

Abstract
Background
Analyses of mitochondrial (mt) genome sequences in recent years challenge the current working hypothesis of Nematoda phylogeny proposed from morphology, ecology and nuclear small subunit rRNA gene sequences, and raise the need to sequence additional mt genomes for a broad range of nematode lineages.
Results
We sequenced the complete mt genomes of three Ascaridia species (family Ascaridiidae) that infest chickens, pigeons and parrots, respectively. These three Ascaridia species have an identical arrangement of mt genes to each other but differ substantially from other nematodes. Phylogenetic analyses of the mt genome sequences of the Ascaridia species, together with 62 other nematode species, support the monophylies of seven high-level taxa of the phylum Nematoda: 1) the subclass Dorylaimia; 2) the orders Rhabditida, Trichinellida and Mermithida; 3) the suborder Rhabditina; and 4) the infraorders Spiruromorpha and Oxyuridomorpha. Analyses of mt genome sequences, however, reject the monophylies of the suborders Spirurina and Tylenchina, and the infraorders Rhabditomorpha, Panagrolaimomorpha and Tylenchomorpha. Monophyly of the infraorder Ascaridomorpha varies depending on the methods of phylogenetic analysis. The Ascaridomorpha was more closely related to the infraorders Rhabditomorpha and Diplogasteromorpha (suborder Rhabditina) than they were to the other two infraorders of the Spirurina: Oxyuridorpha and Spiruromorpha. The closer relationship among Ascaridomorpha, Rhabditomorpha and Diplogasteromorpha was also supported by a shared common pattern of mitochondrial gene arrangement.
Conclusions
Analyses of mitochondrial genome sequences and gene arrangement has provided novel insights into the phylogenetic relationships among several major lineages of nematodes. Many lineages of nematodes, however, are underrepresented or not represented in these analyses. Expanding taxon sampling is necessary for future phylogenetic studies of nematodes with mt genome sequences.
Keywords: Mitochondrial genome, Ascaridia, Nematode, Gene arrangement, Phylogeny
Background
Nematodes (also called roundworms) are an extremely diverse group of bilateral animals with an estimate of 1–10 million species although only ~25,000 species have been described [1]. Nematode taxonomy has traditionally been formed from morphology and ecology, e.g. the early systems proposed by Schneider (1866) [2] on somatic musculature and by Cobb (1919) [3] on stoma armature, and the system proposed by Filipjev (1929) [4] on the presence and absence of zooparasitism. Chitwood (1937) [5] proposed initially the bipartite phylogenetic and taxonomic system based on the presence and absence of phasmids; this system was further elaborated later by Maggenti (1963, 1983) [6,7] based on pharyngeal structure and excretory systems. However, due to the lack of homologous characters and informative fossil records, and the extensive convergent evolution, it has been extremely difficult to derive a consistent phylogenetic and taxonomic framework for the phylum Nematoda from morphological and ecological characters [8-12]. In the past two decades, nematode phylogeny and taxonomy have been revised with analyses of the nuclear small subunit (SSU) rRNA gene sequences [12-14]. The current working hypothesis of Nematoda phylogeny incorporated evidence from morphology, ecology and SSU rRNA gene sequence analyses [13,14].
The mitochondrial (mt) genomes of bilateral animals usually contain 37 genes and a control region on a circular chromosome, ~16 kb in size [15-17]. Sequences of individual mt genes and whole mt genomes are widely used to infer phylogenetic relationships among animals at different taxonomic levels [18,19]. In a number of cases, arrangement of mt genes has also been used to resolve long-standing phylogenetic relationships that could not be resolved by other means [20-22].
Recently, mt genome sequences have also been analyzed to understand the phylogenetic relationships among nematodes. Kang et al. [23] inferred the Nematoda phylogeny with the mt genome sequences of 25 species; Park et al. [24] and Sultana et al. [25] expanded further the analysis to include 36 and 41 species respectively. A major difference between mt genome phylogenies and the current working hypothesis of the Nematoda is on the relationship among four infraorders: Ascaridomorpha, Rhabditomorpha, Spiruromorpha and Oxyuridomorpha. In the current working hypothesis, Ascaridomorpha, Spiruromorpha and Oxyuridomorpha are in the suborder Spirurina whereas Rhabditomorpha is in the suborder Rhabditina [13,14]. Furthermore, Ascaridomorpha is most closely related to Spiruromorpha, whereas Oxyuridomorpha is sister to Ascaridomorpha + Spiruromorpha + Rhigonematomorph. In the phylogenies inferred from mt genome sequences, however, Ascaridomorpha is most closely related to Rhabditomorpha; in most analyses, Oxyuridomorpha is sister to Ascaridomorpha + Rhabditomorpha, whereas Spiruromorpha is sister to the group that contains Ascaridomorpha, Rhabditomorpha and Oxyuridomorpha [23,24]. Due to limited taxon sampling, however, the novel phylogenetic relationships inferred from mt genome sequences in these studies need to be interpreted with caution and further test with more taxa from a wide range of lineages is necessary.
In this study, we sequenced the complete mt genomes of three Ascaridia species in the family Ascaridiidae of the infraorder Ascaridomorpha. Ascaridia species are among the most prevalent and pathogenic parasitic nematodes found in domestic and wild birds and have a worldwide distribution [26]. These Ascaridia species share an identical arrangement of mt genes to each other but differ substantially from those of other nematodes. We inferred the phylogenetic relationships with the complete mt genome sequences of the Ascaridia species and 62 other nematode species that have been sequenced to date. Our analyses support the close relationship between Ascaridomorpha and Rhabditomorpha, and provide novel insights into other phylogenetic relationships in the Nematoda.
