

Abstract
The combination rilpivirine (RPV)/emtricitabine (FTC)/tenofovir (TDF) is a once-daily, single-tablet regimen (STR) containing one nonnucleoside reverse-transcriptase inhibitor associated with two nucleos(t)ide reverse transcriptase inhibitors. It is approved by regulatory agencies (eg, US Food and Drug Association, European Medicines Agency) in all countries in which it is manufactured, except Switzerland, as first-line highly active antiretroviral therapy (HAART) for the treatment of naïve patients with HIV infection and a viral load HIV-RNA level of ≤100,000 copies/mL. Two large trials (ECHO and THRIVE) comparing RPV with efavirenz, along with different background regimens, led to approval of the drug, while a more recent trial (STaR) explored the use of STR. RPV showed noninferiority to efavirenz in all the studies, including superiority as an STR in patients with HIV-RNA ≤100,000 copies/mL in the STaR study. A positive CD4 cell response was observed in all the studies, both in the RPV and efavirenz groups. The incidence of virologic failures was higher for RPV, but was mostly referred to patients with HIV-RNA >100,000 copies/mL. There were fewer adverse events (AEs) with the RPV-based regimens versus efavirenz-based regimens, with a lower discontinuation rate because of AEs, especially psychiatric–neurological AEs, and a significantly lower rate of blood-lipid abnormalities. In the SPIRIT study (a switch study), significantly greater improvements from baseline in serum total cholesterol, low-density lipoprotein cholesterol, and trygliceride were demonstrated in patients switching to RPV/FTC/TDF from a ritonavir-boosted protease inhibitor (PI/r)-based regimen, than in those who continued treatment with a PI/r regimen. RPV’s better tolerability, associated with its once-daily STR formulation, is key to improving patients’ adherence and quality of life, which are among the most important factors affecting the therapeutic efficacy of an antiretroviral regimen. In summary, RPV/FTC/TDF STR is a valuable treatment option for the majority of antiretroviral-naïve HIV-infected patients. Furthermore, the use of this STR in the therapeutic switch, like in the SPIRIT study, can result in another valuable option by which to reduce AEs and improve patients’ quality of life.
Keywords: rilpivirine or RPV, HIV, treatment-naïve, once-daily, single-tablet regimen
Introduction
Highly active antiretroviral therapy (HAART) plays a key role in mitigating the HIV/AIDS epidemic by reducing morbidity and mortality. Furthermore, extensive use of treatment may reduce the incidence of HIV infection, as the risk of transmission of the virus is lower in those receiving antiretrovirals than in naïve patients.1 However, HAART regimens are often complex and require careful consideration on drug–drug interactions and toxicities, all of which potentially lead to poor patient compliance/adherence and, as a consequence, to the emergence of resistance that may limit the long-term efficacy.2
Current combination treatment options for naïve HIV patients
According to international guidelines,1 all adults with HIV infection should be offered HAART regardless of CD4 cell count. Although there is no CD4 cell-count threshold at which starting therapy is contraindicated, the strength of the recommendation and the quality of the evidence supporting initiation of therapy increase as the CD4 cell count decreases and when specific clinical conditions, such as AIDS-defining illnesses, coinfections with hepatitis B virus/hepatitis C virus, or pregnancy are present.3,4 The options for initial therapy in naïve adults with confirmed drug-susceptible virus continue to expand, with new drugs and coformulations available (Table 1).1 Because therapy can be expected to last indefinitely, regimen choice must consider patient convenience, potential toxicities, and tolerability, all of which may affect adherence. The aim of therapy is the constant, lifelong suppression of HIV replication, with the aims of preventing emergence of resistance, facilitating optimal immune recovery, and improving health. Interactions are a growing challenge as persons with HIV age and require additional medications for comorbid conditions.5–7
Table 1.
Recommended and alternative initial antiretroviral regimens, including strength of recommendations and quality of evidence
| Recommended regimens | Alternative regimens | Comments | |
|---|---|---|---|
| NNRTI plus | Efavirenz/tenofovir/emtricitabine (AI). | Nevirapine plus tenofovir/emtricitabine or abacavir/lamivudine (BI). | Severe hepatotoxicity and rash with nevirapine are more common in initial therapy when CD4 cell count is >250/μL in women and >400/μL in men. |
| NRTIs | Efavirenz plus abacavir/lamivudine (AI) in HLA-B 5701-negative patients with baseline plasma HIV-RNA <100,000 copies/mL. | ||
| PI/r plus | Darunavir/r plus tenofovir/emtricitabine (AI). | Darunavir/r plus abacavir/lamivudine (BIII). | Other alternative PIs include fosamprenavir/r and saquinavir/r, but indications to use these options for initial treatment are rare. |
| NRTIs | Atazanavir/r plus tenofovir/emtricitabine (AI). | ||
| Atazanavir/r plus abacavir/lamivudine (AI) in patients with plasma HIV-RNA <100,000 copies/mL. | Lopinavir/r plus tenofovir/emtricitabine (BI) (or abacavir/lamivudine) (BI). | ||
| InSTI plus NRTIs | Raltegravir plus tenofovir/emtricitabine (AI). | Raltegravir plus abacavir/lamivudine (BII). | Raltegravir is given twice daily. |
Notes: Fixed-dose combinations are recommended when available and appropriate. Current fixed-dose combinations available are: efavirenz/tenofovir/emtricitabine; tenofovir/emtricitabine; abacavir/lamivudine; lopinavir/ritonavir.
