

Abstract
Age is the most significant risk factor for atherosclerosis; however, the link between age and atherosclerosis is poorly understood. During both aging and atherosclerosis progression, the blood vessel wall stiffens owing to alterations in the extracellular matrix. Using in vitro and ex vivo models of vessel-wall stiffness and aging, we show that stiffening of extracellular matrix within the intima promotes endothelial cell permeability—a hallmark of atherogenesis. When cultured on hydrogels fabricated to match the elasticity of young and aging intima, endothelial monolayers exhibit increased permeability and disrupted cell-cell junctions on stiffer matrices. In parallel experiments, we showed a corresponding increase in cell-cell junction width with age in ex vivo aortas from young (10 weeks) and old (21 to 25 months) healthy mice. To investigate the mechanism by which matrix stiffening alters monolayer integrity, we found that cell contractility increases with increased matrix stiffness, mechanically destabilizing cell-cell junctions. This increase in endothelial permeability results in increased leukocyte extravasation, which is a critical step in atherosclerotic plaque formation. Mild inhibition of Rho-dependent cell contractility using Y-27632, an inhibitor of Rho-associated kinase, or siRNA restored monolayer integrity in vitro and in vivo. Our results suggest that extracellular matrix stiffening alone, which occurs during aging, can lead to endothelial monolayer disruption and atherosclerosis pathogenesis. Because previous therapeutics designed to decrease vascular stiffness have been met with limited success, our findings could be the basis for the design of therapeutics that target the Rho-dependent cellular contractile response to matrix stiffening, rather than stiffness itself, to more effectively prevent atherosclerosis progression.
INTRODUCTION
Vascular stiffening accompanies a variety of cardiovascular pathologies including hypertension (1) and atherosclerosis (2). The blood vessel wall also stiffens with age (3). During aging, vessel stiffness increases owing to changes in the micro-scale architecture of the extracellular matrix (ECM) within the vessel wall, namely the increases in elastin fragmentation, collagen deposition, and matrix protein cross-linking (4, 5). This matrix stiffening decreases arterial distensibility and capacitance and can increase mechanical strain on the heart. Macroscopic measurements of arterial stiffness are often used for clinical diagnosis and can independently predict cardiovascular events, such as coronary heart disease and stroke (6) and mortality in elderly patients (7). Although vascular stiffening occurs ubiquitously with age and is a predictor of cardiovascular risk, little is known about how vessel stiffness affects endothelial cells within blood vessels where arteriosclerosis initiates.
Recent evidence suggests that matrix stiffness affects cell behaviors (8), including cell spreading and adhesion (9), migration (10), and differentiation (11). It has been shown to alter vascular smooth muscle cell phenotype (12) and promote intimal hyperplasia (13). Our own data suggest that changes in matrix stiffness can alter cell-cell contact (14). Because cell-cell contact is directly linked to monolayer integrity and permeability, we hypothesized that increased matrix stiffness with age may disrupt barrier function of the endothelium. Increased endothelial permeability to lipoproteins and immune cells is considered the initiating step of atherosclerosis pathogenesis, and the accumulation of debris in the intima results in the formation of atherosclerotic plaques (15–17). Notably, decreasing permeability decreases plaque formation (18, 19). Endothelial permeability is controlled in part by the dynamic opening and closing of endothelial cell-cell junctions (20), which are directly affected by interactions between endothelial cells and the ECM (21). Although the intima stiffens during aging and atherosclerosis progression, and endothelial permeability occurs with age and is regarded as one of the first steps in atherogenesis, the relationship between age-related blood-vessel stiffening, endothelial cell function, and monolayer integrity has not been investigated in depth.
Because endothelial permeability is known as an early event in atherogenesis, we explored the effects of age-related matrix stiffness on endothelial barrier function using both in vitro and ex vivo models of intimal stiffening and aging. We found that stiffening of the matrix increased monolayer permeability. This increase in permeability was the result of upregulated cell contractility, which mechanically disrupted cell-cell junctions and promoted leukocyte transmigration—a critical step in atherosclerotic plaque formation. Pharmacological inhibition of cell contractility in vivo in mice reversed the effects of matrix stiffness on endothelial permeability by restoring tight cell-cell junctions, and also decreased leukocyte transmigration. These findings suggest that matrix stiffening alone, which occurs during the natural aging process, can directly cause endothelial cell permeability and atherosclerosis progression.
RESULTS
Increased matrix stiffness promotes endothelial monolayer permeability by destabilizing cell-cell junctions
We first investigated the effect of substrate stiffness on the barrier function of endothelial monolayers. To mimic the stiffness of young and aged intima, synthetic hydrogel substrates (22) were fabricated that approximated and exceeded the Young’s Modulus of 2.7 ± 1.1 kPa reported previously for the subendothelial matrix in bovine carotid arteries (23). Bovine aortic endothelial cells (BAECs) were seeded on polyacrylamide substrates of 2.5, 5 and 10 kPa. Endothelium permeability was measured on the basis of 40 kDa FITC-dextran movement across the monolayer (Fig. 1A; fig. S1). 40 kDa FITC-dextran was chosen as its hydrodynamic radius (~4.8 nm) approximates that of albumin (~3.6 nm) (24), a model protein commonly used in permeability studies (25) and whose permeation through the endothelium is associated with cardiovascular pathologies such as diabetes (26) and atherosclerosis (27). To validate our permeability measurements, we treated BAECs on gels with known barrier-altering agonists. Barrier-disruptive agonists vascular endothelial growth factor (VEGF), tumor necrosis factor (TNF)-α, and thrombin (21) increased permeability, whereas barrier-enhancing agonist BW245C (28) decreased permeability (Fig. 1B). Importantly, endothelial monolayer permeability increased as a function of matrix stiffness (Fig. 1C). These data suggest that matrix stiffness alone, without any externally added agonists, promotes increased endothelial monolayer permeability.
Fig. 1.
