

Summary
Background:
Chondrodysplasia punctata (CDP) is a rare, heterogeneous congenital skeletal dysplasia, characterized by punctate or dot-like calcium deposits in cartilage observed on neonatal radiograms. A number of inborn metabolic diseases are associated with CDP, including peroxisomal and cholesterol biosynthesis dysfunction and other inborn errors of metabolism such as: mucolipidosis type II, mucopolysacharidosis type III, GM1 gangliosidosis. CDP is also related to disruption of vitamin K-dependent metabolism, causing secondary effects on the embryo, as well as fetal alcohol syndrome (FAS), chromosomal abnormalities that include trisomies 18 and 21, Turner syndrome.
Case Report:
This article presents clinical data and diagnostic imaging findings of two newborn babies with chondrodysplasia punctata. Children presented with skeletal and cartilage anomalies, dysmorphic facial feature, muscles tone abnormalities, skin changes and breathing difficulties. One of the patients demonstrated critical stenosis of spinal canal with anterior subluxation of C1 vertebra relative to C2. The aim of this article is to present cases and briefly describe current knowledge on etiopathogenesis as well as radiological and clinical symptoms of diseases coexisting with CDP.
Conclusions:
Radiological diagnostic imaging allows for visualization of punctate focal mineralization in bone epiphyses during neonatal age and infancy.
Determining the etiology of chondrodysplasia punctata requires performing various basic as well as additional examinations, including genetic studies.
Keywords: chondrodysplasia punctata, skeletal dysplasia, newborn, congenital malformation
Background
Punctuate mineralizations – chondroplasia punctuata (CDP) – are observed in several diseases of various etiologies, some of which are well recognized, while others require further studies. Chondrodysplasia punctuata is a rarely occurring skeletal dysplasia characterized by stippled, punctuate calcifications around joints and within cartilages. Calcifications are most often located in the epiphyses of long bones and soft tissues around joints and vertebral column. They constitute radiological signs of chondrodysplasia punctuata. CDP occurs in hereditary metabolic diseases [1,2], chromosomal aberrations [3], may be one of the symptoms of fetal alcohol syndrome (FAS) [4], may occur in children of mothers with vitamin K deficiency [5] or mothers treated with warfarin [6]. There are also reports in the literature regarding the association of CDP with autoimmunological disease diagnosed in a mother (e.g. systemic lupus erythematosus) [1,7,8].
Foci of calcification formed during osteogenesis are observed in newborns, although they may be seen as early as during fetal life. Due to epiphyseal ossification, calcifications are no longer visible at the preschool and later age (they often disappear in the 2nd or 3rd year of life).
Foci of mineralization and calcifications of epiphyseal cartilage visible on plain x-ray pictures are secondary to early improper calcium deposition and also occur in cartilages that usually do not undergo calcification, such as trachea, larynx, bronchi, distal ends of the ribs.
Phenotypically, children exhibit a variety of symptoms depending on the type of CDP and etiology of this disorder: dysmorphic facial features, nose hypoplasia, sunken nasal bridge, symmetrical or asymmetrical shortening of proximal ends of limbs, other bone anomalies, ichthyosis [1,2,9,10].
The goal of this work is to present clinical data and results of imaging studies of two newborn babies with chondrodysplasia punctuata and to shortly illustrate current knowledge on etiopathogenesis as well as radiological and clinical symptoms of diseases associated with CDP.
Case Report
Case 1
A five-day-old male baby, born in the 39th week of pregnancy was admitted to Neonatology Clinic due to hypoplastic anterior nares and breathing problems. On physical examination we noted craniofacial anomaly in a form of flattening and shortening of nasal bridge, improper setting of anterior nares and mongoloid eyelids. Moreover, we observed arched feet, overlapping fingers, a transverse groove on the left palm and shortened distal phalanges. There were eruptions on the skin resembling newborn toxic erythema. Severely impaired nasal patency caused increased breathing effort. Breathing problems required transient use of oropharyngeal tube and oxygen therapy. In neurological examination we noted decreased muscle tone in the lower limbs. Serological studies excluded congenital cytomegalovirus (CMV) and protozoan Toxoplasma gondii infection. Cytogenetic study revealed normal karyotype.