Abbreviations: InSTI, integrase strand transfer inhibitor; NNRTI, nonnucleoside reverse-transcriptase inhibitor; NRTI, nucleos(t)ide reverse-transcriptase inhibitor; PI, protease inhibitor; /r, ritonavir-boosted ; AI, strong support for the recommendation - evidence from one or more randomized controlled clinical trials; BI, moderate support for the recommendation - evidence from one or more randomized controlled clinical trials; BII, moderate support for the recommendation - evidence from nonrandomized clinical trials or cohort or case control studies; BIII, moderate support for the recommendation - recommendation based on the panel’s analysis of the accumulated available evidence; RNA, ribonucleic acid.
Initial therapy continues to be based on a combination of two nucleos(t)ide reverse-transcriptase inhibitors (NRTIs) and a potent third agent, generally a non-NRTI (NNRTI), a ritonavir-boosted protease inhibitor (PI/r), or an integrase strand transfer inhibitor (InSTI). Coformulations of drugs or complete regimens in fixed-dose combinations (FDCs) are preferred to enhance convenience and medication adherence.4,8,9
Overview of pharmacology
Rilpivirine (RPV) is a diarylpyrimidine NNRTI that inhibits HIV reverse transcriptase by binding to it noncompetitively.10 NNRTIs bind reverse transcriptase near the active site of the enzyme, within a small hydrophobic pocket, inducing changes in enzyme conformation depending on the specific drug’s chemical structure, its size, and its mode of binding.11 These conformational changes inhibit the catalysis of viral RNA to DNA by the transcriptase, thereby decreasing replication of HIV The second-generation NNRTIs etravirine and RPV are flexible and adapt to changes in the NNRTI-binding pocket associated with resistance mutations, a characteristic that may help to explain their high barrier to resistance.12,13 RPV does not inhibit human cellular DNA polymerases α, β or γ,10 which may be advantageous, as inhibition of these enzymes is implicated in the major mithocondrial toxicity observed with NRTI use.14
In a multiple-dose-escalating study, orally administered RPV was rapidly absorbed. After 7 days of RPV 25 mg once daily, the mean maximum plasma RPV concentration (Cmax) was 263 ng/mL, the mean plasma area under the concentration–time curve from time 0 to 24 hours (AUC0–24) was 3659 ng h/mL, and the median time to Cmax was 4 hours,15 which is consistent with a general estimate of 4–5 hours across various studies.10,16 At steady state, the average plasma concentration of RPV was 152 ng/mL. Across dosage groups, RPV was detectable in plasma in most patients for up to 168 hours after administration.15 RPV should always be taken with a meal to ensure adequate exposure.10,16 The reduction in RPV bioavailability when taken after fasting is ≈40% when compared with ingestion after a normal caloric or high-fat, high-caloric meal. There is no difference in RPV exposure when taken with a standard versus a high-fat breakfast. However, compared with administration with a meal, bioavailability was reduced by 50% when taken with only a protein-rich nutritional drink.10,16 In vitro, RPV is ≈99.7% bound to plasma proteins, chiefly to albumin, whereas its distribution outside plasma is unknown.10,16
RPV undergoes oxidative metabolism in the liver, chiefly mediated by the hepatic CYP3A system.10,16 It is excreted mostly in the feces, with minimal excretion via the kidneys.17 RPV has a terminal elimination half-life of ≈50 hours.10,16
As RPV is metabolized by CYP3A enzymes, drug interactions are possible when it is coadministered with inducers or inhibitors of these enzymes.10
The coadministration with the following drugs is contraindicated: anticonvulsants (strong CYP3A inducers), rifampicin, rifabutin, antifungals, proton pump inhibitors, systemic dexamethasone (except as a single dose), and St John’s wort.16
RPV has been shown to cause a prolongation of the QTc interval at supratherapeutic doses, so caution is required when it is used with drugs known to be associated with Torsades de pointes.10,16
Coadministration of drugs raising gastric pH should be avoided as plasma concentrations can be decreased by higher gastric pH.10 No adjustment of the dosage is required when paracetamol, atorvastatin, raltegravir, ribavirin, maraviroc, methadone, and oral contraceptives are coadministered.10
Tenofovir (TDF) and emtricitabine (FTC), the two NRTIs coformulated with RPV have long been available as a once-daily FDC. FTC and TDF are phosphorylated by cellular enzymes to the active moieties FTC triphosphate and TDF diphosphate, respectively, which competitively inhibit HIV reverse transcriptase, leading to DNA chain termination.18,19 Both of these active moieties are weak inhibitors of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ. Both drugs may be taken without food restrictions. TDF is well tolerated but has been associated with kidney injury, which appears to increase in incidence with long-term administration and concurrent PI/r use.20–22
Therapeutic efficacy
The efficacy of oral RPV as a component of combination antiretroviral therapy in antiretroviral-naïve patients with HIV infection was evaluated in a Phase II, randomized multinational, dose-ranging study23 and in the Phase III ECHO (Efficacy Comparison in Treatment-naïve HIV-infected Subjects of TMC278 and Efavirenz) (NCT00540449)24 and THRIVE (TMC278 against HIV in a once-daily regimen versus efavirenz) (NCT00543725)25 trials.
