. 2010;49:177–180. doi: 10.1002/anie.200905173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang GT, Romesberg FE. J Am Chem Soc. 2008;130:14872–14882. doi: 10.1021/ja803833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE. J Am Chem Soc. 2008;130:2336–2343. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leconte AM, Romesberg FE. In: Protein Engineering. K C, RajBhandary UL, editors. Springer-Verlag; Berlin: 2009. pp. 291–314. [Google Scholar]
- 8.Malyshev DA, Pfaff DA, Ippoliti SI, Hwang GT, Dwyer TJ, Romesberg FE. Chem Eur J. 2010;16:12650–12659. doi: 10.1002/chem.201000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malyshev DA, Seo YJ, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2009;131:14620–14621. doi: 10.1021/ja906186f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda S, Fillo JD, Henry AA, Rai P, Wilkens SJ, Dwyer TJ, Geierstanger BH, Wemmer DE, Schultz PG, Spraggon G, Romesberg FE. J Am Chem Soc. 2007;129:10466–10473. doi: 10.1021/ja072276d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda S, Leconte AM, Romesberg FE. J Am Chem Soc. 2007;129:5551–5557. doi: 10.1021/ja068282b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]
- 13.Seo YJ, Hwang GT, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2009;131:3246–3252. doi: 10.1021/ja807853m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo YJ, Matsuda S, Romesberg FE. J Am Chem Soc. 2009;131:5046–5047. doi: 10.1021/ja9006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seo YJ, Romesberg FE. ChemBioChem. 2009;10:2394–2400. doi: 10.1002/cbic.200900413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, Henry AA, Romesberg FE, Schultz PG. Angew Chem Int Ed. 2002;41:3841–3844. doi: 10.1002/1521-3773(20021018)41:20<3841::AID-ANIE3841>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 17.Hirao I. Curr Opin Chem Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 18.Hirao I, Kimoto M, Mitsui T, Fujiwara T, Kawai R, Sato A, Harada Y, Yokoyama S. Nat Methods. 2006;3:729–735. doi: 10.1038/nmeth915. [DOI] [PubMed] [Google Scholar]
- 19.Hirao I, Mitsui T, Kimoto M, Yokoyama S. J Am Chem Soc. 2007;129:15549–15555. doi: 10.1021/ja073830m. [DOI] [PubMed] [Google Scholar]
- 20.Kimoto M, Kawai R, Mitsui T, Yokoyama S, Hirao I. Nucleic Acids Res. 2009;37:e14. doi: 10.1093/nar/gkn956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitsui T, Kitamura A, Kimoto M, To T, Sato A, Hirao I, Yokoyama S. J Am Chem Soc. 2003;125:5298–5307. doi: 10.1021/ja028806h. [DOI] [PubMed] [Google Scholar]
- 22.Keefe AD, Cload ST. Curr Opin Chem Biol. 2008;12:448–456. doi: 10.1016/j.cbpa.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 23.Fritzsche W, Bier F. International Symposium on DNA-Based Nanodevices; American Institute of Physics Conference Proceedings; 2008. [Google Scholar]
- 24.Vrabel M, Pohl R, Votruba I, Sajadi M, Kovalenko SA, Ernsting NP, Hocek M. Org Biomol Chem. 2008;6:2852–2860. doi: 10.1039/b805632c. [DOI] [PubMed] [Google Scholar]
- 25.Wilson MA, Filzen G, Welmaker GS. Tetrahedron Lett. 2009;50:4807–4809. [Google Scholar]
- 26.Ludwig J, Eckstein F. J Org Chem. 1989;54:631–635. [Google Scholar]
- 27.Boosalis MS, Petruska J, Goodman MF. J Biol Chem. 1987;262:14689–14696. [PubMed] [Google Scholar]
- 28.Datta K, LiCata VJ. Nucleic Acids Res. 2003;31:5590–5597. doi: 10.1093/nar/gkg774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen E, Jørgensen PT, Sofan MA, Pederson EB. Synthesis. 1994:1037–1038. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.