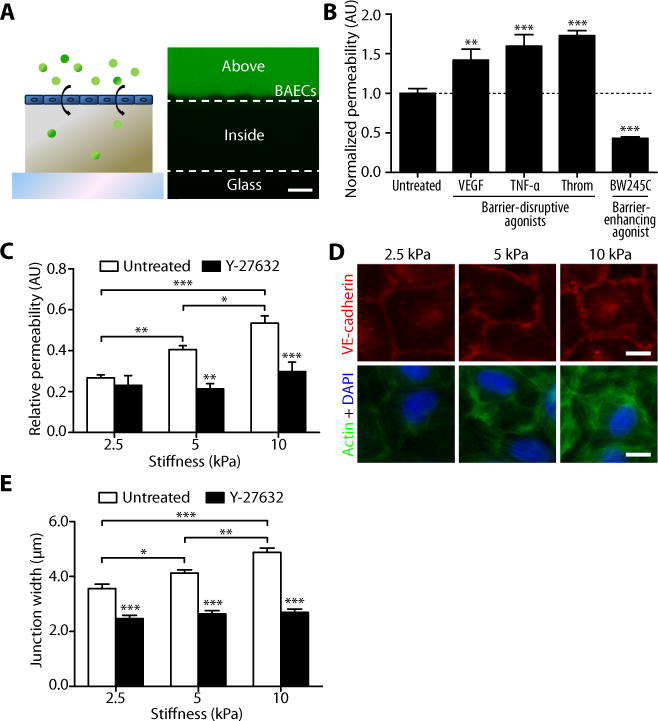
Matrix stiffness increases endothelial permeability and cell-cell junction width. (A) A cartoon shows FITC-dextran permeation through an endothelial monolayer into a polyacrylamide gel. On the right is a representative confocal cross-section of dextran capture in a 2.5-kPa polyacrylamide gel, depicting the method used to measure permeability. Dashed lines represent the top and bottom edges of the gel. Permeability values of cell (BAEC)-seeded gels were calculated as the pixel intensity within the gel divided by the pixel intensity above the gel normalized to that calculated in cell-free gels. Scale bar, 20 μm. (B) Effects of known barrier-disruptive (VEGF, TNF-α, or thrombin) and barrier-enhancing (BW245C) agonists on the permeability of BAECs cultured on 5-kPa gels. Permeability values are normalized to that of untreated cells. Data are means ± SEM. **P < 0.01, ***P < 0.001 (Dunnett’s test) compared to untreated cells. (C) Relative permeability of BAECs cultured on substrates matching (2.5 kPa) and exceeding (5 and 10 kPa) the stiffness of bovine arterial intima with (n = 8 gels, three independent experiments) or without (n = 16–20 gels, ten independent experiments) Y-27632 treatment. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Tukey’s test) compared to respective untreated conditions unless otherwise indicated by brackets. (D) Fluorescent images showing VE-cadherin (red), actin (green), and nuclei (blue) of confluent BAECs on gels. (E) VE-cadherin junction-width measurements of BAECs on gels with or without 30 min Y-27632 treatment (n = 60 junction measurements, two independent experiments). Scale bar, 10 μm. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Tukey’s test) compared to respective untreated conditions unless otherwise indicated by brackets.
To investigate the mechanism by which substrate stiffness contributes to changes in permeability, we probed the integrity of the cell-cell junctions within the endothelial monolayer by fluorescently imaging vascular endothelial (VE)-cadherin (Fig. 1D), a cell-cell adhesion molecule that mediates endothelium stabilization (29). Using methods similar to those that have been described previously (30) where lines were drawn perpendicularly to cell junctions to determine pixel intensities (fig. S2), we measured the separation width of intercellular junctions on polymers of varying stiffnesses (2.5 to 10 kPa). These measurements indicated that the distance between cells increased as a function of matrix stiffness (Fig. 1E). Together, these data suggest that increased matrix stiffness disrupts the formation of cell-cell junctions in vitro, which increases endothelial permeability.
Age-related intimal stiffening disrupts endothelial cell-cell junction integrity
Because blood vessel stiffness increases with age, and because our in vitro data indicated that increased matrix stiffness destabilizes cell-cell junctions (Fig. 1, C and E), we investigated the effects of aging and vessel stiffness on endothelial permeability and cell-cell junction integrity in a mouse model. We first characterized the intimal stiffness of de-endothelialized thoracic aortas of young (10 to 11 weeks) and old (21 to 25 months) mice using atomic force microscopy (AFM) (fig. S3). There was a significant increase in indentation modulus of the subendothelium with age (Fig. 2A). This measured modulus for young mice (31.9 ± 4.5 kPa, mean ± SEM) was higher than the value found for 18 to 30 month-old bovine carotid intima (2.7 ± 0.6 kPa, mean ± SEM) (23), presumably owing to the close proximity of the first internal elastic lamina relative to the lumen of the mouse vessel, which prevents the intima from deforming fully under indentation. The bovine carotid intima is more similar to the human carotid intima in its structure. Despite the difference between the moduli found for bovine and mouse intima, our data confirm that there is a relative increase in ECM stiffness in blood vessels with age.
Fig. 2.
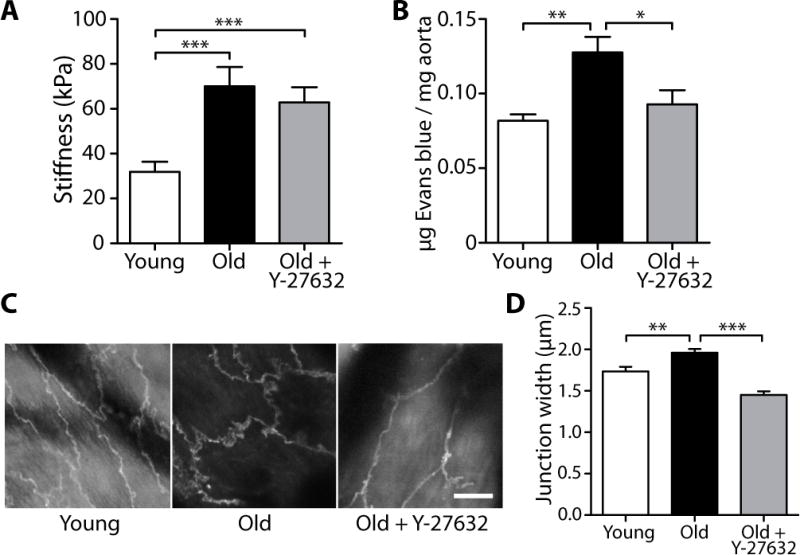
Aging increases subendothelial stiffness and endothelial intercellular junction separation in mice. (A) AFM indentation measurements of thoracic aorta of untreated young (10 to 11 weeks, n = 61 indent sites, 3 aortas) and old (20 to 25 months, n = 55 indent sites, 5 aortas) mice, as well as Y-27632-treated old (20 months, n = 76 indent sites, 4 aortas) mice. Data are means ± SEM. ***P < 0.001 (Dunn’s test). (B) Permeability measurements of thoracic aorta of untreated young (9 weeks, n = 4 aortas) and old (19 to 22 months, n = 6 aortas) mice, as well as Y-27632-treated old (20 months, n = 4 aortas) mice. Data are means ± SEM. *P < 0.05, **P < 0.01 (Dunn’s test). (C) Two-photon microscopy of VE-cadherin immunofluorescent staining in endothelial cells of intact, untreated young (10 week) and old (16 month) thoracic mouse aortas, as well as Y-27632-treated old (24 month) thoracic aortas. Scale bar, 10 μm. (D) VE-cadherin junction width measurements of thoracic aortic endothelial cells from untreated young (10 weeks, n = 240 width measurements, 4 aortas) and old (16 to 24 months, n = 390 width measurements, 7 aortas) mice, as well as Y-27632-treated old (21 to 24 months, n = 156 width measurements, 3 aortas) mice. Data are means ± SEM. **P < 0.01, ***P < 0.001 (Dunn’s test).