Radiological symptoms
X-ray bone pictures visualized symmetrical stippled calcifications located bilaterally, mainly in the paravertebral lumbosacral region. Lesions of similar morphology were also visible at a level of hip joints, maxillary angle, around wrists and feet (Figures 1 and 2). There were numerous anomalies of vertebral bodies (Figure 3).
Figure 1.
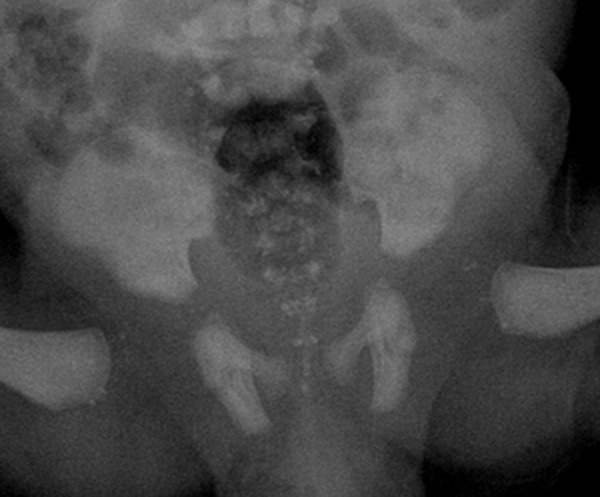
Radiograph of the pelvis – bilateral punctate calcifications in the region of hip joints and lumbosacral spine.
Figure 2.
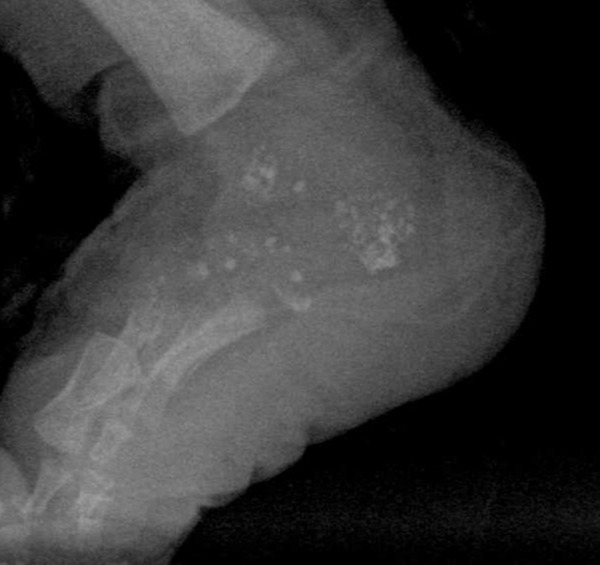
Radiograph demonstrates stippled calcifications in the tarsal bones and in the surrounding soft tissues.
Figure 3.
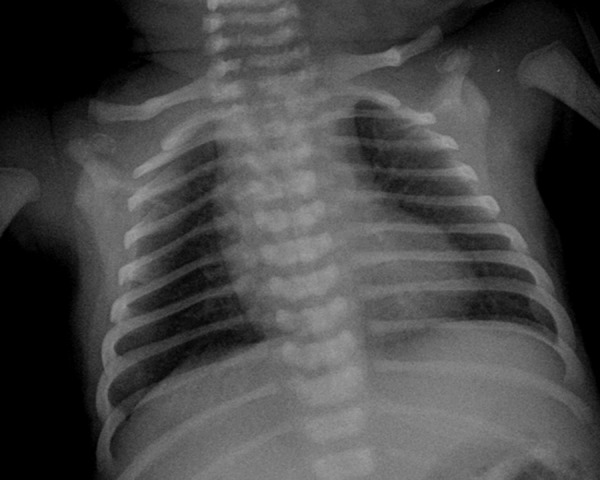
Plain chest radiograph showing thoracic spine defects.
Computed tomography (CT) of facial bones showed significantly smaller piriform aperture, right-sided nasal septum deviation and vertical position of nasal bones.
Based on the results of radiological studies and clinical picture we suggested the diagnosis of chondrodysplasia punctuata of brachytelephalangic type.
At the age of 31 days patient was transferred to the Department of Pediatric Otolaryngology at another hospital in order to continue the diagnosis and verify indications for surgical correction of nasal deformation.