Results
Mitochondrial genomes of three Ascaridia species
The complete mt genomes of A. galli, A. columbae and Ascaridia sp. (GHL-2012) were 13,977 bp, 13,931 bp and 13,862 bp long, respectively (GenBank accession numbers JX624728, JX624729 and JX624730). Each mt genome contains 12 protein-coding genes, 22 tRNA genes and two rRNA genes on a circular chromosome (Figure 1). As in most other nematodes, atp8 gene is not present in the mt genome of A. galli, A. columbae and Ascaridia sp. (GHL-2012); all mt genes are transcribed from the same direction. TTT is used as the initiation codon in nad6 gene in the mt genome of A. galli. TTT was previously reported to be the initiation codon for cox2, cytb, nad4, nad3, nad2 and cox3 genes in the mt genome of Strongyloides stercoralis (Strongyloididae), but not for any other Ascaridida nematodes. All of the 12 protein-coding genes have complete termination codons, TAA or TAG. The tRNA genes in mt genomes of A. galli, A. columbae and Ascaridia sp. (GHL-2012) range from 51 to 76 bp. The tRNA-Ser(AGN) and tRNA-Ser(UCN) have truncated secondary structures; these two tRNAs lack a DHU stem but possess a TΨC loop. In all other 20 tRNA genes, the TΨC arm and variable loop are replaced with a TV replacement loop. There are two non-coding regions (NCR) in the mt genomes of A. galli, A. columbae and Ascaridia sp. (GHL-2012) (Table 1). The longer NCR (NCRL) is between trnC gene and trnN gene, and is 610 bp (A. galli), 563 bp (A. columbae) and 566 bp [Ascaridia sp. (GHL-2012)] respectively. The shorter NCR (NCRS) is between nad4 and trnM gene, and is 157 bp (A. galli), 91 bp (A. columbae) and 101 bp [Ascaridia sp. (GHL-2012)] respectively. A NCR at this location, i.e. between nad4 and trnM, is also present in the mt genomes of most other nematodes [27,28].
Figure 1.
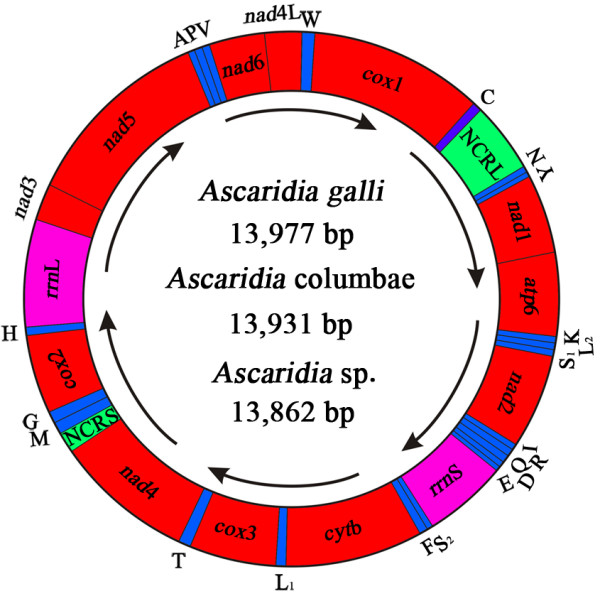
The mitochondrial genomes of three Ascaridia species. All genes are on the same DNA strand and are transcribed clockwise. Protein-coding and rRNA genes are indicated with the standard nomenclature. tRNA genes are indicated with the one-letter code of their corresponding amino acids. There are two tRNA genes for leucine: L1 for codons CUN and L2 for UUR; and two tRNA genes for serine: S1 for codons AGN and S2 for UCN. “NCRL” refers to the large non-coding region. “NCRS” refers to a small non-coding region.
Table 1.
List of annotated mitochondrial genes and regions of Ascaridia galli, Ascaridia columbae and Ascaridia sp
| Gene/region |
Position/length (bp) |
Start/stop codon |
Anticodons | ||||
|---|---|---|---|---|---|---|---|
| A. galli | A. columbae | Ascaridia sp. | A. galli | A.columbae | Ascaridia sp. | ||
|
cox1 |
1-1563 (1563) |
1-1563 (1563) |
1-1581 (1581) |
ATG/TAA |
ATG/TAA |
GTG/TAG |
|
| tRNA-Cys (C) |
1565-1620 (56) |
1565-1620 (56) |
1562-1616 (55) |
|
|
|
GAT |
| Non-coding region (NCL) |
1621-2230 (610) |
1623-2183 (563) |
1617-2182 (566) |
|
|
|
|
| tRNA-Asn (N) |
2231 -2284 (54) |
2184-2241 (58) |
2183-2239 (57) |
|
|
|
GTT |
| tRNA-Tyr (Y) |
2286-2344 (59) |
2246-2303 (58) |
2240-2295 (56) |
|
|
|
GTA |
|
nad1 |
2342-3217 (876) |
2301-3176 (876) |
2298-3167 (870) |
TTG/TAA |
TTG/TAA |
GTT/TAG |
|
|
atp6 |
3217-3813 (597) |
3176-3772 (597) |
3170-3766 (597) |
ATA/TAA |
ATA/TAA |
TTG/TAG |
|
| tRNA-Lys (K) |
3816-3876 (61) |
3790-3853 (64) |
3772-3833 (62) |
|
|
|
TTT |
| tRNA-Leu UUR (L2) |
3876-3931(56) |
3853-3907 (55) |
3831-3886 (56) |
|
|
|
TAA |
| tRNA-Ser AGN (S1) |
3932-3982 (51) |
3908-3962 (55) |
3887-3939 (53) |
|
|
|
GCT |
|
nad2 |
3986-4828 (843) |
3966-4808 (843) |
3943-4779 (837) |
TTG/TAA |
TTG/TAA |
TTG/TAG |
|
| tRNA-Ile (I) |
4832-4891 (60) |
4823-4884 (62) |
4781-4841 (61) |
|
|
|
GAT |
| tRNA-Arg (R) |
4896-4950 (55) |
4888-4944 (57) |
4845-4900 (56) |
|
|
|
ACG |
| tRNA-Gln (Q) |
4951-5005 (55) |
4945-4998 (54) |
4901-4954 (54) |
|
|
|
TTG |
| tRNA-Asp (D) |
5005-5063 (59) |
5011-5070 (60) |
4971-5025 (55) |
|
|
|
GTC |
| tRNA-Glu (E) |
5065-5125 (61) |
5069-5126 (58) |
5025-5083 (59) |
|
|
|
TTC |
|
rrnS |
5123-5823 (701) |
5124-5826 (703) |
5084-5774 (691) |
|
|
|
|
| tRNA-Ser UCN (S2) |
5826-5880 (55) |
5837-5891 (55) |
5781-5835 (55) |
|
|
|
TGA |
| tRNA-Phe (F) |
5883-5939 (57) |
5898-5953 (56) |
5841-5897 (57) |
|
|
|
GAA |
|
cytb |
5964-7067 (1104) |
5975-7075 (1101) |
5919-7019 (1101) |
ATG/TAA |
GTT/TAA |
GTT/TAG |
|
| tRNA-Leu CUN (L1) |
7067-7122 (56) |
7077-7134 (58) |
7020-7075 (56) |
|
|
|
TAG |
|
cox3 |
7144-7914 (771) |
7154-7918 (765) |
7097-7864 (768) |