Using a different set of young (9 weeks) and old (19 to 22 months) mice than those used in the stiffness measurements (Fig. 2A), we measured the permeability of thoracic aortas with an Evans blue dye assay. We found that Evans blue deposition within the vessel wall increased with age (Fig. 2B). Additionally, endothelial cell-cell junctions within intact aortas were visualized based on VE-cadherin localization using two-photon microscopy (Fig. 2C). Intercellular distance, measured as the widest VE-cadherin separation width between two adjacent cells, was greater in aged, stiffer aortas than in younger, more compliant vessels (Fig. 2D), similar to the effects measured in vitro on stiffer matrices (Fig. 1E). These data suggest that, with age, the intima stiffens, causing endothelial cell-cell junctions to separate and increase permeability.
Endothelial permeability and cell-cell junctions are regulated by cell contractility
Previous work suggests that endothelial permeability is regulated in part by the cellular cytoskeleton and cell contractility (31). The Rho signaling pathway is a well-established mediator of the sustained contraction of the cytoskeleton (21, 32). Evidence in other cell types suggests that matrix stiffness induces changes in Rho activation (33, 34). When activated, Rho and its downstream effector Rho-associated kinase (ROCK) stimulate cell contractility by modulating actomyosin contraction and cell-matrix adhesion. To explore the intracellular mechanisms by which the mechanical properties of the matrix affect monolayer permeability, we hypothesized that stiffer substrates induce Rho activation, leading to increased cell contractility, the physical separation of cell-cell junctions, and increased permeability. Using a commercially available Rho detection kit, we found increased Rho activity in endothelial cells on 10 kPa substrates compared to those on 2.5 kPa substrates (Fig. 3A). To ensure that Rho activation was not altered by the concentration of acrylamide within the hydrogel substrates but rather by stiffness, we made two formulations of gels with different acrylamide concentrations that were both approximately 2.5 kPa in stiffness (Fig. 4A). Rho activity was unchanged (Fig. 4B), suggesting that polymer composition does not activate Rho independently of stiffness. To determine if increased Rho activity resulted in increased cell contractility, we used traction force microscopy to quantify the forces exerted by endothelial cells as a function of matrix stiffness. Endothelial cells exerted stronger traction forces on 10 kPa substrates than on 2.5 kPa substrates (Fig. 3B), and the total magnitude of cellular force increased as a function of matrix stiffness (Fig. 3C).
Fig. 3.
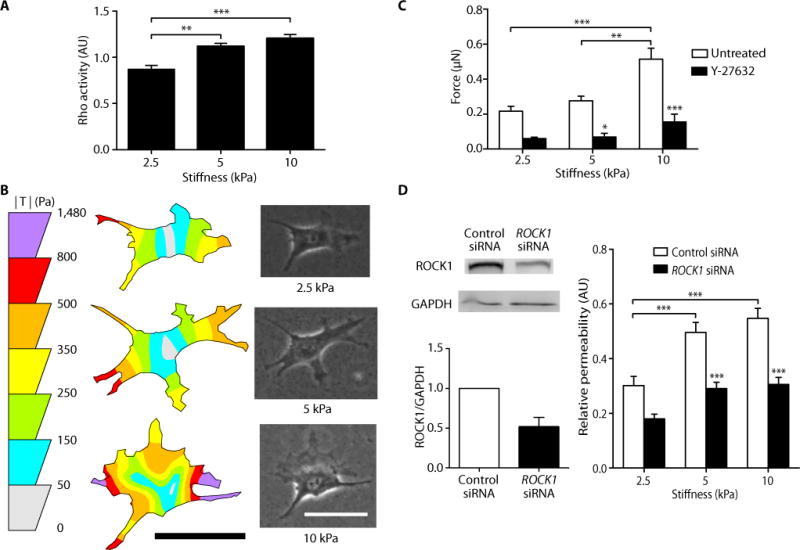
Matrix stiffness increases Rho activity and cell contractility, and inhibition of cell contractility restores barrier integrity. (A) Rho activity of BAECs cultured on polyacrylamide gels of different stiffness (three independent experiments, each performed in duplicate). Data are means ± SEM. **P < 0.01, ***P < 0.001 (Tukey’s test). (B) Representative traction force distribution maps and phase images of BAECs on gels. Scale bars, 50 μm. (C) Traction force microscopy measurements of cell contractility with (n = 9–12 cells) or without (n = 19–34 cells) Y-27632 treatment. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Tukey’s test) compared to respective untreated conditions, unless otherwise indicated by brackets. (D) RNAi of ROCK1 decreases stiffness-induced endothelial permeability. (Left) Western blot analysis and densitometry quantification of RNAi of ROCK1 (n = 2). (Right) Relative permeability of BAECs cultured on gels of varying stiffnesses with (n = 13–15 gels) or without (n = 13 gels) siRNA knockdown of ROCK1. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Tukey’s test) compared to respective control siRNA, unless otherwise indicated by brackets.
Fig. 4.
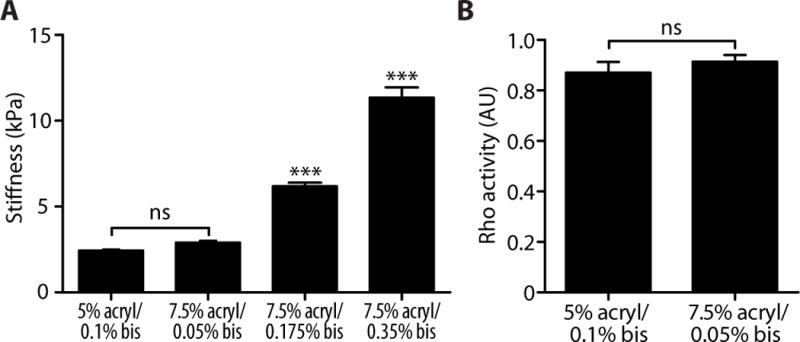
Gels made from different acrylamide concentrations that are similar in stiffness elicit similar levels of Rho activation. (A) Stiffness measurements of polyacrylamide gels of varying percentages of acrylamide and bisacrylamide. Data are means ± SEM (n = 7–8 measurements, 2 gels per group). ***P < 0.001, ns (not significant), (Tukey’s test) compared to 5% acrylamide/0.1% bisacrylamide. (B) Intracellular Rho activity of bovine aortic endothelial cells seeded on polyacrylamide gels of similar stiffness. Data for the 5% acrylamide/0.1% bisacrylamide is re-plotted from the 2.5 kPa group of Fig. 3A. Data are means ± SEM (n = 3 independent experiments, each performed in duplicate). ns, not significant (Student’s t test).