Case 2
A male baby born in asphyxia in the 38th week of pregnancy was admitted to the Neonatology Clinic on the first day of life in severe general condition for selective head cooling using “cool cup” method. On physical examination we noted dysmorphic facial features, nasal deformation, lowset ears, thoracic cavity malformation, petechiae, skin cyanosis. In neurological examination we noted loss of spontaneous limb movements, absence of deep-tendon reflexes and significant general hypotonia. Severe breathing difficulties required treatment with tracheostomy. Audiological consultation confirmed bilateral sensorineural hearing loss. Serological studies excluded congenital cytomegalovirus infection and toxoplasmosis. The karyotype was normal.
Radiological signs
Plain chest x-ray visualized numerous stippled calcifications located bilaterally around the vertebrae (Figure 4). Point calcifications were also visible at a level of laryngeal and tracheal cartilage (Figure 5) and bilaterally at the level of feet.
Figure 4.
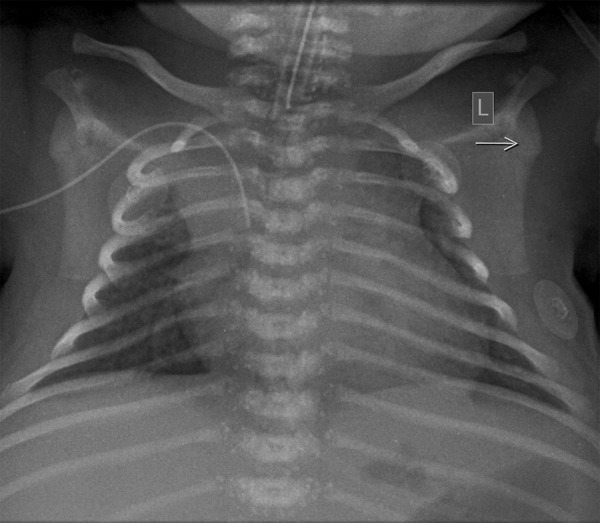
Chest radiograph – bilateral diffuse calcifications in the region of thoracic spine and in the tracheal cartilages.
Figure 5.
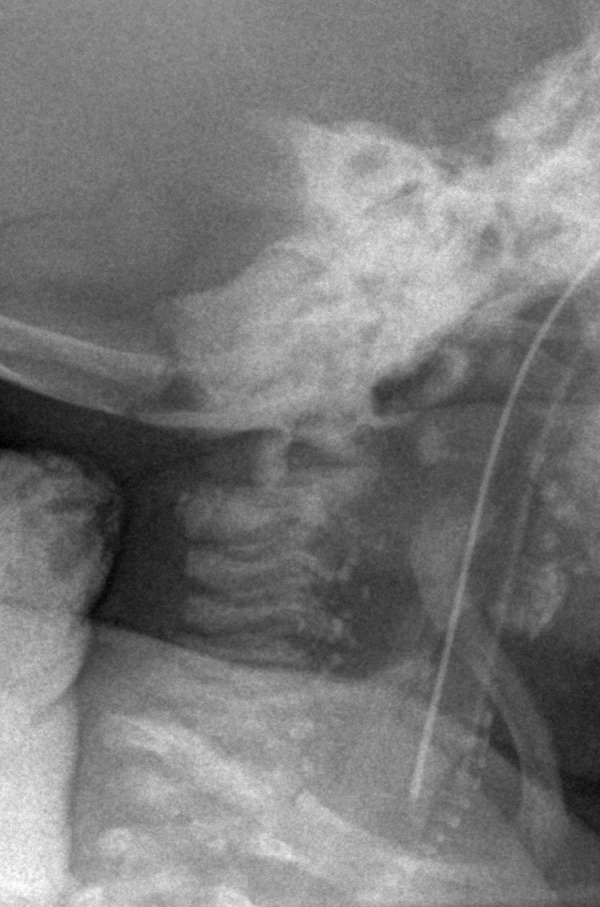
Lateral radiograph of cervical spine.
Brain MRI showed abnormal signal intensity in the white matter, myelinated posteroinferior part of brainstem, loss of myelin in the supratentorial region, presence of blood in posterior horns of lateral ventricles. Severe spinal canal stenosis with spinal compression ensuing from abnormal anterior dislocation of C1 vertebra was noted in coronal cross-section (Figure 6).
Figure 6.