In the Phase II, dose-ranging study, patients received once-daily RPV 25 mg (n = 93), 75 mg (n = 95), or 150 mg (n = 91) or once-daily efavirenz 600 mg (n = 89). Patients and investigators were blinded to the RPV dosage, whereas the open design was used for efavirenz. Background regimens for this study were zidovudine/lamivudine or TDF/FTC.23 In the Phase II study, RPV reduced plasma HIV-RNA levels over the short term more than placebo.15 Patients with an HIV-RNA level of >5000 copies/mL and a CD4 cell count of 75–500 cells/mcL (n = 47) were randomized 3:1 to receive an oral solution of RPV (25, 75, or 150 mg) or placebo once daily as monotherapy for 7 days before commencement of standard antiretroviral therapy. There was a significantly greater reduction in HIV-RNA levels with all RPV dosages (median −1.199 log10 copies/mL in RPV groups combined) compared to placebo (+0.002 log10 copies/mL) (P < 0.01).15
ECHO and THRIVE were independent, randomized double-blind double-dummy, multinational trials of almost identical design.24,25 Included patients underwent a 6-week screening period after which they were randomized to 96 weeks of treatment with RPV 25 mg once daily or efavirenz 600 mg once daily, plus a fixed-dose background regimen.24,25 In the ECHO trial, the backbone was an FDC of TDF/FTC, while backbones in the THRIVE study were, at the physician’s discretion, TDF/FTC, zidovudine/lamivudine, and abacavir/lamivudine. Randomization was stratified by HIV-RNA level at screening (≤ 100,000, 100,000–500,000, or ≥500,000 copies/mL)24,25 and in the THRIVE trial, by backbone regimen as well.25 The primary analysis in both trials was a logistic regression analysis (adjusted for stratification factors) that predicted response rates at 48 weeks based on an intent-to-treat time-to-loss-of-virological-response (ITT-TLOVR) algorithm.24,25 Noninferiority of RPV to efavirenz was established if the lower limit of the two-sided 95% confidence interval (CI) for the difference in response rates was within a 12% margin. Patients who never achieved an HIV-RNA level of <50 copies/mL, had a level of ≥50 copies/mL at two consecutive assessments, or discontinued treatment prematurely were classified as nonresponders.24,25 At 48 weeks, ≥83% of RPV and efavirenz recipients in both trials achieved the goal of an HIV-RNA level of <50 copies/mL (Figure 1A).24,25 In the primary analyses, the predicted response rates in the RPV group met the noninferiority criterion versus efavirenz. There were no significant between-group difference in response rates in either trial.
Figure 1.
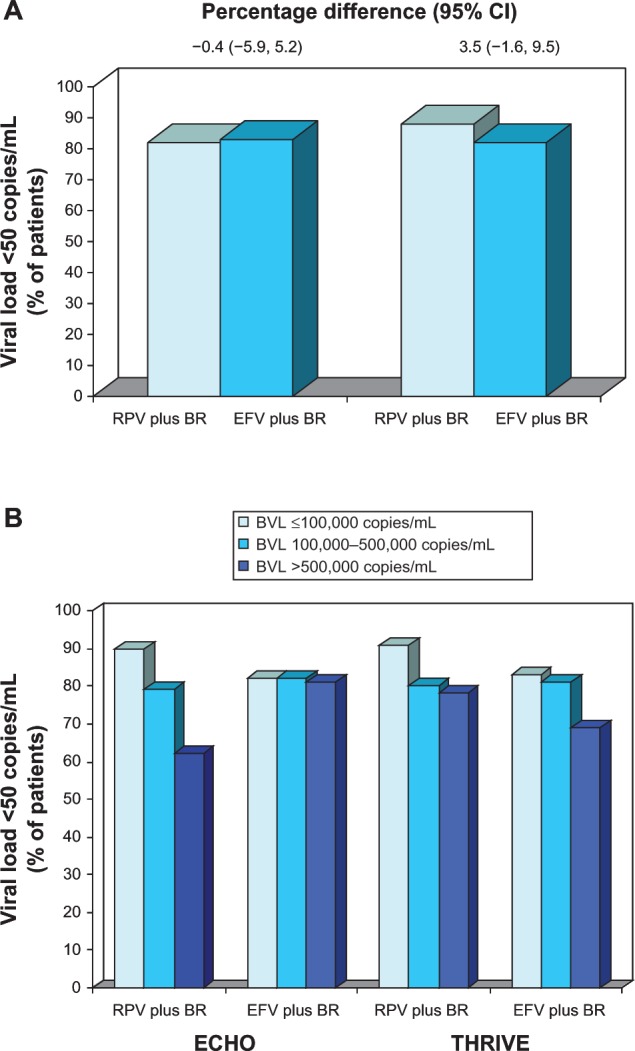
Efficacy of oral rilpivirine as a component of combination therapy in antiretroviral-naïve patients with HIV infection.
Notes: Response rates (A) in the full modified intent-to-treat populations (primary end point) and (B) in descriptive subgroup analyses according to baseline HIV-RNA level. Results are from the randomized, double-blind, double-dummy, multinational ECHO (n = 690)24 and THRIVE (n = 678)25 trials, which compared rilpivirine 25 mg once daily with efavirenz 600 mg once daily, plus background regimens. For the primary analysis, the noninferiority margin (rilpivirine vs efavirenz) for the lower bound of the 95% CI was −12%. In the ECHO and THRIVE trials, 344 and 354, 265 and 254, and 81 and 70 patients, respectively, had baseline HIV-RNA levels of ≤100,000, 100,000–500,000, and >500,000 copies/mL.
Abbreviations: BR, background regimen; BVL, baseline viral load; CI, confidence interval; EFV, efavirenz; RPV, rilpivirine; RNA, ribonucleic acid.
In both trials, there was a virological response to treatment with RPV or efavirenz within 2–4 weeks from baseline and this increased steadily during the first 6 months of treatment, with no apparent between-treatment difference in the timing of response.24,25 Response rates were >80% by 24 weeks in all treatment groups.24,25 Similar results were obtained when the analyses were performed on the per-protocol populations.24,25
In descriptive subgroup analyses, response rates in RPV recipients were higher in those with a lower baseline HIV-RNA level (≤100,000 copies/mL) than in those with higher viral loads (Figure 1B).24,25
The proportion of virological failures (VFs) in Phase III trials was higher among patients treated with RPV (10%) compared with those who received efavirenz (6%).24–26
A subanalysis based on baseline viral load demonstrated that the proportion of VFs was the same for RPV and efavirenz (5%) recipients with baseline HIV-RNA ≤100,000 copies/mL, but was higher in RPV (17%) compared to efavirenz (7%) among those patients with a baseline HIV-RNA >100,000 copies/mL.