To further investigate the role of the Rho pathway in regulating increased cellular contractility and monolayer permeability in response to matrix stiffness, we inhibited ROCK, a downstream target of Rho, using the pharmacological inhibitor Y-27632. When ROCK was inhibited by exposing cells to Y-27632 for 30 min, cellular force decreased to a similar level across all matrices (Fig. 3C). Furthermore, endothelial permeability on 5 and 10 kPa substrates decreased to levels found on 2.5 kPa substrates (Fig. 1C). Similarly, endothelial permeability decreased with addition of siRNA targeting ROCK1 (Fig. 3D). In our mouse model, Y-27632 treatment of old (20 months) mice also decreased the permeability of the thoracic aorta endothelium to Evans blue dye compared to untreated old mice (Fig. 2B). These data indicate that age-related stiffness-induced permeability is mediated by increased contractility and that decreasing contractility restores monolayer integrity.
Measurements of cell-cell junction separation width in Y-27632-treated cells demonstrated that decreasing ROCK activity decreases the average cell-cell separation distance to about 2 μm (Fig. 1E). Likewise, cell-cell separation decreased significantly in Y-27632-treated old (21 to 24 months) mice compared to untreated old mice (Fig. 2D). Y-27632 treatment did not change intimal stiffness (Fig. 2A), suggesting that Y-27632 acts intracellularly to inhibit contractility, but does not cause local matrix rearrangement and stiffness changes. Taken together, these data indicate that increased endothelial monolayer permeability in response to matrix stiffness is the result of greater cell contractility, which destabilizes and widens intercellular junctions. Importantly, our data suggest that the adverse effect of age-related matrix stiffening can be reversed in vivo by inhibiting Rho pathway–mediated cell contractility.
Increased matrix stiffness promotes leukocyte transmigration
Because VE-cadherin rearrangement and monolayer permeability are known to regulate leukocyte transmigration into the vessel wall (35, 36), we asked whether stiffer substrates and the subsequent widening of junctions promote increased transmigration of leukocytes, a crucial step in the formation of atherosclerotic plaques. Primary human leukocytes were seeded on top of TNF-α–stimulated endothelial cells that were first seeded on polyacrylamide matrices. Leukocytes that had transmigrated were defined as those in which at least 50% of their cell body appeared beneath the endothelial monolayer (Fig. 5A). Notably, the number of transmigrated leukocytes increased as a function of matrix stiffness (Fig. 5B), and Y-27632 treatment of endothelial monolayers decreased transmigration (Fig. 5B), which suggests that decreased cell-cell separation inhibits leukocyte transmigration. Of the cells that transmigrated, between 87 and 98% migrated through the monolayer at cell-cell junctions (Fig. 5C), indicating that paracellular transmigration dominated over transcellular transmigration.
Fig. 5.
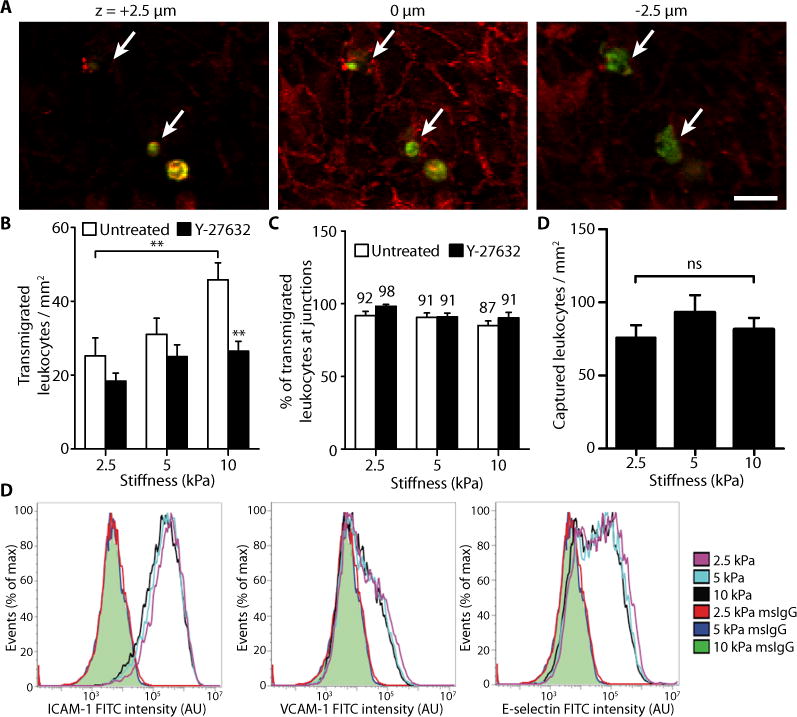
Matrix stiffness enhances leukocyte transmigration. (A) Representative fluorescent images of leukocytes (green) transmigrating through an endothelial monolayer (VE-cadherin, red). Images were taken at focal planes above the monolayer (+2.5 μm), at the monolayer (0 μm), and below the monolayer (−2.5 μm). Arrows indicate transmigrated cells. Scale bar, 20 μm. (B) Histogram showing the effect of matrix stiffness on the number of transmigrated leukocytes. Endothelial cells were either untreated (n = 19 fields of view, three independent experiments) or pre-treated with Y-27632 (n = 17–18 fields of view). Data are means ± SEM. **P < 0.01 (Tukey’s test) compared to respective untreated condition, unless otherwise indicated by brackets. (C) Percentage of leukocytes that transmigrated through intercellular junctions. Numbers above bars indicate the nominal values. Data are means ± SEM. (D) The effect of substrate stiffness on the number of captured leukocytes. Data are means ± SEM (n = 79 to 96 fields of view per stiffness). ns, not significant (Tukey’s test). (E) Flow cytometry analysis of HUVEC expression of ICAM-1, VCAM-1, and E-selectin on gels after a 6-h treatment with TNF-α. Shaded green curves represent overlapping mouse IgG isotype controls (msIgG) for each of the three matrices. Three independent experiments were performed. No statistically significant differences (Tukey’s test) were found between the three stiffnesses for all three adhesion molecules.