TTG/TAG |
TTG/TAA |
TTG/TAA |
|
| tRNA-Thr (T) |
7889-7944 (56) |
7899-7956 (58) |
7845-7900 (56) |
|
|
|
TAG |
|
nad4 |
7939-9174 (1236) |
7951-9186 (1236) |
7901-9130 (1230) |
GTG/TAG |
ATA/TAG |
TTG/TAG |
|
| Non-coding region(NCR) |
9175-9331 (157) |
9187-9277 (91) |
9131-9231 (101) |
|
|
|
|
| tRNA-Met (M) |
9345-9407 (63) |
9288-9352 (65) |
9232-9294 (63) |
|
|
|
CAT |
| tRNA-Gly (G) |
9407-9462 (56) |
9355-9410 (56) |
9296-9351 (56) |
|
|
|
TCC |
|
cox2 |
9463-10158 (696) |
9411-10106 (696) |
9364-10050 (687) |
TTG/TAG |
TTG/TAG |
ATG/TAG |
|
| tRNA-His (H) |
10163-10218 (56) |
10113-10167 (55) |
10049-10105 (57) |
|
|
|
GTG |
|
rrnL |
10211-11166 (956) |
10167-11121 (955) |
10103-11055 (953) |
|
|
|
|
|
nad3 |
11167-11502 (336) |
11119-11454 (336) |
11053-11391 (339) |
TTG/TAA |
ATA/TAA |
TTG/TAA |
|
|
nad5 |
11502-13082 (1581) |
11451-13031 (1581) |
11394-12968 (1575) |
ATA/TAA |
ATA/TAG |
ATT/TAG |
|
| tRNA-Ala (A) |
13085-13139 (55) |
13031-13085 (55) |
12968-13022 (55) |
|
|
|
TGC |
| tRNA-Pro (P) |
13140-13197 (58) |
13094-13149 (56) |
13026-13082 (57) |
|
|
|
TGG |
| tRNA-Val (V) |
13197-13252 (56) |
13149-13205 (57) |
13082-13137 (56) |
|
|
|
TAC |
|
nad6 |
13253-13687 (435) |
13206-13640 (435) |
13138-13572 (435) |
TTT/TAA |
TTG/TAA |
TTG/TAG |
|
|
nad4L |
13702-13941 (240) |
13656-13895 (240) |
13588-13827 (240) |
ATT/TAA |
GTT/TAA |
ATT/TAA |
|
| tRNA-Trp (W) | 13919-13976 (58) | 13873-13930 (58) | 13805-13862 (58) | TCA | |||
Gene rearrangement in the mitochondrial genomes of Ascaridia species
A. galli, A. columbae and Ascaridia sp. (GHL-2012) have an identical gene arrangement in their mt genomes. The arrangement of mt genes in these three species, however, is substantially different from those of other nematodes (Figure 2). The mt genomes of the 62 species of nematodes that have been sequenced to date showed 25 types of gene arrangements (GA1–GA25, Figure 2), of which GA3 is the most common type and has been found in 32 species of nematodes. In comparison to the GA3 type, at least three rearrangement events occurred in the Ascaridia species: trnM was translocated, and a block of 11 genes (trnN, trnY, nad1, atp6, trnK, trnL2, trnS1, nad2, trnI, trnR and trnQ) swapped position with another block of 12 genes (trnG, cox2, trnH, rrnL, nad3, nad5, trnA, trnP, trnV, nad6, nad4L and trnW) (Figure 3).
Figure 2.

Mitochondrial gene arrangement in three Ascaridia species (GA1) compared with those in other nematodes (GA2-GA25). The circular mt genomes were linearized at the 5′ end of cox1 gene for illustration purpose. Non-coding regions were not shown.
Figure 3.
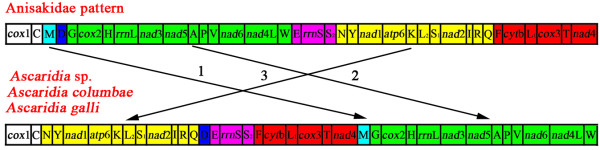
Rearrangement of mitochondrial genes in three Ascaridia species (pattern GA1) relative to the most common pattern of mt gene arrangement observed in nematodes (GA3).
Phylogeny of nematodes inferred from mitochondrial genome sequences
Monophylies of the order Rhabditida and the subclass Dorylaimia
Of the 65 species of nematodes included in the phylogenetic analyses in this study, 54 species were from the order Rhabditida of the class Chromadorea, and 11 species were from the subclass Dorylaimia of the class Enoplea. Both Rhabditida and Dorylaimia were monophyletic in all of the trees inferred with Bayesian, ML and MP methods from concatenated amino acid sequences deduced from the sequences of the 12 mt protein-coding genes. The monophyly of the order Rhabditida was strongly supported with a posterior probability (PP) of 1 in Bayesian analysis (Figure 4), a bootstrapping frequency (BF) of 100% in ML analysis (Figure 5), and a BF of 94% in MP analysis (Figure 6). The monophyly of the subclass Dorylaimia was strongly supported in Bayesian and ML analyses (PP = 1, Figure 4; BF = 97, Figure 5), and was moderately supported in MP analysis (BF = 67%, Figure 6).
Figure 4.

Phylogenetic relationships among 65 species of nematodes inferred from Bayesian analysis of deduced amino acid sequences of 12 mitochondrial proteins.Taenia multiceps and T. hydatigena (GenBank accession numbers FJ495086 and FJ518620) were used as the outgroup. Posterior probability (PP) values were indicated at nodes.
Figure 5.

Phylogenetic relationships among 65 species of nematodes inferred from maximum likelihood (ML) of deduced amino acid sequences of 12 mitochondrial proteins.Taenia multiceps and T. hydatigena (GenBank accession numbers FJ495086 and FJ518620) were used as the outgroup. Bootstrapping frequency (BF) values were indicated at nodes.