Among patients with VF who had evaluable post-baseline resistance data at week 48, 63% (39 of 62) and 54% (15 of 28) in the RPV and efavirenz groups, respectively, had treatment-emergent NNRTI resistance-associated mutations (RAMs). NRTI RAMs occurred at a significantly (P = 0.003; post hoc analysis) higher rate in RPV 68% (42 of 62) than efavirenz 32% (9 of 28) recipients.
In RPV recipients with ≤100,000 HIV-RNA copies/mL at baseline, eight of 16 (50%) patients had any NNRTI and/or NRTI RAM, compared with 36 of 46 (78%) patients with >100,000 HIV-RNA copies/mL at baseline.26 Specific NNRTI and NRTI RAMs occurring in ≥2 patients with VF at week 48 are shown in Figure 2.
Figure 2.
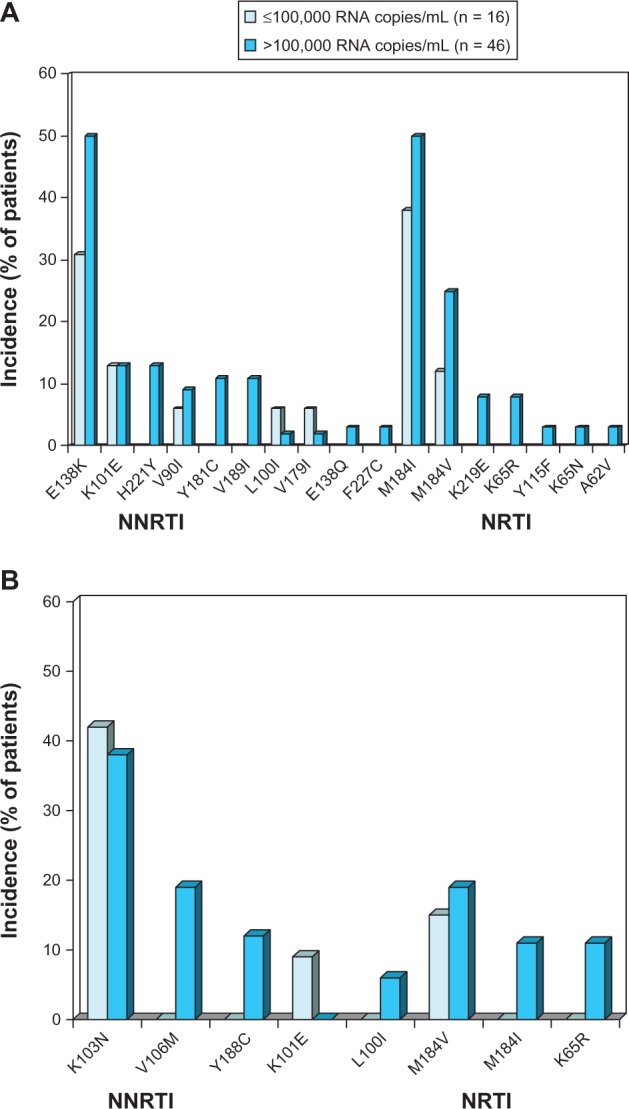
Resistance-associated mutations occurring in ≥2 patients at time of virological failure with (A) once-daily oral rilpivirine 25 mg plus a background regimen or (B) efavirenz 600 mg plus a background regimen.
Notes: Data are from a pooled analysis26 of the Phase III ECHO24 and THRIVE25 trials. Patients with evaluable post-baseline resistance data at 48 weeks were included and the data are presented according to baseline HIV viral load.
Abbreviations: NNRTI, nonnucleoside reverse-transcriptase inhibitor; NRTI, nucleoside/nucleotide-reverse transcriptase inhibitor; RNA, ribonucleic acid.
Although the resistance profile for RPV has not been completely defined, the presence of a single NNRTI RAM seems to only marginally affect susceptibility to the drug. By far, E138 K was the most frequently selected (45%) mutation in antiretroviral-naïve patients that failed on RPV therapy in the ECHO24 and THRIVE25 studies. Interestingly, this substitution was generally associated with M184I mutation (34%), which confers lamivudine and FTC resistance.26 The combination E138K/M184I confers a 6.7-fold reduced phenotypic susceptibility to RPV compared with a 2.8-fold reduction for E138K alone. Mutation K103N, which is associated with clinical resistance to efavirenz and nevirapine, does not reduce susceptibility to RPV.
Drug-resistance interpretation systems (ie, Stanford [http://hivdb.stanford.edu/], Agence Nationale de Recherhes sur le Sida [www.hivfrenchresistance.org] [French National Agency for AIDS Research]) have recently incorporated predictions of virological response to RPV. The Drug Resistance Platform of the Spanish AIDS Research Network has weighted NNRTI RAMs27 so that, for considering resistance to RPV, at least two mutations must be present. Mutations with the greatest impact on RPV susceptibility are at four codons K101E/P/T, E138A/G/K/R, Y181C/I/V, and M230 L. Changes in the other nine positions display a lower impact (V90I, L100I, V106A/I, V108I, V179F/I/L, Y188I, G190E, H221Y, and F227C/L). However, in the presence of M184I, the selection of either E138K or K101E is sufficient to induce high-level RPV resistance.