To determine if increased transmigration may also be due to differential expression of endothelial inflammatory molecules associated with leukocyte adhesion and transmigration, we also investigated the number of captured leukocytes under shear (to mimic blood flow) and the relative expression levels of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin—three endothelial cell inflammatory markers responsible for mediating leukocyte adhesion to the endothelium. Our data revealed no differences in leukocyte attachment to TNF-α–stimulated endothelial cells as a function of matrix stiffness (Fig. 5D; fig. S4). Additionally, the expression levels of ICAM-1, VCAM-1, and E-selectin in TNF-α-treated endothelial cells did not change as a function of matrix stiffness (Fig. 5E). Mouse IgG isotype controls were used to ensure non-specific binding of antibodies was not different between stiffnesses. These results suggest that the increase in transmigration on stiffer matrices is probably not from a differential upregulation of inflammatory molecules associated with leukocyte attachment, but rather an increase in cell-cell junction separation, which promotes transmigration.
DISCUSSION
Vessel stiffening accompanies the progression of both aging and atherosclerosis and is used to predict risk of cardiovascular diseases (6, 7); yet, the relationship between age-related changes in ECM mechanical properties that result in blood vessel stiffening and endothelial cell permeability is relatively unknown. In the present study, we demonstrate that increased vessel stiffness promotes disruption of endothelial barrier integrity, a hallmark of atherogenesis. Moreover, we demonstrate that by inhibiting endothelial cellular contractility via the Rho/ROCK pathway, we can prevent increased permeability with age. Because increased permeability of the endothelium permits cholesterol uptake in the vessel wall (16), these results demonstrate one possible therapeutic approach to preventing cholesterol accumulation within the vessel wall is to inhibit increased endothelial cell contractility.
Recently, others have also shown that endothelial cell contractile pathways modulate permeability (37) and leukocyte transmigration (18, 38). Krishnan et al.(37) showed that human umbilical vein endothelial cells (HUVECs) generated strong traction forces on stiff substrates (11 kPa) and formed intercellular gaps when treated with permeability-inducing agent thrombin; whereas cells on more compliant substrates (1.2 kPa) were less contractile and did not form gaps in response to thrombin. Sun et al. (18) showed that non-muscle myosin light-chain kinase deficiency, which reduces endothelial contractility, attenuates endothelial permeability and monocyte transmigration in an atherosclerotic mouse model. Importantly, we reversed the negative effects of matrix stiffening in both our mouse and in in vitro models by pharmacologically inhibiting Rho-dependent cell contraction (Fig. 3C). In addition to matrix stiffening, ROCK inhibition has also been shown to inhibit the negative effects of barrier-disruptive agonists that increase cell contractility, such as thrombin (39) and TNF-α (40). These data collectively indicate that inhibiting increased endothelial cell contractility may inhibit the progression of atherosclerosis.
To assess cardiovascular risk clinically, non-invasive techniques, such as pulse wave velocity tests, have been implemented to indirectly measure patient arterial stiffness (41). Because stiffness is known to correlate with the risk of cardiovascular disease, there is now substantial interest in the discovery or design of therapeutics that target vessel stiffening to prevent and treat cardiovascular disease. Thus far, several drugs have been identified, including angiotensin-converting enzyme inhibitors and calcium channel blockers (42). However, their mechanisms of action within the endothelium are poorly understood and often confounded owing to their actions on other aspects of the cardiovascular system, like blood pressure and heart rate (43). More recently, alagebrium, a drug that disrupts advanced glycation end-product (AGE) crosslinks that stiffen vessel walls, has shown mild success in treating patients with chronic (44) or diastolic heart failure (45) and improving endothelial flow-mediated dilation in patients with systolic hypertension (46). Therefore, there is evidence clinically that decreasing vessel stiffness can improve patient health.
Here, we present another approach to mitigate the deleterious effects of vessel stiffening on cardiovascular health. Rather than exclusively targeting vessel stiffness, we propose that also targeting the endothelial cells’ response to vessel stiffening may also prevent or slow atherogenesis. In some cases, directly targeting the stiffening within vessel walls is beneficial because decreasing vessel stiffness decreases the load on the heart and can decrease risk of heart failure. However, in the case of atherosclerosis, a therapy which also targets the endothelium may have certain advantages over using “de-stiffening” agents alone. ROCK inhibitors, as were used here, work quickly through intracellular signaling pathways rather than on the matrix within the vessel wall, which may require long-term treatment before significant changes occur. Because decreasing cellular contractility decreases permeability, which is one of the first steps in atherogenesis, ROCK-inhibition therapy may be more suitable for patients who are at-risk or in the very early stages of arteriosclerosis. In those patients with advanced atherosclerosis, inhibiting endothelial contractility may slow the growth of atherosclerotic lesions by preventing additional cholesterol uptake and inflammatory cell transmigration, but may not directly reduce the size of lesions that are already present. Therefore, ROCK-inhibition therapy will likely be best used as a preventative treatment for atherosclerosis and should be used in conjunction with additional therapies for the treatment of other cardiovascular complications, like heart failure, that are also due in part to vessel stiffening.
Currently, fasudil is the only ROCK inhibitor approved for clinical use (47). Originally used to treat cerebral vasospasm, fasudil has shown promise in treating a variety of cardiovascular diseases, including pulmonary hypertension (48), atherosclerosis (49), and aortic stiffness (50). Ongoing phase II clinical trials using fasudil are being performed to understand the relationship between Rho-dependent cell signaling and carotid atherosclerosis (ClinicalTrials.gov Identifier: NCT00670202). There is also some evidence that statins, which include the widely marketed Crestor and Lipitor brands, decrease Rho/ROCK activity through pleiotropic effects (47). As such, statins, in addition to their known ability to lower cholesterol levels in the blood, may also be effective in lowering cholesterol accumulation in the vessel wall by restoring barrier integrity. Overall, our data indicate that pharmacological ROCK inhibition may prevent atherosclerosis progression by preventing increased endothelial monolayer permeability and leukocyte transmigration due to vessel wall stiffening.
MATERIALS AND METHODS
Cell culture and gel synthesis
Bovine aortic endothelial cells (BAECs) were maintained in Medium 199 (Invitrogen) with 10% FetalClone III (HyClone) and supplements. Human umbilical vein endothelial cells (HUVECs) were maintained in Medium 200 (Invitrogen) supplemented with low serum growth supplement (Invitrogen) and 5% fetal bovine serum (Invitrogen). Polyacrylamide gels were prepared as described previously (51) and coated with 0.1 mg/ml rat tail collagen type I (BD Biosciences). 2.5, 5, and 10 kPa gels were made according to the ratios 5%:0.1%, 7.5%:0.175%, and 7.5%:0.35% acrylamide:bisacrylamide, respectively. All in vitro experiments were performed two days after the cells reached confluence.