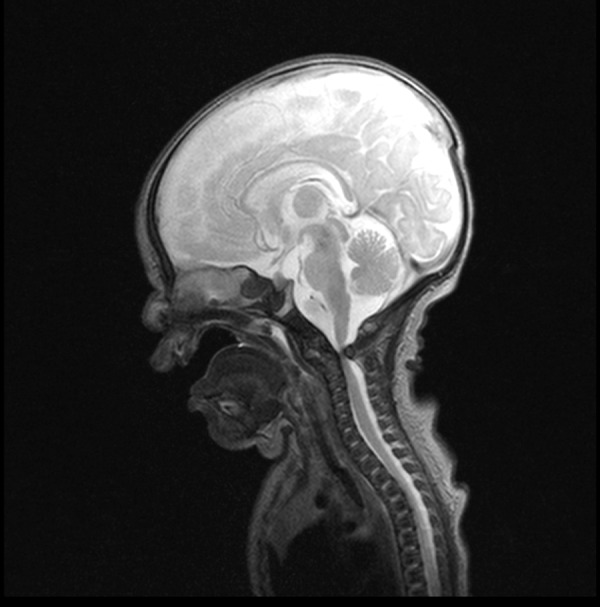
MR examination – sagittal plane, T2-weighted image – anterior subluxation of C1 vertebra relative to C2 with severe spinal canal stenosis causing severe cord compression.
CT examination of craniocervical junction and cervical spine confirmed dislocation of C1 body in atlantoaxiliary joint with anterior displacement of C1 vertebra in relation to C2, visualized disrupted bone structure of vertebral bodies in cervical spine and presence of severely mineralized foci at the level of larynx (Figures 7A, 7B and 8).
Figure 7.

Axial CT scans – punctate calcifications in cervical vertebrae, around left humeral joint (A) and in the cartilages of the trachea (B).
Figure 8.
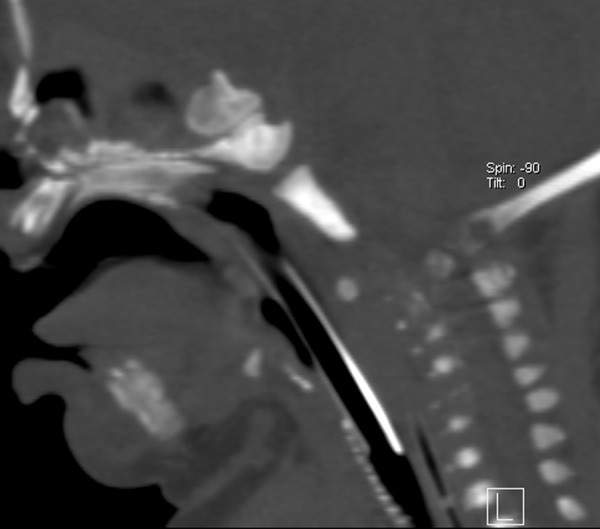
Sagittal CT scan of cervical spine demonstrates punctate calcifications of tracheal and laryngeal cartilages.
Child was not referred to neurosurgical treatment due to high surgical risk.
Metabolic diagnostics revealed normal levels of ammonia, lactic acid, pyruvic acid and very long-chain fatty acids (VLCFA). Diagnosis of type 1 rhizomelic chondrodysplasia punctuata was suggested on the basis of imaging studies and clinical picture.
Molecular analysis of patient DNA (coding regions together with adjacent intron sequences of PEX7 gene) was conducted. Result of molecular examination did not confirm clinical diagnosis of type 1 rhizomelic chondrodysplasia punctuata. Child remains under clinical follow-up.
Discussion
Chondrodysplasia punctuata (CDP) was first described by Conradi in 1914 [11]. Minute foci of calcification are seen in a variety of diseases of diverse etiologies. CDP is associated with inborn metabolic diseases in the course of peroxisome dysfunction and disturbances of cholesterol biosynthesis as well as other inborn metabolic errors, including type 2 mucolipidosis, type 3 mucopolysaccharidosis and gangliosidosis GM1 [1,2]. Vitamin K-dependent metabolic abnormalities [5], use of warfarin during pregnancy [6], autoimmunological disease in a mother (systemic lupus erythematosus) [1,7,8], fetal alcohol syndrome FAS [4], chromosomal aberrations: trisomy 18 and 21 and Turner syndrome [3], which give symptoms typical for CDP, exert harmful influence on the embryo.