Overall, the change in sensitivity to RPV ranges from 3.7- to 554-fold in the presence of a combination of two or three RAMs.10
VFs with RPV compared to efavirenz (EFV) were more likely to show cross-resistance with all NNRTIs.10 After VF with RPV, 89% of patients were resistant to etravirine and efavirenz and 63% were resistant to nevirapine, whereas none of the efavirenz recipients with VF were cross-resistant to etravirine.10
Response rates were higher in patients who were more adherent to RPV- or efavirenz-based regimens (86%–90% in those with >95% adherence versus 62%–73% in those with ≤95% adherence).24,25
RPV was associated with a positive immunological response. In both ECHO and THRIVE trials, the CD4 cell count steadily increased over time. At 48 weeks, the CD4 mean increment in RPV recipients was 190 cells/mcL (ECHO) and 189 cells/mcL (THRIVE), while in efavirenz-treated patients, the same values were 180 cells/mcL and 171 cells/mcL, respectively.
In generalized additive modeling, adherence to treatment, systemic exposure to the NNRTI, and lower baseline viral load were the most important predictors of virological response (persistence of an HIV-RNA level of <50 copies/mL) to both RPV and efavirenz at 48 weeks.28 In descending order of importance, the prognostic variables in the final RPV model were: adherence; plasma RPV Ctrough; baseline viral load; baseline fold change in viral load with RPV; baseline CD4 cell count; undectable plasma level of RPV at any time point (a proxy of poor adherence); and trial (ECHO or THRIVE). For efavirenz, in descending order of importance, the variables in the final model were: undetectable plasma level of efavirenz at any time point; adherence; baseline viral load; plasma efavirenz Ctrough; baseline fold change in viral load with efavirenz; and background regimen.28
The results of a post hoc pooled analysis of the ECHO and THRIVE trials restricted to the subset of patients receiving RPV or efavirenz plus FTC/TDF showed that RPV plus FTC/TDF was noninferior to efavirenz plus FTC/TDF at both 48 and 96 weeks.29 ITT-TLOVR response rates were high, both at 48 weeks (83.5% vs 82.4%) and at 96 weeks (77% in both groups). Response rates in RPV/FTC/TDF recipients were higher in those with a lower baseline HIV-RNA level (≤100,000 copies/mL) than in those with higher viral loads at 48 and 96 weeks (Figure 3).30,31
Figure 3.
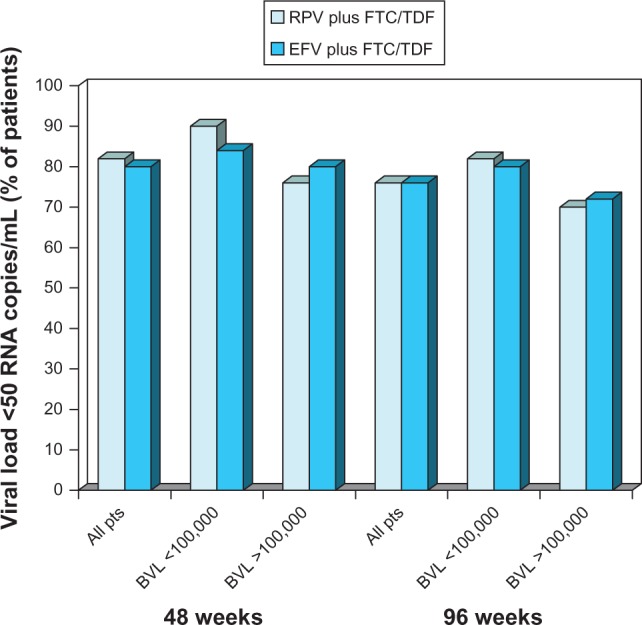
Virological response in antiretroviral-naïve patients with HIV infection receiving oral rilpivirine plus emtricitabine/tenofovir.
Notes: Proportion of patients with confirmed HIV-RNA levels <50 copies/mL at 48 weeks (intent-to-treat time-to-loss-of-virological-response analysis) and 96 weeks (intent-to-treat snapshot analysis) in all patients stratified according to baseline HIV-RNA levels. Results are shown for patients receiving rilpivirine plus emtricitabine/tenofovir (n = 550) or efavirenz plus emtricitabine/tenofovir (n = 546) in pooled subset analyses29 of the ECHO24 and THRIVE25 trials.
Abbreviations: BVL, baseline viral load; EFV, efavirenz; FTC/TDF, emtricitabine/tenofovir; pts, patients; RPV, rilpivirine; RNA, ribonucleic acid.
Rationale for combination of the individual components in RPV/FTC/TDF
The single-tablet formulation of RPV, FTC, and TDF (Complera™/Eviplera™; Gilead, Foster City, CA, USA) was developed on the grounds that, compared with multiple-tablet regimens, single-tablet regiment (STRs) are preferred by patients and are likely to be associated with increased adherence. RPV/FTC/TDF comprises FTC 200 mg, TDF 300 mg, and RPV 25 mg, and has met bioequivalence criteria, being the geometric least squares mean plasma Cmax, AUC from time 0 to infinity, and AUC time 0 to the last dose for each of the constituent drugs of the combined tablet, within an 80–125 percentage margin of the 90% CI compared to the same parameters calculated for the separate formulations of each drug.32
The STR RPV/FTC/TDF has been studied in clinical trials, both in naïve patients starting their first HAART and as a simplification regimen for chronically treated subjects.