Measurement of in vitro and in vivo endothelial permeability
For in vitro studies, a 10-μM solution of 40-kDa FITC-conjugated dextran (Sigma-Aldrich) was added to BAECs for 5 min. For barrier-altering agonist experiments, BAECs were either pretreated with 1 μM BW245C (Sigma-Aldrich) for 30 min or 100 ng/ml recombinant bovine TNF-α (R&D Systems) for 4 h which was then washed out before being immersed in FITC-dextran for 5 min. For human VEGF (50 ng/ml, R&D Systems) or bovine thrombin (4 U/ml, Calbiochem) experiments, drugs were administered simultaneously in FITC-conjugated dextran for 5 min. For Y-27632 experiments, BAECs were pretreated with 10 μM Y-27632 (Sigma-Aldrich) for 30 min which was then washed out before the addition of FITC-dextran. Confocal z-slices were obtained on a Leica TCS SP2 equipped with a 40 × dipping lens. To calculate relative permeability, fluorescent intensities of images were measured using ImageJ software (NIH, v. 1.42q). Dextran accumulation was determined on the basis of the fluorescent intensity within the gel normalized by the fluorescent intensity above the BAEC monolayer. This value was then normalized against dextran accumulation values determined for polyacrylamide gels without cells, as described in the SOM.
All animal protocols were approved by the Cornell University Institutional Animal Care and Use Committee. For in vivo studies, endothelial permeability was measured with an Evans blue assay (18). Briefly, C57BL/6 mice were anesthetized on isoflurane, and 0.1 mg/kg Y-27632 in PBS or PBS only was administered retro-orbitally and allowed to circulate for 1 h while mice were allowed to wake. Mice were again anesthetized and 20 mg/kg Evans blue dye was administered retro-orbitally and allowed to circulate for 1 h while mice were allowed to wake. After re-anesthetizing, 10 ml PBS was perfused through the left ventricle, and the thoracic aorta was excised and cleaned of connective tissue. After drying at 80° C for 18 h, aortas were weighed. Aortas were then immersed in 150 μl formamide and incubated at 60° C for 24 h to extract Evans blue. Absorbance was read at 620 nm. The amount of dye was determined using standard curves and normalized to aorta dry weight.
Subendothelium and polyacrylamide gel stiffness measurements
C57BL/6 mice were deeply anesthetized on isoflurane and euthanized. In the Y-27632 group, 0.1 mg/kg Y-27632 in PBS was administered retro-orbitally and allowed to circulate for 1 h while mice were allowed to wake. The thoracic aorta was dissected, and individual sections were cut from the aorta, opened longitudinally, and gently scraped 10–15 times with a cotton-tipped applicator to remove the endothelium. Stiffness of the subendothelium was measured by AFM, as described in the Supplementary Methods. Stiffnesses of polyacrylamide gels were measured using steel ball indentation, as described in the Supplementary Methods.
In vitro and ex vivo VE-cadherin junctional gap width quantification
BAECs on polyacrylamide gels were immunostained with goat polyclonal VE-cadherin primary antibody (C-19, Santa Cruz Biotechnology) and Alexa Fluor 568 donkey anti-goat secondary antibody (Invitrogen). Actin was stained with FITC-conjugated phalloidin (Sigma-Aldrich) and nuclei with DAPI (Sigma-Aldrich). Images were captured on a Zeiss Axio Observer.Z1m microscope with a Hamamatsu ORCA-ER camera.
C57BL/6 mice were anesthetized on isoflurane, and 0.1 mg/kg Y-27632 in PBS or PBS only was administered retro-orbitally and allowed to circulate for 1 h while mice were allowed to wake. The mice were then deeply anesthetized with isoflurane, overdosed on pentobarbital and intracardially perfused with cold 1% paraformaldehyde at an average flow rate of 10 ml/min, which matches the blood flow in mice to minimize artifacts from perfusion (52). Aortas were extracted and post-fixed in 1% paraformaldehyde for 1 h. Sections of the descending thoracic aorta approximately 1 mm long were stained with a biotinylated monoclonal antibody against mouse CD144 (13-1221, eBioscience). Secondary label was Texas Red streptavidin (SA-5006, Vector Laboratories, or S872, Invitrogen). Aortic sections were opened longitudinally and covered with a coverslip. Endothelial cells were imaged using a locally-built two-photon-excited fluorescence microscope using 830-nm excitation (MIRA-HP, Coherent) with a 63 ×, 1.2 NA objective (Zeiss) and 645/65-nm emission filter (Chroma Technology).
Junctional gap width measurements were analyzed using ImageJ and a custom-written MATLAB algorithm. Briefly, using fluorescent images of VE-cadherin, a line was drawn perpendicular to the widest gap per junction, and the intensity profiles were recorded using ImageJ. A two-Gaussian curve was then fit to the intensity profiles in MATLAB. Gap widths were defined as the width of the two-Gaussian fit 20% above background pixel intensity (fig. S3).
Measurement of RhoA activity
BAEC intracellular RhoA activity was quantified using a RhoA G-LISA kit (Cytoskeleton) according to the manufacturer’s protocol.
Traction force microscopy
BAECs plated on polyacrylamide gels embedded with 0.5-μm diameter fluorescent beads (Invitrogen) were allowed to adhere for 24 h. Isolated cells were imaged in phase, and the fluorescent bead field beneath the cell was imaged immediately after. A second fluorescent image of the bead field was taken after cells were removed with 0.05% trypsin/EDTA (Invitrogen). Bead displacements were used to compute cellular traction vectors, T, and total magnitudes of force, |F|, using the LIBTRC analysis library developed by M. Dembo (53). |F| (Equation 1) is the sum of the traction field magnitudes over the entire cell area,
| (Eqn 1) |
where T(x, y) = [Tx(x, y), Ty(x, y)] is the continuous field of cellular traction vectors defined at spatial position (x, y) over the entire cell area (54).
RNA interference
BAECs at 90% confluency on tissue culture plastic were transfected with 10 nM non-targeting (control) siRNA or siRNA targeting ROCK1 using 2 μg/ml Lipofectamine 2000 (Invitrogen). The non-targeting sequence was 5′-AAUCAUCAAGUCUUAACCCGUACUC-3′. The sequence targeting ROCK1 was 5′-CAGAAGUGCAGAACGUCAAACAUAA-3′. Both the control siRNA and the siRNA targeting ROCK1 (accession number NM_001191227.1) were synthesized by Invitrogen. ROCK1 interference was quantified by Western blot densitometry. Primary antibodies against ROCK1 and GAPDH were purchased from Santa Cruz Biotechnology and Millipore, respectively.