Clinical symptoms may manifest as: psychomotor impairment, dysmorphic facial features (nose hypoplasia, sunken nasal bridge, large and prominent forehead), skin lesions (ichthyosis), alopecia areata, cataract, hearing impairment, arthropathies with contractures, congenital heart disease. A typical feature noted in radiological studies involves presence of punctate calcifications in the cartilages of trachea, larynx, bronchi and nose, as well as around joints and vertebra during neonatal period and infancy.
Calcifications usually disappear around 2nd–3rd year of life. Bone mineralization is usually insufficient. There are also defects of the spine – butterfly vertebrae and abnormal vertebral curvature – most often kyphoscoliosis. Limb length may be abnormal due to symmetrical or asymmetrical shortening of proximal limb bones.
Table 1 presents a compilation of clinical and radiological symptoms as well as genetic data regarding specific comorbidities coexisting with chondrodysplasia punctuata based on the available literature [1–6,9,11–14].
Table 1.
Compilation of literature clinical and radiological symptoms as well as genetic data regarding children with chondrodysplasia punctata manifestations.
| Clinical symptoms | Radiological signs | Inheritance | Gene | Locus | Etiology | |
|---|---|---|---|---|---|---|
| Rhizomelic chondrodysplasia punctuata (RCDP) | Osteoarticular system: symmetrical rhizomelic shortening of proximal limb bones; articular contractures; deformation of feet; shortened metacarpal bones, most often IV; hypoplastic distal phalanges. Face: flattened nasal bridge; micrognathia, cleft palate. Other: skin lesions - ichthyosis; cataract; impaired psychomotor development; epileptic episodes; deafness; congenital heart disease; |
Skeletal system: symmetrical shortening of proximal fragments of long bones; punctate calcifications in cartilages of axial skeleton; vertebral fissures visible in frontal plane; irregular reduction in height of vertebral bodies; trapezoid iliac bones; CNS: microcephaly |
Autosomal recessive | PEX7 (RCDP1) DHAPAT (RCDP2) AGPS (RCDP3) |
6q22-q24 1q42 2q31 |
Peroxisomal metabolism dysfunction; PTS2 dysfunction (peroxisomal targeting signal); inability of proxisomal receptors to recognize and bind proteins; Dysfunction of plasmalogens and peroxisomal enzymes: dihydroxyacetone phosphate acyltransferase acyl-CoA and acyl-CoA synthase |
| Zellweger syndrome | Face: nasal hypoplasia, large, prominent forehead. Other: camptodactyly; hepatosplenomegaly; severe muscle hypotonia; club foot; epileptic seizures; nystagmus; retinopathy; cataract; deafness; liver cirrhosis |
Osteoarticular system: punctate calcifications in skeletal cartilages and around joints, particularly hip and knee joints; point calcifications in the patella. CNS: brain hypomyelination; leukoencephalopathy; polimicrogyria; pachygyria; agyria Other: cortical renal cysts |
Autosomal recessive | PEX1 PEX2 PEX3 PEX5 PEX6 PEX10 PEX12 PEX13 |
7q21 8q 6q 12p13.3 6p.21.1 1p.36 17q11.2 2p15 |
Deficiency of peroxisomal enzymes Elevated VLCFA level |
| Chondrodysplasia punctuata: Conradi-Hunermann type (CDPX2) | Face: flattened nasal bridge; large and prominent forehead, asymmetrical face; Osteoarticular system: symmetrical or asymmetrical shortening of limbs; articular contractures; deformation of feet; polydactyly; short neck. Ocular symptoms: uni- or bilateral cataract. Skin lesions: ichthyosis; linear areas of skin atrophy; pigmented nevi; hair thinning. Other: delayed psychomotor development; deafness |
Diffuse paravertebral calcifications as well as around joints, in the cartilages of larynx, trachea and nose; symmetrical or asymmetrical shortening of long bones; articular contractures; scoliosis; vertebral anomalies; platyspondylia; deformed feet | X-linked dominant; possible lethal disease in males, hemizygote (Xp 22.32) | EBP | Xp11.23 p11.22 | 3β-hydroxysteroid-Δ8,Δ7 isomerase deficiency; Impaired cholesterol biosynthesis |
| Chondrodysplasia punctuata: Brachytelephalangic type (CDPX1) | Face: nasal hypoplasia with flattened nasal bridge; cleft palate. Osteoarticular system: hypoplastic or small fingers of the hands and feet; polydactyly; short height. Other: cataract, hearing impairment or deafness; delayed motor development; normal mental development or slight mental impairment; ichthyosis, congenital heart disease |