The Single-Tablet Regimen study (STaR) is the first study comparing the efficacy, safety, and tolerability of the two STRs so far marketed in Europe: RPV/FTC/TDF and EFV/FTC/TDF. It is a multicenter, international, randomized, open-label, Phase IIIb, 96-week study, the primary endpoint of which is virological efficacy as defined by the proportion of patients with HIV-RNA <50 copies/mL at week 48. The study aims to demonstrate noninferiority of RPV/FTC/TDF compared to EFV/FTC/TDF with a noninferiority margin of 12%. The snapshot analysis of 48-week results demonstrated noninferiority for the overall population. Patients treated with STR RPV/FTC/TDF presented virologic suppression in 86% of cases and virologic failure in 8% and were categorized as missing data in 6%, the same figures for patients treated with EFV/FTC/TDF were 86%, 6%, and 13%, respectively. A subanalysis performed according to the baseline HIV-RNA value demonstrated that the RPV/FTC/TDF STR was superior to EFV/FTC/TDF in subjects with a baseline HIV-RNA ≤100,000 copies/mL (virologic response 89% vs 82%) and noninferior in subjects with higher HIV-RNA (100,000–500,000 copies/mL: 83% vs 82%; ≥ 500,000 copies/mL: 72% vs 80%). The proportions of VFs were very low in patients with HIV-RNA ≤100,000 copies/mL (5% vs 3%) and HIVR-NA 100,000–500,000 copies/mL (10% vs 9%), but rose in those subjects with higher HIV-RNA levels (HIV-RNA ≥ 500,000 copies/mL: 25% vs 16%).33
The STaR study, in contrast to the previous ECHO and THRIVE studies, seems to indicate that the risk of VF with the combination of RPV/FTC/TDF rises when the baseline HIV-RNA is very high (>500,000) and, therefore, the currently approved use (patients with HIV-RNA ≤100,000) can be considered safe. On the other hand, the same study indicates that, for lower HIV-RNA values, the STR combination of RPV/FTC/TDF may be superior to EFV/FTC/TDF. One of the driving forces of this result is certainly the elevated tolerability of the new drug combination, but the use of the STR itself may have played a relevant role by enhancing patients’ adherence. Although RPV has so far been approved only as first-line treatment for antiretroviral therapy-naïve patients, the drug is currently being considered in other clinical scenarios, such as switch and simplification strategies.
The SPIRIT (Switching boosted PI to Rilpivirine in combination with Truvada as an STR) study34 is a multicenter, international, randomized, open-label, Phase IIIb, 48-week study aiming to demonstrate the noninferiority of a RPV/FTC/TDF switch compared to a continuing PI/r + 2 NRTIs therapy. According to a 24-week snapshot analysis, switching to RPV/FTC/TDF was noninferior to remaining on PI/r + 2 NRTI (93.7% vs 89.9%); moreover, at the same time point, lower rates of VF were observed in subjects switching to RPV/FTC/TDF (0.9%) compared to those remaining on PI/r + 2 NRTI (5.0%).34
The possibility of switching to RPV/FTC/TDF (STR) from EFV/FTC/TDF (STR) has been explored by the GS-264-111 study.35 The virological results of the trial showed that the transient efavirenz inductive effects on RPV metabolism do not appear to be clinically relevant in suppressed patients. Overall, virological suppression was maintained in 100% of cases, analyzed after 24 weeks. RPV mean Ctrough was within historic range in 2 weeks.35
Altogether, the switch studies indicate that the RPV/FTC/TDF STR is a favorable choice also in a planned simplification strategy. Its use in this indication is not yet approved, but, on the basis of the results of these studies, an application to regulatory agencies (eg, US Food and Drug Association, European Medicines Agency) has been filed and the approval of the new indication is expected by the mid-2013.
Safety and tolerability
General
In the ECHO and THRIVE trials, the RPV-based treatment regimen was generally well tolerated, with a more favorable profile with respect to psychiatric, neurological, and metabolic adverse events (AEs) compared to the efavirenz-based regimen.24,25 In the pooled analysis, conducted after a median treatment duration of 55.7 and 55.6 weeks in the RPV and efavirenz groups, 2% and 4% of patients, respectively, discontinued treatment because of AEs.10
The most common AEs leading to discontinuation were any rash (0.1% of RPV vs 1.8% of efavirenz patients) and depression (0.3% vs 0.6% of patients, respectively). Pregnancy led to discontinuation in 0.4% of patients in each group.36
The incidence of any rash at least possibly related to treatment was significantly lower in the RPV group than in the efavirenz group. The incidence of rash was higher in the first 4 weeks of treatment. The majority of rashes were of severity grade 1 or 2 and there were no grade 4 rashes.36
RPV recipients were also less likely to have psychiatric AEs (24% vs 29%; P = 0.03) and treatment-related psychiatric AEs (15% vs 23%; P = 0.006). The between-group differences in neurological and psychiatric AEs were chiefly due to the higher proportion of efavirenz recipients reporting dizziness or abnormal dreams and nightmares. Grades 3 or 4 neurological and psychiatric AEs were infrequent (≤1% of patients in any group).37
AEs of severity grade 2–4 occurring at 48 weeks in ≥2% of patients in at least one treatment group in the ECHO and THRIVE trials are shown in Figure 4. The most frequent grade 2–4 AEs were depressive disorders, headache, rash, and insomnia in RPV recipients, and rash, dizziness, abnormal dreams, depressive disorders, headache, insomnia, and nausea in efavirenz recipients.10 Occurrence of treatment-related grade 2–4 AEs was time dependent. Analyzing the first 12 weeks of treatment, the incidence of these AEs was 9.6% and 22.3% (weeks 0–4), 1.6% and 3.4% (weeks 5–8) and 1.2% and 2.5% (weeks 9–12) in RPV and efavirenz recipients, respectively.38
Figure 4.