Neutrophil transmigration and rolling adhesion
All human subject protocols have been approved by the Institutional Review Board for Human Participants at Cornell University. Fresh peripheral human blood was collected into vacutainer tubes containing heparin and allowed to equilibrate at room temperature. Blood was layered over 1-Step Polymorphs (Accurate Chemical) and separated by centrifugation. The neutrophil layer was collected, washed, and resuspended in flow buffer (0.5% human serum albumin (Sigma-Aldrich), 10 mM HEPES and 2 mM CaCO3 in Hank’s Balanced Salt Solution). HUVEC monolayers pre-treated with 0.1 ng/ml recombinant human TNF-α (R&D Systems) for 6 h were either untreated or treated with 10 μM Y-27632 for 1 h. Neutrophils (50,000 cells/cm2) were then allowed to adhere for 5 min. Gels were gently washed twice with flow buffer to remove unattached neutrophils. HUVEC VE-cadherin was stained as described previously, and neutrophils were stained with a FITC-conjugated antibody against human CD45 (Invitrogen). Images were taken on a Zeiss LSM710 confocal microscope with a 25 × objective. Transmigrated neutrophils were defined as CD45-positive cells with at least 50% of its cell body located beneath the endothelial layer. For rolling experiments, neutrophils were flowed over TNF-α–stimulated HUVECs and quantified, as described in the Supplementary Methods.
Flow cytometry
HUVECs on gels were treated with 0.1 ng/ml TNF-α for 6 h. Gels were rinsed with PBS and inverted onto accutase (MP Biomedicals). Cells were dislodged by pipetting and pelleted by centrifugation. After blocking with 1% goat serum, cells were incubated with primary antibodies against ICAM-1, VCAM-1, or E-selectin, or mouse IgG1 isotype control (R&D Systems). Secondary label was Alexa Fluor 488 anti-mouse antibody (Invitrogen). Labeled cells were resuspended and analyzed using an Accuri C6 flow cytometer.
Statistical analysis
All analyses were completed using JMP 8 (SAS Institute, Cary, NC) or GraphPad Prism 5 (GraphPad Software, Inc.). Data in Fig. 1B were analyzed by parametric one-way ANOVA with post-hoc Dunnett’s test to compare treated conditions to untreated control. Data in Fig. 1C, Fig. 1E, Fig. 3, Fig. 4A, and Fig. 5 were analyzed by parametric one-way ANOVA with post-hoc Tukey’s test to compare untreated conditions. Two-way ANOVA with post-hoc Tukey’s test was then used to compare Y-27632 or ROCK1-targeted siRNA data to its respective control. Data in Fig. 2 were analyzed by non-parametric Kruskal-Wallis ANOVA with post-hoc Dunn’s test. Data in Fig. 4B and fig. S1C were analyzed using Student’s t test. All data are means ± SEM. P values < 0.05 were considered significant.
Supplementary Material
Materials and Methods
Fig. S1. Endothelial permeability measurements.
Fig. S2. VE-cadherin junctional gap-width measurement.
Fig. S3. Histology and AFM indentation of mouse thoracic aorta.
Fig. S4. Leukocytes captured on HUVEC monolayers grown on polyacrylamide gels under flow.
References
Acknowledgments
We thank the Cornell University Life Sciences Core Laboratories Center Microscopy and Imaging Facility. Funding: The American Heart Association, the American Federation for Aging Research, and the National Institutes of Health funding (grant number HL097296) to C.A.R. and the L’Oréal USA Fellowship for Women in Science and National Institutes of Health funding (grant number F32AG031620) to N.N.
Footnotes
Author contributions: J.H. performed in vitro permeability studies, Rho assays, junction-width quantification, and traction force microscopy. N.N. performed two-photon microscopy of VE-cadherin in mouse aortas. N.N. and J.H. performed in vivo permeability measurements of mouse aortas to Evans blue dye. K.R. and J.H. performed the leukocyte adhesion and transmigration assays. J.M.P. performed AFM indentation testing. J.P.C. performed in vitro VE-cadherin staining, traction force microscopy, and gel-stiffness measurements. C.R.M. performed flow cytometry experiments. C.A.R. and J.H. analyzed the data and wrote the paper. C.B.S., M.R.K., and C.A.R. designed the study. All authors discussed the results and commented on the manuscript.
Competing interests: The authors declare no competing interests.
REFERENCES AND NOTES
- 1.Laurent S, Boutouyrie P. Recent advances in arterial stiffness and wave reflection in human hypertension. Hypertension. 2007;49:1202–1206. doi: 10.1161/HYPERTENSIONAHA.106.076166. [DOI] [PubMed] [Google Scholar]
- 2.Fernandes VR, Polak JF, Cheng S, Rosen BD, Carvalho B, Nasir K, McClelland R, Hundley G, Pearson G, O’Leary DH, Bluemke DA, Lima JA. Arterial stiffness is associated with regional ventricular systolic and diastolic dysfunction: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:194–201. doi: 10.1161/ATVBAHA.107.156950. [DOI] [PubMed] [Google Scholar]
- 3.McEniery CM, Yasmin, Hall IR, Qasem A, Wilkinson IB, Cockcroft JR. Normal vascular aging: differential effects on wave reflection and aortic pulse wave velocity: the Anglo-Cardiff Collaborative Trial (ACCT) J Am Coll Cardiol. 2005;46:1753–1760. doi: 10.1016/j.jacc.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 4.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 5.Greenwald SE. Ageing of the conduit arteries. J Pathol. 2007;211:157–172. [Google Scholar]
- 6.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. 2006;113:657–663. doi: 10.1161/CIRCULATIONAHA.105.555235. [DOI] [PubMed] [Google Scholar]
- 7.Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, Havlik R, Lakatta EG, Spurgeon H, Kritchevsky S, Pahor M, Bauer D, Newman A. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111:3384–3390. doi: 10.1161/CIRCULATIONAHA.104.483628. [DOI] [PubMed] [Google Scholar]
- 8.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 9.Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA. 2006;103:10889–10894. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 12.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204:198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 13.Lemarie CA, Tharaux PL, Lehoux S. Extracellular matrix alterations in hypertensive vascular remodeling. J Mol Cell Cardiol. 2010;48:433–439. doi: 10.1016/j.yjmcc.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 14.Reinhart-King CA, Dembo M, Hammer DA. Cell-cell mechanical communication through compliant substrates. Biophys J. 2008;95:6044–6051. doi: 10.1529/biophysj.107.127662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 16.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K, Forudi F, Saidel GM, Penn MS. Alterations in internal elastic lamina permeability as a function of age and anatomical site precede lesion development in apolipoprotein E-null mice. Circ Res. 2005;97:450–456. doi: 10.1161/01.RES.0000181026.94390.c9. [DOI] [PubMed] [Google Scholar]