Figure 6.

Phylogenetic relationships among 65 species of nematodes inferred from maximum parsimony (MP) of deduced amino acid sequences of 12 mitochondrial proteins.Taenia multiceps and T. hydatigena (GenBank accession numbers FJ495086 and FJ518620) were used as the outgroup. Bootstrapping frequency (BF) values were indicated at nodes.
Phylogenetic relationships within the order Rhabditida
The 54 species of nematodes in the order Rhabditida included in this study were from three suborders: Rhabditina (24 species), Spirurina (26 species), and Tylenchina (4 species). The monophyly of the suborder Rhabditina was strongly supported in Bayesian and ML analyses (PP = 1, Figure 4; BF = 99%, Figure 5), and was moderately supported in MP analysis (BF = 79%, Figure 6). The Spirurina and the Tylenchina, however, were not monophyletic in all of the three phylogenetic analyses in this study.
Of the 24 species from the suborder Rhabditina, 23 species were from the infraorder Rhabditomorpha and one species from the infraorder Diplogasteromorpha. Species of these two infraorders were most closely related in all of the phylogenetic analyses in this study. The Rhabditomorpha, however, was paraphyletic with respect to the Diplogasteromorpha. Three species from the superfamily Rhabitoidea of the Rhabditomorpha were more closely related to Pristionchus pacificus (Diplogasteromorpha) than they were to the other 20 species from the Rhabditomorpha. The close relationship between the species of the superfamily Rhabitoidea and P. pacificus was strongly supported in Bayesian analysis (PP = 1, Figure 4), and was moderately supported in ML and MP analyses (BF = 83%, Figure 5; BF = 82%, Figure 6). In addition to the Rhabitoidea, three other superfamilies of the infraorder Rhabditomorpha were also represented in our analyses: Ancylostomatoidea (4 species), Strongyloidea (10 species), and Trichostrongyloidea (6 species). The Trichostrongyloidea was monophyletic with strong support in all of the three phylogenetic analyses in this study (PP = 1, Figure 4; BF = 88%, Figure 5; BF = 88%, Figure 6). The Rhabitoidea was monophyletic with strong support in Bayesian analysis (PP = 0.99) and weak support in MP analysis (BF = 52%, Figure 6), but was paraphyletic in ML analysis with weak support (BF = 51%, Figure 5). The Ancylostomatoidea and the Strongyloidea were paraphyletic in all of the three phylogenetic analyses in this study; there was strong support for the paraphylies of these two superfamilies in Bayesian and MP analyses (PP > 0.96, Figure 4; BF = 99%, Figure 5).
The 26 species of the suborder Spirurina included in this study were from three infraorders: Ascaridomorpha (14 species), Oxyuridomorpha (2 species) and Spiruromorpha (10 species). The Oxyuridomorpha and the Spiruromorpha were both monophyletic with strong support in all of the three phylogenetic analyses (PP = 1, Figure 4; BF = 100%, Figure 5; BF = 100%, Figure 6). The Ascaridomorpha, however, was monophyletic only in ML analysis with moderate support (BF = 84%, Figure 5), and was paraphyletic in Bayesian analysis with strong support (PP = 0.99, Figure 4) and MP analysis with weak support (BF = 52%, Figure 6). In both Bayesian and MP analyses, the 10 species of the superfamily Ascarodoidea of the infraorder Ascaridomorpha were more closely related to those of the Rhabditina and Steinernema carpocapsae (Tylenchina) than they were to the three Ascaridia species and Cucullanus robustus, which were also from the infraorder Ascaridomorpha. The three Ascaridia species, for which mt genomes were sequenced in this study, form a monoplyletic group with strong support in all of the three phylogenetic analyses (Figures 4, 5, 6). Cucullanus robustus was sister to Ascaridomorpha + Rhabditina + Steinernema carpocapsae in Bayesian analysis with strong support (PP = 1, Figure 4). Cucullanus robustus, however, was sister the Ascaridia species with moderate support in ML analysis (BF = 83%, Figure 5) and weak support in MP analysis (55%, Figure 6).
The four species of the suborder Tylenchina included in this study were from two infraorders: Panagrolaimorpha (2 species), and Tylenchomorpha (2 species). Both of these infraorders were paraphyletic in all of the three analyses (PP > 0.99, Figure 4; BF > 57%, Figure 5; BF > 51%, Figure 6).
Phylogenetic relationships within the subclass Dorylaimia
The 11 species of the subclass Dorylaimia included in this study were from three orders: Mermithida (7 species), Dorylaimida (1 species), and Trichinellida (3 species). The Mermitida and the Trichinellida were both monophyletic with strong support in all of the three analyses (PP = 1, Figure 4; BF = 100%, Figure 5; BF = 100%, Figure 6). Among the three orders, the Mermithida and the Dorylaimida were more closely related than either of them to the Trichocephalida, with strong support in Bayesian analysis (PP = 1, Figure 4) and moderate support in ML analysis (BF = 73%, Figure 5). The Dorylaimida was grouped with the Trichocephalida in MP analysis but the support to this grouping was weak (BF = 40%, Figure 6).