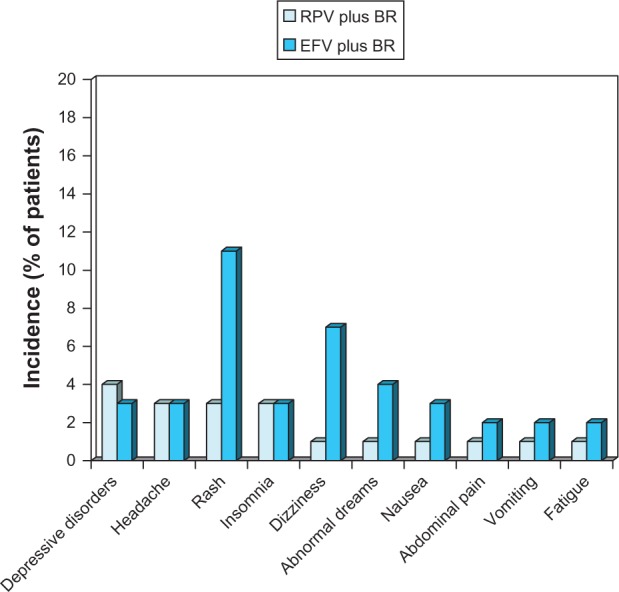
Tolerability of oral rilpivirine as a component of combination therapy in antiretroviral-naïve patients with HIV infection.
Notes: Pooled descriptive data10 from the ECHO24 and THRIVE25 trials, which compared rilpivirine 25 mg once daily with efavirenz 600 mg/day, plus background regimens. Adverse events (grade ≥ 2 level of severity) occurring in ≥2% of patients in at least one group during 48 weeks of treatment.
Abbreviations: BR, background regimen; EFV, efavirenz; RPV, rilpivirine.
In the ECHO and THRIVE trials, there were five deaths, one in the RPV group (grade 3 bronchopneumonia) and four in the efavirenz group (grade 3 Burkitt’s lymphoma, grade 4 cerebral toxoplasmosis/respiratory failure, grade 4 dysentery, grade 4 cerebrovascular accident), none of which was considered related to treatment.36
In the subgroups with FTC/TDF as a backbone, at 96 weeks the tolerability profile of RPV was more favorable than that of EFV, according to pooled data from the ECHO and THRIVE trials.38 Rates of treatment-related AEs of grade 2–4 severity well known to be associated with NNRTI use occurred in significantly fewer RPV/FTC/TDF recipients than EFV/FTC/TDF recipients, especially in those with HIV-RNA ≤100,000 copies/mL.29
The majority of treatment-related AEs occurred during the first year of treatment.36 For example, treatment-related AEs of grade 2–4 severity occurred in 17% and 33% of RPV/FTC/TDF and EFV/FTC/TDF recipients, respectively, during the first year of treatment, and only in 2.5% and 3.7% of patients, respectively, during the second year of treatment.29
From clinical trial results, it seems unlikely that RPV at therapeutic dosages will increase the QTc interval.39 The actual 25 mg dosage of RPV has been selected because, in the Phase II dose-ranging study,23 an increment of QTc interval was observed. The increment in QTc interval corrected according to Fridericia’s formula (QTcF) was gradual up to week 48, was slightly more pronounced with either efavirenz or RPV 75 mg or 150 mg than with RPV 25 mg, and stabilized in all groups from week 48 up to week 96. The alteration was seen in patients receiving lamivudine/zidovudine but not in those treated with FTC/TDF.23 In a confirmatory thorough QT study in HIV-negative volunteers, compared with placebo, the upper limit of the 90% CI for time-matched differences in QTcF was <10 milliseconds at all time points for the RPV 25 mg once daily and efavirenz 600 mg/day groups, whereas this limit was exceeded for the active control, moxifloxacin 400 mg/day.39 The QTc interval of the electrocardiogram, corrected using QTcF interval increased over time in both groups; mean (95% CI) increases were +11 (10–13) milliseconds and 13 (12–14) milliseconds, respectively. There were no QTcF intervals >500 milliseconds.36
Laboratory abnormalities
In the ECHO trial, there was no significant change from baseline to 48 weeks in serum calcifediol (hydroxylated vitamin D [25(OH)D]) levels in RPV recipients, whereas 25(OH)D levels were significantly (P < 0.0001) lower in efavirenz recipients.40 In the RPV group, severe 25(OH)D deficiency (serum 25[OH]D level of <25 nmol/L) was observed in 4.8% of patients at baseline and 4.5% at 48 weeks compared to 5.2% and 9.0% of efavirenz recipients (P = 0.032).40
There was a small decrease from baseline to week 48 in basal cortisol of 13.1 nmol/L for RPV and a small increase of 9.0 nmol/L for efavirenz. The proportions of patients with at least two consecutive (treatment-emergent) abnormal cortisol responses to an adrenocorticotropic hormone test (≤500 nmol/L) during the trial were 1.7% (11 of 643) in the RPV group and 0% in the efavirenz group. No patient had signs or symptoms of adrenal insufficiency or discontinued the study secondary to abnormal adrenocorticotropic hormone test results. There were no clinically relevant changes from baseline to week 48 in androstenedione, dehydroepiandrosterone sulfate, luteinizing hormone, total testosterone, or progesterone.36
Serum creatinine was elevated in 1% and 2% of RPV and efavirenz recipients.10 In both ECHO and THRIVE trials, RPV recipients showed an initial increase in mean serum creatinine levels (4–9 mmol/L) that thereafter remained stable up to 48 weeks. Mean values remained close to baseline in efavirenz recipients.24,25 There were no clinically significant changes in the glomerular filtration rate measured using the serum creatinine (ECHO and THRIVE) or the Hoek formula based on serum cystatin C concentrations (THRIVE). There were also no renal-related AEs or renal-related discontinuations in either treatment group.24,25
RPV was associated with significantly smaller mean changes from baseline in aspartate aminotransferase, alanine aminotransferase, total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and triglyceride levels than efavirenz (Figure 5). Both mean LDL cholesterol and triglyceride levels did not increase above baseline values with RPV, but did with efavirenz. There was no between-group statistical difference in the change from baseline to week 48 of the total/HDL cholesterol ratio.36
Figure 5.