- 18.Sun C, Wu MH, Yuan SY. Nonmuscle Myosin Light-Chain Kinase Deficiency Attenuates Atherosclerosis in Apolipoprotein E-Deficient Mice via Reduced Endothelial Barrier Dysfunction and Monocyte Migration. Circulation. 2011;124:48–57. doi: 10.1161/CIRCULATIONAHA.110.988915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rozenberg I, Sluka SH, Rohrer L, Hofmann J, Becher B, Akhmedov A, Soliz J, Mocharla P, Boren J, Johansen P, Steffel J, Watanabe T, Luscher TF, Tanner FC. Histamine H1 receptor promotes atherosclerotic lesion formation by increasing vascular permeability for low-density lipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:923–930. doi: 10.1161/ATVBAHA.109.201079. [DOI] [PubMed] [Google Scholar]
- 20.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 21.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 22.Reinhart-King CA. Endothelial cell adhesion and migration. Methods Enzymol. 2008;443:45–64. doi: 10.1016/S0076-6879(08)02003-X. [DOI] [PubMed] [Google Scholar]
- 23.Peloquin J, Huynh J, Williams RM, Reinhart-King CA. Indentation measurements of the subendothelial matrix in bovine carotid arteries. J Biomech. 2011;44:815–821. doi: 10.1016/j.jbiomech.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J. 2004;87:4259–4270. doi: 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper JA, Del Vecchio PJ, Minnear FL, Burhop KE, Selig WM, Garcia JG, Malik AB. Measurement of albumin permeability across endothelial monolayers in vitro. J Appl Physiol. 1987;62:1076–1083. doi: 10.1152/jappl.1987.62.3.1076. [DOI] [PubMed] [Google Scholar]
- 26.Mogensen CE. Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N Engl J Med. 1984;310:356–360. doi: 10.1056/NEJM198402093100605. [DOI] [PubMed] [Google Scholar]
- 27.Pedrinelli R, Penno G, Dell’Omo G, Bandinelli S, Giorgi D, Di Bello V, Nannipieri M, Navalesi R, Mariani M. Transvascular and urinary leakage of albumin in atherosclerotic and hypertensive men. Hypertension. 1998;32:318–323. doi: 10.1161/01.hyp.32.2.318. [DOI] [PubMed] [Google Scholar]
- 28.Murata T, Lin MI, Aritake K, Matsumoto S, Narumiya S, Ozaki H, Urade Y, Hori M, Sessa WC. Role of prostaglandin D2 receptor DP as a suppressor of tumor hyperpermeability and angiogenesis in vivo. Proc Natl Acad Sci USA. 2008;105:20009–20014. doi: 10.1073/pnas.0805171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 30.van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol. 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 31.Birukova AA, Arce FT, Moldobaeva N, Dudek SM, Garcia JG, Lal R, Birukov KG. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomedicine. 2009;5:30–41. doi: 10.1016/j.nano.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 33.Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8:175–176. doi: 10.1016/j.ccr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turowski P, Martinelli R, Crawford R, Wateridge D, Papageorgiou AP, Lampugnani MG, Gamp AC, Vestweber D, Adamson P, Dejana E, Greenwood J. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121:29–37. doi: 10.1242/jcs.022681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VW, Carman CV, Brain JD, Fredberg JJ, Butler JP, van Nieuw Amerongen GP. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol. 2011;300:C146–154. doi: 10.1152/ajpcell.00195.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stroka KM, Aranda-Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood. 2011;118:1632–1640. doi: 10.1182/blood-2010-11-321125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 40.Nwariaku FE, Rothenbach P, Liu Z, Zhu X, Turnage RH, Terada LS. Rho inhibition decreases TNF-induced endothelial MAPK activation and monolayer permeability. J Appl Physiol. 2003;95:1889–1895. doi: 10.1152/japplphysiol.00225.2003. [DOI] [PubMed] [Google Scholar]
- 41.Blacher J, Asmar R, Djane S, London GM, Safar ME. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension. 1999;33:1111–1117. doi: 10.1161/01.hyp.33.5.1111. [DOI] [PubMed] [Google Scholar]
- 42.Oliver JJ, Webb DJ. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events. Arterioscler Thromb Vasc Biol. 2003;23:554–566. doi: 10.1161/01.ATV.0000060460.52916.D6. [DOI] [PubMed] [Google Scholar]
- 43.Hope SA, Hughes AD. Drug effects on the mechanical properties of large arteries in humans. Clin Exp Pharmacol Physiol. 2007;34:688–693. doi: 10.1111/j.1440-1681.2007.04661.x. [DOI] [PubMed] [Google Scholar]
- 44.Willemsen S, Hartog JW, Hummel YM, Posma JL, van Wijk LM, van Veldhuisen DJ, Voors AA. Effects of alagebrium, an advanced glycation end-product breaker, in patients with chronic heart failure: study design and baseline characteristics of the BENEFICIAL trial. Eur J Heart Fail. 2010;12:294–300. doi: 10.1093/eurjhf/hfp207. [DOI] [PubMed] [Google Scholar]
- 45.Little WC, Zile MR, Kitzman DW, Hundley WG, O’Brien TX, Degroof RC. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J Card Fail. 2005;11:191–195. doi: 10.1016/j.cardfail.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 46.Zieman SJ, Melenovsky V, Clattenburg L, Corretti MC, Capriotti A, Gerstenblith G, Kass DA. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens. 2007;25:577–583. doi: 10.1097/HJH.0b013e328013e7dd. [DOI] [PubMed] [Google Scholar]
- 47.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–629. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91:391–392. doi: 10.1136/hrt.2003.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, Liao JK, Creager MA. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. 2006;99:1426–1432. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, Kimura M, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Liao JK, Higashi Y. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol. 2007;49:698–705. doi: 10.1016/j.jacc.2006.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Califano JP, Reinhart-King CA. A Balance of Substrate Mechanics and Matrix Chemistry Regulates Endothelial Cell Network Assembly. Cell Molec Bioeng. 2008;1:122–132. [Google Scholar]
- 52.Huo Y, Guo X, Kassab GS. The flow field along the entire length of mouse aorta and primary branches. Ann Biomed Eng. 2008;36:685–699. doi: 10.1007/s10439-008-9473-4. [DOI] [PubMed] [Google Scholar]
- 53.Dembo M, Wang YL. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J. 1999;76:2307–2316. doi: 10.1016/S0006-3495(99)77386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reinhart-King CA, Dembo M, Hammer DA. The dynamics and mechanics of endothelial cell spreading. Biophys J. 2005;89:676–689. doi: 10.1529/biophysj.104.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Endothelial permeability measurements.
Fig. S2. VE-cadherin junctional gap-width measurement.
Fig. S3. Histology and AFM indentation of mouse thoracic aorta.
Fig. S4. Leukocytes captured on HUVEC monolayers grown on polyacrylamide gels under flow.
References