Diffuse punctate calcifications in proximal epiphyses of femurs, at the level of wrists and feet, in soft tissues surrounding joints, paravertebrally, in the area of lumbar spine, sacrum and coccyx; punctate calcifications in the cartilages of trachea and larynx; scoliosis; hypoplasia of metacarpal and metatarsal bones; hypoplasia of distal phalanges of hands and feet, triangular distal phalanges | X-linked recessive; deletion of the short arm of X chromosome | ARSE | Xp22.3 | Dysfunction of vitamin K-dependent arylsulfatase E |
| Sheffield type of chondrodysplasia punctuata | Face: sunken nasal bridge and flattened nasal tip. Other: short statue; mental impairment |
Punctate calcifications at a level of calcaneus in early childhood; in older children calcaneus is improperly mineralized, hypoplastic and abnormal; punctuate calcifications in the area of sacral and coccygeal bone; vertebral fissures visible in frontal and sagittal planes | Unknown | |||
| Chromosomal aberrations Trisomy 18 Trisomy 21 Turner syndrome |
Nasal hypoplasia; facial dysmorphic features; clinodactyly; brachydactyly; short statue; mental impairment; congenital abnormalities of internal organs | Punctate calcifications in the epiphyses, in the sacral and coccygeal bones, bones of the tarsum and calcaneus, in the cartilages of the pelvis, in the patella; vertebral anomalies; scoliosis, kyphoscoliosis CNS: microcephaly |
Chromosomal aberrations | 47,XX/XY,+18 47,XX/XY,+21 45,X |
||
| Fetal alcohol syndrome (FAS) | Face: nasal hypoplasia; hypertelorism; mongoloid eyelids; micrognathia; flattened nasal bridge; high-set palate with cleft soft palate; dysplastic ears. Other: camptodactyly; syndactyly; clinodactyly; abnormal palmar groove; short neck; ocular abnormalities; pulmonary artery stenosis; congenital heart disease, urinary system abnormalities | Skeletal system: symmetrical punctuate calcifications: in cartilages, calcaneus and tarsal bones, in proximal epiphyses of femurs, in the sacral bone; scoliosis; hemivertebrae; Klippel-Feil syndrome; hypoplastic distal phalanges CNS: agenesis or hypoplasia of corpus callosum; delayed brain myelination; microcephaly; widened cranial sutures |
||||
| Warfarin embryopathy | Hypoplasia of nasal bones; sunken nasal bridge; large and prominent forehead; additional fontanelles; anomalies of auditory and visual systems (cataract), abnormal central nervous system development; hypotonia. | Skeletal system: punctuate calcifications in the axial skeleton, mainly in proximal epiphyses of long bones, wrists, calcaneal bones, ribs; scoliosis CNS: meningomyelocele; Dandy-Walker malformation; mikrocephaly. |
Abnormalities of vitamin K metabolism; use of warfarin by the mother during pregnancy | |||
| Vitamin K deficiency in mothers of children with CDP | Flattened nasal bridge; nasal hypoplasia; brachydactyly; hypoplasia of distal phalanges | Hypoplasia of distal phalanges with punctuate calcifications in the epiphyses of long bones; spinal anomalies (clefting); scoliosis; hemivertebrae CNS: agenesis of corpus callosum; subependymal cysts |
Persistent vomiting during early pregnancy; abnormalities of gastrointestinal system with malabsorption syndromes; use of phenytoin by the mother during pregnancy. | |||
| Autoimmune diseases in mothers of children with CDP (i.a. SLE) | Face: nasal hypoplasia with flattened nasal bridge; long and flattened philtrum; Other: short limbs; brachydactyly; clinodactyly; camptodactyly; large anterior and posterior fontanelle; short neck |
Punctuate calcifications in the epiphyses of long bones, around joints, in the spine, tarsal bones and in the trachea and larynx; fissures visible in vertebral bodies in frontal and sagittal planes; rhizomelic shortening of long bones; hypoplasia of distal phalanges; shortened metacarpal bones with calcifications in proximal parts; abnormal, thoracic hyperkyphosis; scoliosis; hypoplasia of craniofacial bones CNS: dolichocephaly |
Detrimental influence of antibodies on the embryo (i.a. antiphospholipid antibodies in the course of SLE) |
Normal karyotypes found in both neonates presented in this publication allowed for exclusion of trisomy 18 and 21 as well as Turner syndrome as a cause of CDP. Based on medical history, we also excluded FAS syndrome, autoimmunological diseases (systemic lupus erythematosus) in children’s mothers, vitamin K deficiency and warfarin use. Zellweger syndrome was excluded based on normal VLCFA levels and magnetic resonance brain imaging.