Discussion
Phylogenetic relationships among nematodes have been revised in the past two decades using sequences of nuclear small subunit (SSU) rRNA gene [12-14]. The current working hypothesis of Nematoda phylogeny incorporated evidence from both morphology, ecology and nuclear SSU rRNA gene sequence analyses [14,15]. Recently, Kang et al. [23] tested the hypothesis of the Nematoda phylogeny with the mt genome sequences of 25 species. Kang et al. [23] showed strong support to the monophyly of the class Chromadorea, represented by 16 species from the order Rhabditida; this study, however, could not establish the monophyly of the class Enoplea, represented by nine species from the subclass Dorylaimia. Park et al. [24] and Sultana et al. [25] expanded the taxon sampling and inferred phylogenies with mt genome sequences of 36 and 41 species of nematodes respectively. Park et al. [24] showed strong support for the monophylies of both Chromadorea and Enoplea. Sultana et al. [25], however, could not establish the monophyly of the class Enoplea in most of their analyses. A major difference between the mt genome phylogenies and the current working hypothesis of the Nematode phylogeny is on the monophyly of the suborder Spirurina, which includes Clade III nematodes proposed in Blaxter et al. [12]. In the current working hypothesis, the suborder Spirurina is monophyletic, which includes the infraorders Ascaridomorpha, Spiruromorpha and Oxyuridomorpha, whereas the infraorder Rhabditomorpha is in the suborder Rhabditina [13,14]. Further, Ascaridomorpha is most closely related to Spiruromorpha, whereas Oxyuridomorpha is sister to the group that includes Ascaridomorpha + Spiruromorpha. In the phylogenies inferred from mt genome sequences, however, Ascaridomorpha is most closely related to Rhabditomorpha; in most analyses, Oxyuridomorpha is sister to Ascaridomorpha + Rhabditomorpha, whereas Spiruromorpha is sister to the group that contains Ascaridomorpha, Rhabditomorpha and Oxyuridomorpha. Kang et al. [23], Park et al. [24] and Sultana et al. [25] contained much less taxa compared to previous phylogenetic studies on nematodes with nuclear SSU rRNA gene sequences (e.g. >200 taxa in Meldal et al. [14]). The novel phylogenetic relationship inferred from mt genome sequences, thus, needs to be tested further with more taxa from a wide range of nematode lineages.
In the present study, we sequenced the complete mt genomes of three Ascaridia species from the family Ascaridiidae of the superfamily Heterakoidea, which was not represented in previous studies (Kang et al. [23]; Park et al. [24]; Sultana et al. [25]). Furthermore, we inferred the phylogenetic relationships among 65 nematode species, for which complete mt genomes have been sequenced to date. Our analyses support the division of nematodes into two classes, Chromadorea and Enoplea, represented by the order Rhabditida and the subclass Dorylaimia, respectively, in the present study. Both the Rhabditida and the Dorylaimia were monophyletic with strong support, regardless the methods of phylogenetic analysis used. Within the order Rhabditida, the suborder Rhabditina was monophyletic; the other two suborders, Spirurina and Tylenchina, however, were both paraphyletic in all of the three phylogenetic analyses in this study. Monophyly of the suborder Spirurina was well supported in nuclear SSU rRNA gene sequence analyses [12,14], but was rejected in mt genome sequence analyses in Kang et al. [23], Park et al. [24], Sultana et al. [25] and the present study. Phylogenetic analyses of mt genome sequences in all of these four studies support strongly a close relationship between Ascaridomorpha and Rhabditomorpha to the exclusion of Oxyuridomorpha and Spiruromorpha. Apparently, additional markers for phylogenetic inference are required to resolve the controversy on the Spirurina between mt genome sequence phylogeny and nuclear SSU rRNA phylogeny.
Gene arrangement in mt genomes provides a source of information for phylogenetic inference, independent from mt genome sequences [18]. It is noteworthy that 10 species of Ascaridomorpha and 20 species of Rhabditomorpha included in our phylogenetic analyses share a common pattern of gene arrangement, GA3, in their mt genomes (Figure 2). GA3 is also present in Pristionchus pacificus, the only species from the infraorder Diplogasteromorpha, which is closely related to the species from the superfamily Rhabitoidea of the Rhabditomorpha (Figures 4, 5, 6). Indeed, GA3 is the most common pattern of mt gene arrangement observed in nematodes to date; nevertheless, this pattern is present only in species of the Ascaridomorpha, the Rhabditomorpha and the Diplogasteromorpha. The possibility that GA3 is ancestral to the phylum Nematoda can be rejected with confidence thanks to the discovery of mt gene arrangement pattern GA23 in three Trichinellida species [29,30]. GA23 shares obvious similarity with that of many arthropods and would resemble the ancestral pattern of mt gene arrangement of the phylum Nematoda more than any other patterns found in the nematodes. There is no evidence either that GA3 is ancestral to the class Chromadorea. Given the strong support to the close relationship between Ascaridomorpha and Rhabditomorpha + Diplogasteromorpha indicated by mt genome sequence analyses in the present study, the most parsimonious explanation is that GA3 is a shared derived character (i.e. synapomorphy) of the clade Ascaridomorpha + Rhabditomorpha + Diplogasteromorpha. The other six patterns, GA1, GA2, GA4, GA5, GA6 and GA25, observed in either Ascaridomorpha and Rhabditomorpha or in species closely related to these two infraorders, can be derived from GA3 by only a few rearrangement events (Figure 2), e.g. GA1, observed in the three Ascaridia species, can be converted from GA3 by three rearrangement events (see above).
Monophyly of the suborder Tylenchina could not be established previously in nuclear SSU RNA gene sequence analyses [14]. No species from this suborder were included in two previous analyses of mt genome sequences [23,24]. The present study included four species from the Tylenchina and indicated the paraphyly of this suborder. The positions of the four species of the Tylenchina change depending on the methods of phylogenetic inference used in this study. For instance, Steinernema carpocapsae is sister to the suborder Rhabditina in Bayesian analysis (Figure 4) and MP analysis (Figure 6); it is, however, sister to Ascaridomorpha + Rhabditomorpha + Diplogasteromorpha in ML analysis (Figure 5). Further test with more species from this suborder apparently is needed in order to clarify the phylogenetic position of the suborder Tylenchina. Indeed, a very recent study by Sultana et al. [25] included six species from the Tylenchina and showed the paraphyly of this suborder with strong support. The paraphyly of the infraorder Rhabditomorpha with respect to the Diplogasteromorpha will also need further test with expanded taxon sampling as only one species, Pristionchus pacificus, from the Diplogasteromorpha was included in the present study. No species from the subclass Enoplia, which has two orders and eight suborders [13], was included in our analyses. For the subclass Chromadoria, all of the species included in this study were from the order Rhabditida whereas the other five orders, Plectida, Araeolaimida, Monhysterida, Desmodorida, and Chromadorida, were not represented. Expanding taxon sampling from these lineages of nematodes is clearly the next step for phylogenetic studies of nematodes with mt genome sequences.