Chondrodysplasia punctuata is a rare skeletal dysplasia characterized by occurrence of stippled point calcifications around joints and in cartilages of larynx, trachea, bronchi and nose. Respiratory system anomalies ensuing from abnormal cartilage structures lead to respiratory problems of varying intensity, which may be the cause of death during infancy. Respiratory problems observed in both presented patients required transient use of oropharyngeal tube and oxygen therapy in the first case and tracheostomy in the other.
Differential diagnosis included two types of chondrodysplasia punctuata.
In the first case, shortened distal phalanges and presence of point calcifications at the level of wrists, feet and mandibular angles suggested the brachytelephalangic type of chondrodysplasia punctuata.
In the second case, a diagnosis of type 1 rhizomelic chondrodysplasia punctuata was suggested based on severe general condition, severe general hypotonia, bilateral sensorineural hearing loss and congenital anomaly of C1 vertebral body as well as critical spinal stenosis.
Spinal canal stenosis is a frequent sign of bone dysplasia, while it rarely occurs in chondrodysplasia punctuata. Several dozen cases were published regarding coexistence of spinal canal stenosis and vertebral instability with CDP [9,13–22]. Literature described stenosis of cervical part of spinal canal and dislocation of C1 vertebral body in the atlantoaxillary joint in the following chondrodysplasias: rhizomelic type (RCDP1), brachytelephalangic type (CDPX1), Conradi-Hunermann type (CDPX2).
In the second case molecular analysis was performed to confirm or exclude PEX7 gene mutation associated with type 1 RCDP1, in which coexistence of cervical spine instability was most frequently observed. In this case, we did not demonstrate PEX7 gene mutation, which does not rule out the remaining subtypes of chondrodysplasia: CDPX1 or CDPX2. However, it requires performing other genetic studies.
Conclusions
Radiological imaging studies allow for visualization of point foci of improper mineralization of bone epiphyses during the newborn period and infancy.
Determining the etiology of chondrodysplasia punctuata requires various basic and additional examinations, including genetic testing.
References:
- 1.Chitayat D, Keating S, Zand DJ, et al. Chondrodysplasia Punctata Associated With Maternal Autoimmune Disease: Expanding the Spectrum From Systemic Lupus Erythematosus (SLE) to Mixed Connective Tissue Disease (MCTD) and Scleroderma Report of Eight Cases. Am J Med Genet. 2008;Part A 146A:3038–53. doi: 10.1002/ajmg.a.32554. [DOI] [PubMed] [Google Scholar]
- 2.Irving MD, Chitty LS, Mansour S, et al. Chondrodysplasia punctata: a clinical diagnostic and radiological review. Clin Dysmorph. 2008;17:229–41. doi: 10.1097/MCD.0b013e3282fdcc70. [DOI] [PubMed] [Google Scholar]
- 3.Morrisom SC. Punctate epiphyses associated with Turner syndrome. Pediatr Radiol. 1999;29:478–80. doi: 10.1007/s002470050622. [DOI] [PubMed] [Google Scholar]
- 4.Leicher-Duber A, Schumacher R, Spranger J. Stippled epiphyses in fetal alcohol syndrome. Pediatr Radiol. 1990;20:369–70. doi: 10.1007/BF02013185. [DOI] [PubMed] [Google Scholar]
- 5.Alessandri, Jean-Luc D, Ramful, et al. Binder fenotype and brachytelephalanic chondrodysplasia punctata secondary to maternal vitamin K deficiency. Clin Dysmorph. 2010:85–87. doi: 10.1097/MCD.0b013e328335c14a. [DOI] [PubMed] [Google Scholar]
- 6.Menger H, Lin AE, Toriello HV, et al. Vitamin K Deficiency Embryopathy: A Phenocopy of the Warfarin Embryopathy Due to a Disorder of Embryonic Vitamin K Metabolism. Am J Med Genet. 1997;72:129–34. [PubMed] [Google Scholar]