Conclusions
We sequenced the complete mt genomes of three Ascaridia species (family Ascaridiidae) that infest chickens, pigeons and parrots, respectively. These species have an identical arrangement of mt genes to each other but differ substantially from other nematodes. Phylogenetic analyses of the mt genome sequences of the Ascaridia species, in combination with 62 other nematode species, support the monophylies of the subclass Dorylaimia; the orders Rhabditida, Trichinellida and Mermithida; the suborder Rhabditina; and the infraorders Spiruromorpha and Oxyuridomorpha. Analyses of mt genome sequences, however, reject the monophylies of the suborders Spirurina and Tylenchina, and the infraorders Rhabditomorpha, Panagrolaimomorpha and Tylenchomorpha. Monophyly of the infraorder Ascaridomorpha varies depending on the methods of phylogenetic analysis. The Ascaridomorpha was more closely related to the infraorders Rhabditomorpha and Diplogasteromorpha (suborder Rhabditina) than they were to the other two infraorders of the Spirurina: Oxyuridorpha and Spiruromorpha. The closer relationship among Ascaridomorpha, Rhabditomorpha and Diplogasteromorpha was also supported by a shared common pattern of mitochondrial gene arrangement. Analyses of mitochondrial genome sequences and gene arrangement in the current study and several previous studies have provided novel insights into the phylogenetic relationships among several major lineages of nematodes. Many lineages of nematodes, however, are underrepresented or not represented in these analyses. Expanding taxon sampling is necessary for future phylogenetic studies of nematodes with mt genome sequences.
Methods
Ethics statement
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Approval No. LVRIAEC2011-009). The animals, from which the nematodes were collected post-mortem, were handled in strict accordance with good animal practices required by the Animal Ethics Procedures and Guidelines of the People's Republic of China.
Sample collection and DNA extraction
Adult Ascaridia galli were obtained from naturally infected chickens in Hunan province, China. Adult A. columbae and an undescribed Ascaridia sp. [hereafter Ascaridia sp. (GHL-2012)] were obtained from naturally infected pigeons and parrots in Guangdong province, China. These nematodes were washed in physiological saline, identified preliminarily to species based on host preference, morphological characters and predilection sites [31-33], fixed in 70% ethanol, and stored at −20°C.
Total genomic DNA was isolated from individual nematodes using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (Wizard® SV Genomic DNA Purification System, Promega). In order to independently verify the identity of the specimen, the region spanning ITS-1, 5.8S rRNA gene and ITS-2 was amplified from each individual nematode by PCR using previously reported primers [34] and sequenced directly.
PCR amplification of the mitochondrial genomes of Ascaridia species
We amplified fragments (400–500 bp) of cox1, nad1 and nad4 genes by PCR using conserved primers (Table 2). The amplicons were sequenced from both directions using BigDye terminator v3.1 on ABI PRISM 3730 platform. We then designed specific primers from these fragments for long PCR amplification (Table 3). We amplified the entire mt genome of individual specimens of A. galli, A. columbae and Ascaridia sp. (GHL-2012) by long PCR in three or four overlapping fragments, respectively. The three overlapping long-PCR fragments for A. galli were between cox1 and nad1 (~2.5 kb), between nad1 and nad4 (~7.5 kb), and between nad4 and cox1 (~5.0 kb). The four overlapping long-PCR fragments for A. columbae and Ascaridia sp. (GHL-2012) were between cox1 and nad1 (~2.5 kb), between nad1 and nad3 (~5 kb), between nad3 and cox2 (~2.5 kb) and between cox2 and cox1 (~5.0 kb). Each long-PCR reaction (25 μl) contains 2.5 μL 2 mM MgCl2, 4.0 μL 0.2 mM each of dNTPs, 2.5 μL 2.5 μl 10× rTaq buffer, 0.25 μL 2.5 μM of each primer, 0.25 μL 1.25 U rTaq polymerase (Takara), and 2 μl of total genomic DNA. The PCR conditions were: 92°C for 2 min (initial denaturation), then 92°C for 10 sec (denaturation), 48–51°C for 30 sec (annealing) and 60°C for 10 min (extension) for 10 cycles, followed by 92°C for 10 sec, 48–51°C for 30 sec, and 60°C for 10 min for 20 cycles and a final extension at 60°C for 10 min. PCR amplicons were column-purified and then sequenced using a primer walking strategy [35].
Table 2.
Primers used to amplify short-PCR fragments from Ascaridia galli, Ascaridia columbae and Ascaridia sp
| Name of primer | Sequence (5′ to 3′) | Reference |
|---|---|---|
|
For A. galli |
|
|
|
p cox 1 |
|
|
| AGCox1F |
GAAGTTTGTATTTGACTGGTAAGAA |
This study |
| AGCox1R |
CAGTGAGACCACCAATAGTAAACAA |
This study |
|
p nad 1 |
|
|
| JB11 |
AGATTCGTAAGGGGCCTAATA |
[36] |
| JB12 |
ACCACTAACTAATTCACTTTC |
[36] |
|
p nad 4 |
|
|
| AGNad4F |
CTTATTATTTAATTTTTTATGCTGCT |
This study |
| AGNad4R |
AAGCGGCTAAAGCCTTAGCATCACT |
This study |
|
For A. columbae and Ascaridia sp. |
|
|
|
p cox 2 |
|
|
| ACCox2F |
TTTAAGTTTGTTGTATTATTATGGTTT |
This study |
| ACCox2R |
AAAATACACCAACCACAGGAAAACT |
This study |
|
p nad 1 |
|
|
| JB11 |
AGATTCGTAAGGGGCCTAATA |
[36] |
| JB12 |
ACCACTAACTAATTCACTTTC |
[36] |
|
p cox 3 |
|
|
| ACCox3F |
TGGTATTTTCTGGACTTTTTTTGAT |
This study |
| ACCox3R | CCAAACTACATCTACAAAATGCCAA | This study |
Table 3.