- 7.Tim-aroon T, Jaovisidha S, Wattanasirichaigoon D. A new cases of maternal lupus-associated chondrodysplasia punctata with extensive spinal anomalies. Am J Med Genet. 2011:1487–91. doi: 10.1002/ajmg.a.33995. [DOI] [PubMed] [Google Scholar]
- 8.Kozlowski K, Basel D, Beighton P. Chondrodysplasia punctata in siblings and maternal lupus erythematosus. Clin Genet. 2004;66:545–49. doi: 10.1111/j.1399-0004.2004.00364.x. [DOI] [PubMed] [Google Scholar]
- 9.Khanna J, Braverman NE, Valle D, et al. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata: brief clinical report. Am J Med Genet. 2001;99:63–66. doi: 10.1002/1096-8628(20010215)99:1<63::aid-ajmg1117>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Kumar D, Babu TA, Aparna J. Rhizomelic chondrodysplasia punctata (RCDP): A case report. Curr Pediatr Res. 2012;16(2):164–66. [Google Scholar]
- 11.Timothy J, O’Brien MB, BS, et al. Chondrodysplasia punctata (Conradi Disease) Derm Dep. 1990;29:472–76. doi: 10.1111/j.1365-4362.1990.tb04835.x. [DOI] [PubMed] [Google Scholar]
- 12.Sanfilippo A, Bartoletti S. Brachytelephalanic chondrodysplasia punctata: A difficult diagnosis. Radiol Case Rep. 2010;5(1):308. doi: 10.2484/rcr.v5i1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garnier A, Dauger S, Eurin D, et al. Brachytelephalanic chondrodysplasia punctata with severe spinal cord compression: report of four new cases. Eur J Pediatr. 2007;166:327–31. doi: 10.1007/s00431-006-0239-4. [DOI] [PubMed] [Google Scholar]
- 14.Vogel TW, Menezes AH. Natural history and management of cervical spine disease in chondrodysplasia punctata and coumarin embryopathy. Child Nerv Syst. 2012;28:609–19. doi: 10.1007/s00381-012-1694-z. [DOI] [PubMed] [Google Scholar]
- 15.White AL, Modaff P, Holland-Morris F, et al. Natural history of rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2003:332–42. doi: 10.1002/ajmg.a.20009. [DOI] [PubMed] [Google Scholar]
- 16.Violas P, Fraisse B, Chapuis M, et al. Cervical spine stenosis in chondrodysplasia punctata. J Ped Orth B. 2007;16:443–45. doi: 10.1097/BPB.0b013e3282f05675. [DOI] [PubMed] [Google Scholar]
- 17.Eash DD, Weaver DD, Brunetti-Pierri N. Cervical spine stenosis and possible vitamin K embryopathy in an unusual case of chondrodysplasia punctata and an updated classification system. Am J Med Genet. 2003;122A:70–75. doi: 10.1002/ajmg.a.20242. [DOI] [PubMed] [Google Scholar]
- 18.Bams-Mengerinc AM, Majoie CB, Duran M, et al. MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata. Depart Pediatr Neurol. 2006;66(6):798–803. doi: 10.1212/01.wnl.0000205594.34647.d0. [DOI] [PubMed] [Google Scholar]
- 19.Goh S. Neuroimaging features in a neonate with rhizomelic chondrodysplasia punctata. Pediatr Neurol. 2007;37(5):382–384. doi: 10.1016/j.pediatrneurol.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Herman TE, Lee BC, McAlister WH. Brachytelephalangic chondrodysplasia punctata with marked cervical stenosis and cord compression: report of two cases. Pediatr Radiol. 2002;32(6):452–56. doi: 10.1007/s00247-001-0638-7. [DOI] [PubMed] [Google Scholar]
- 21.Goodman P, Dominguez R. Cervicothoracic myelopathy in Conradi-Hunermann disease: MRI diagnosis. Magn Reson Imaging. 1990;8(5):647–50. doi: 10.1016/0730-725x(90)90144-q. [DOI] [PubMed] [Google Scholar]
- 22.Yang BP, Mindea SA, DiParti AJ. Cervical spinal cord compression in chondrodysplasia punctata. Case illustration. J Neurosurg. 2006;104(3Supl):212. doi: 10.3171/ped.2006.104.3.212. [DOI] [PubMed] [Google Scholar]