Primers used to amplify long-PCR fragments from Ascaridia galli, Ascaridia columbae and Ascaridia sp
| Name of primer | Sequence (5′ to 3′) |
|---|---|
|
For A. galli |
|
| AGC1N1F |
ATAGAAGTTTGTATTTGACTGGTAAGAAGGAGGT |
| AGC1N1R |
CACAATACCAGTAACCAAAGTAGCATAAACAG |
| AGN1N4F |
TAAGTTGTTGAAGAAGGAGCAGGAGAGT |
| AGN1N4R |
CAAAAATGGAAAAGAACACAAAGCAGCA |
| AGN4C1F |
TTTTTATGCTGCTTTGTGTTCTTTTCCA |
| AGN4C1R |
CCAAATAAAGTTGCCAGCCACCTAAA |
|
For A. columbae |
|
| ACC1N1F |
AGTTGGACTGTTTATCCGCCTTTGA |
| ACC1N1R |
ATTTCATAAGACACTCTCTGACCTC |
| ACN1C3F |
GCCAGAGGTCAGAGAGTGTCTTATG |
| ACN1C3R |
CTTGCTTCACTATACTCTATTGCCTGT |
| ACC3C2F |
CAGGCAATAGAGTATAGTGAAGCAA |
| ACC3C2R |
ATAGAAGGCACAGCCCAAGAATGAA |
| ACC2C1F |
TAGTATGTGATGTTTGGGAATGCTT |
| ACC2C1R |
CTTTTACACCAGTAGGCACAGCGAT |
|
For Ascaridia sp. |
|
| ACC1N1F |
AGTTGGACTGTTTATCCGCCTTTGA |
| ACC1N1R |
ATTTCATAAGACACTCTCTGACCTC |
| ASN1C3F |
GGTCAGAGGGTTTCTTATGAGATTGCT |
| ASN1C3R |
CAACCGAACAATCTTTATTACTCAACA |
| ASC3C2F |
TGTTGAGTAATAAAGATTGTTCGGTTG |
| ASC3C2R |
ACCAGGAATGTCACCATACTCATAACT |
| ASC2C1F |
TGGAGGTTGATAATCGTTGTGTTTTGC |
| ASC2C1R | CACAACAACAAAGGCTGAAAAACCATC |
Sequence assembly and mitochondrial genome annotation
Sequence reads of A. galli, A. columbae and Ascaridia sp. (GHL-2012) were assembled with ContigExpress program of the Vector NTI software package version 6.0 (Invitrogen, Carlsbad, CA). The mt genome sequences of the Ascaridia species were aligned with those of other nematode species available in GenBank using Clustal X 1.83 [37] to infer gene boundaries. The open-reading frames and codon usage profiles of protein-coding genes were analysed by the Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the invertebrate mitochondrial genetic code, and subsequently compared with those of Ascaris suum[38] and Contracaecum rudolphii B [28]. Sequences of protein-coding genes were translated into amino acid sequences using the invertebrate mitochondrial genetic code in MEGA 5.0 [39]. Translation initiation and termination codons were identified by comparison with those of the nematodes reported previously [27,28]. The secondary structures of 22 tRNA genes were predicted using tRNAscan-SE [40] with manual adjustment [41]. Tandem repeats in the non-coding regions were found using Tandem Repeat Finder program (http://tandem.bu.edu/trf/trf.html) [42].
Phylogenetic analyses
Considering the high degree of intraspecific variation in nucleotide sequences of mt genes of nematodes [43], we used the deduced amino acid sequences of mt proteins for phylogenetic analyses. Amino acid sequences were deduced from the sequences of the 12 mt protein-coding genes common for all of the 65 nematode species and two outgroup species included in this study. The deduced amino acid sequences of each of the 12 mt proteins of these species were aligned individually and then were concatenated into a single alignment. Ambiguous sites and regions in the alignment were excluded using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) [44] with the default parameters. Two Taenia cestodes (T. multiceps and T. hydatigena, GenBank accession number FJ495086 and FJ518620, respectively) were used as the outgroup.
Phylogenetic analyses were conducted using three methods: Bayesian, maximum likelihood (ML), and maximum parsimony (MP). The MtArt model of amino acid evolution [45] was selected as the most suitable model of evolution by ProtTest 2.4 [46] based on the Akaike information criterion (AIC). Bayesian analysis was with MrBayes 3.1.1 [47]. As MtArt model is not implemented in the current version of MrBayes, an alternative model, MtREV, was used in Bayesian analysis. Four independent Markov chains were run for 1,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generations. The first 2,500 trees represented burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (PP). Maximum likelihood (ML) analysis was performed using PhyML 3.0 [48] with the MtArt model. 100 bootstrap replicates were run and bootstrapping frequencies (BF) were calculated. MP analysis was with PAUP* 4.0b10 [49]; indels were treated as missing characters. A total of 1,000 random addition searches using tree bisection-reconnection (TBR) branch swapping were performed for each MP analysis. Bootstrapping frequency (BF) was calculated from 1,000 bootstrap replicates with 10 random additions per replicate in PAUP. Phylograms were drawn using FigTree v.1.31 (http://tree.bio.ed.ac.uk/software/figtree/).
Competing interests
The authors declare no competing interests.
Authors’ contributions
XQZ, RS and GHL designed the research. GHL and JYL performed the research. DHZ contributed reagents/materials/analyses. GHL, RS and HL analyzed the data. RS, GHL and XQZ wrote the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Guo-Hua Liu, Email: liuguohua5202008@163.com.
Renfu Shao, Email: rshao@usc.edu.au.
Jia-Yuan Li, Email: tiantianljy@126.com.
Dong-Hui Zhou, Email: donghui822002@163.com.
Hu Li, Email: tigerleecau@yahoo.cn.
Xing-Quan Zhu, Email: xingquanzhu1@hotmail.com.
Acknowledgements
This work was supported, in part, by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840) and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006) to XQZ. RS acknowledges the funding support from the Australian Research Council (Discovery Project DP120100240) and the Australian Government (Australia-China Science & Research Fund, ACSRF00980).
References
- Dieterich C, Sommer RJ. How to become a parasite - lessons from the genomes of nematodes. Trends Genet. 2009;25:203–209. doi: 10.1016/j.tig.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Schneider A. Monoden. Berlin: Georg Reimer; 1866. p. 357. [Google Scholar]
- Cobb NA. The orders and classes of nemas. Contrib Sci Nematol. 1919;8:213–216. [Google Scholar]
- Filipjev IN. Classification of freeliving Nematoda and their relations to parasitic forms. J Parasitol. 1929;15:281–282. [Google Scholar]
- Chitwood BG. In: Papers on helminthology, 30 year jubileum. Skrjabin KJ, editor. Moscow: All-Union Lenin Academy of Agricultural Sciences; 1937. “A revised classification of the Nematoda”; pp. 67–79. [Google Scholar]
- Maggenti AR. In: The Lower Metazoa: Comparative Biology and Phylogeny. Dougherty EC, Brown ZN, Hanson ED, Hartman WD, editor. Berkeley: University of California Press; 1963. Comparative morphology in nemic phylogeny; pp. 273–282. [Google Scholar]
- Maggenti AR. Nematode Higher Classification as Influenced by Species and Family Concepts. In concepts in nematodesystematics: Academic Press, London; 1983. [Google Scholar]
- Inglis WG. An outline classification on the Phylum Nematoda. Aust J Zool. 1983;31:243–255. doi: 10.1071/ZO9830243. [DOI] [Google Scholar]
- Anderson RC. Nematode Parasites of Vertebrates: Their Development and Transmission. Wallingford: CAB International; 1992. [Google Scholar]
- Lorenzen S. The Phylogenetic Systematics of Free living Nematodes. London: The Ray Society; 1994. [Google Scholar]
- Adamson M. Phylogenetic analysis of the higher classification of the Nematoda. Can J Zool. 1987;65:1478–1482. doi: 10.1139/z87-230. [DOI] [Google Scholar]
- Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, Vierstraete A, Vanfleteren JR, Mackey LY, Dorris M, Frisse LM, Vida JT, Thomas WK. A molecular evolutionary framework for the phylum Nematoda. Nature. 1998;392:71–75. doi: 10.1038/32160. [DOI] [PubMed] [Google Scholar]
- De Ley P, Blaxter M. In: The Biology of Nematodes. Lee DL, editor. London and New York: Taylor & Francis; 2002. Systematic position and phylogeny; pp. 1–30. [Google Scholar]
- Meldal BH, Debenham NJ, De Ley P, De Ley IT, Vanfleteren JR, Vierstraete AR, Bert W, Borgonie G, Moens T, Tyler PA, Austen MC, Blaxter ML, Rogers AD, Lambshead PJ. An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol Phylogenet Evol. 2007;42:622–636. doi: 10.1016/j.ympev.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Wolstenholme DR. Animal mitochondrial DNA, structure and evolution. Int Rev Cytol. 1992;141:173–216. doi: 10.1016/s0074-7696(08)62066-5. [DOI] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrov DV. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr Comp Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/S0959-437X(98)80035-X. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. 2007;134:153–167. doi: 10.1017/S0031182006001429. [DOI] [PubMed] [Google Scholar]
- Boore JL, Lavrov DV, Brown WM. Gene translocation links insects and crustaceans. Nature. 1998;392:667–668. doi: 10.1038/33577. [DOI] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 2000;17:87–106. doi: 10.1093/oxfordjournals.molbev.a026241. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM, Boore JL. Phylogenetic position of the Pentastomida and (pan) crustacean relationships. Proc Biol Sci. 2004;271:537–544. doi: 10.1098/rspb.2003.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Sultana T, Eom KS, Park YC, Soonthornpong N, Nadler SA, Park JK. The mitochondrial genome sequence of Enterobius vermicularis (Nematoda: Oxyurida)–an idiosyncratic gene order and phylogenetic information for chromadorean nematodes. Gene. 2009;429:87–97. doi: 10.1016/j.gene.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Park JK, Sultana T, Lee SH, Kang S, Kim HK, Min GS, Eom KS, Nadler SA. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics. 2011;12:392. doi: 10.1186/1471-2164-12-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana T, Kim J, Lee SH, Han H, Kim S, Min GS, Nadler SA, Park JK. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol Biol. 2013;13:12. doi: 10.1186/1471-2148-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelqader A, Gauly M, Wollny CB, Abo-Shehada MN. Prevalence and burden of gastrointestinal helminthes among local chickens, in northern Jordan. Prevent Vet Med. 2008;85:17–22. doi: 10.1016/j.prevetmed.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, Zhao GH, Huang SY, Zhu XQ. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012;492:110–116. doi: 10.1016/j.gene.2011.10.043. [DOI] [PubMed] [Google Scholar]
- Lin RQ, Liu GH, Zhang Y, D’Amelio S, Zhou DH, Yuan ZG, Zou FC, Song HQ, Zhu XQ. Contracaecum rudolphii B: gene content, arrangement and composition of its complete mitochondrial genome compared with Anisakis simplex s.l. Exp Parasitol. 2012;130:135–140. doi: 10.1016/j.exppara.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012;6:e1539. doi: 10.1371/journal.pntd.0001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM. Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–637. doi: 10.1093/genetics/157.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan HH, Abouznada NY. Morphology and life history of Ascaridia galli in the Domestic Fowl that are raised in Jeddah. J K A U Science. 1992;4:87–99. [Google Scholar]
- Wehr EE, Hwang JC. The life cycle and morphology of Ascaridia columbae (Gmelin, 1790) Travassos, 1913 (Nematoda:Ascarididae) in the Domestic Pigeon (Columba livia domestica) J Parasitol. 1964;50:131–137. doi: 10.2307/3276047. [DOI] [Google Scholar]
- Kajerova V, Barus V, Literak I. New records of Ascaridia platycerium (Nematodes) in parrots (Psittaciformes) Vet Med-Czech. 2004;49:237–241. [Google Scholar]
- Zhu X, Chilton NB, Jacobs DE, Boes J, Gasser RB. Characterisation of Ascaris from human and pig hosts by nuclear ribosomal DNA sequences. Int J Parasitol. 1999;29:469–478. doi: 10.1016/S0020-7519(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nature Protoc. 2007;2:2339–2344. doi: 10.1038/nprot.2007.358. [DOI] [PubMed] [Google Scholar]
- Bowles J, McManus DP. NADH dehydrogenase 1 gene sequences compared for species and strains of the genus Echinococcus. Int J Parasitol. 1993;23:969–972. doi: 10.1016/0020-7519(93)90065-7. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;130:471–498. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea) Int J Parasitol. 2002;32:145–158. doi: 10.1016/S0020-7519(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes progress and perspectives. Trends Parasitol. 2006;22:78–84. doi: 10.1016/j.pt.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Abascal F, Posada D, Zardoya R. MtArt: a new model of amino acid replacement for Arthropoda. Mol Biol Evol. 2007;24:1–5. doi: 10.1093/molbev/msl136. [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Swofford DL. Paup*: Phylogenetic analysis using parsimony, version 4.0b10. Sunderland, MA: Sinauer Associates; 2002. [Google Scholar]