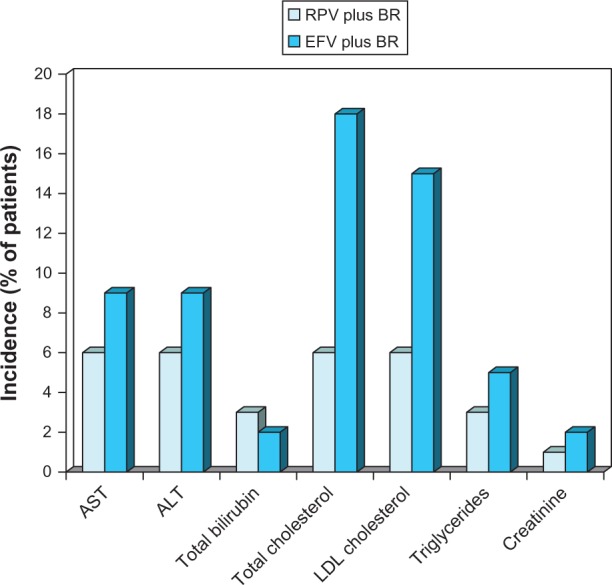
Laboratory abnormalities and changes in blood lipids associated with oral rilpivirine as a component of combination therapy in antiretroviral-naïve patients with HIV infection.
Notes: Pooled descriptive data10 from the ECHO24 and THRIVE trials25 are for severity-level grades 2–4 laboratory abnormalities or changes in blood lipids occurring in ≥2% of patients in at least one group after 48 weeks of treatment. Values shown as <1 at a particular grade level of severity in the original reference were rounded up to 1 when summing to obtain totals for grade 2–4 levels.
Abbreviations: BR, background regimen; EFV, efavirenz; LDL, low-density lipoprotein; RPV, rilpivirine; AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Grade 3–4 severity lipid abnormality rates (week 48) were significantly (P = 0.001) lower in RPV-treated patients compared to efavirenz recipients (total cholesterol: 0.1% vs 3%; LDL cholesterol: 1% vs 4%; triglycerides: 0.3% vs 2%). These differences in lipid abnormalities were independent of background treatment regimens.41 There was no significant between-group difference in the Framingham Coronary Heart Disease relative risk score.41 Similarly, in the 96-week descriptive analysis, in the RPV and efavirenz groups, serum lipid abnormalities of grade 3–4 severity occurred in 0.1% and 3.2% of patients (total cholesterol), 1.5% and 5.1% of patients (LDL cholesterol), and 0.6% and 3.2% of patients (triglycerides), respectively.42
Focusing on the RPV/FTV/TDF and EFV/FTC/TDF subgroups, at 96 weeks the proportion of patients with treatment-related laboratory abnormalities was significantly (P < 0.001) lower for RPV than for EFV, with regard to both grade 2–4 (50% vs 60%) and grade 3–4 (13% vs 21%).36 The most commonly observed abnormalities were hypophosphatemia (14% of RPV/FTC/TDF recipients vs 15% of EFV/FTC/TDF recipients), increased pancreatic amylase (9% vs 10%), and hyperglycemia (8% vs 6%).29 Grade 2–4 lipid abnormalities were also significantly less common with RPV/FTC/TDF than with EFV/FTC/TDF (P < 0.002 for each individual grade of total cholesterol and LDL cholesterol abnormality). The proportion of patients who received lipid-lowering therapy was significantly lower in the first group than in the second (3% vs 6%; P = 0.025).29
In confirmation of these results, in the SPIRIT study, patients switching to RPV/FTC/TDF had significantly (P < 0.001) greater improvements from baseline in serum total cholesterol, LDL cholesterol and trygliceride levels than patients who continued treatment with PI/r regimen.34
Patient perspective
When selecting a first-line antiretroviral therapy regimen, the potential for development of resistance and cross-resistance is an important factor to consider, along with sustained efficacy, favorable tolerability, and convenience.1
Parienti et al investigated treatment adherence with once-daily regimens, assessing eleven randomized, controlled trials with a total of 3029 subjects.43 In this meta-analysis, adherence rates were modestly better with once-daily regimens (+2.9%; 95% CI: 1.0%–4.8%; P , 0.003) than with twice-daily regimens. The effect size was more pronounced for HAART-naïve patients and when all medications were taken once daily. Of note, 48-week virologic suppression with once- and twice-daily regimens was similar (77% vs 76%, respectively). More recent studies comparing once-daily regimens to more frequently dosed regimens continue to support the positive impact of once-daily regimens on HAART adherence.44
Pill burden is another important factor affecting adherence. The ADONE (Adherence to ONE pill) study verified the effect of a reduced number of pills on adherence and quality of life; patients did not change their therapy in terms of active molecules or doses of the same molecules, but simply reduced the daily number of pills in their regimen from three or two to one. This approach was made possible by the use of an FDC pill combining all the drugs previously taken by the patients as separated entities (FTC lamivudine, TDF, efavirenz). One month post-switch to STR, the adherence rate increased significantly to 96.1% from a baseline value of 93.8% (P < 0.01); the increase was steadily maintained throughout the study (96.2% at 6 months). Quality of life also improved over time, from 68.8% to 72.7% (P = 0.042), and was significantly associated with the perception of health status, presence of AEs, and number of reported AEs. Quality of life significantly influenced adherence (P < 0.0001).45
Dosing frequency and pill burden have also been identified as important treatment characteristics for treatment persistence.46 Distinct from but related to medication adherence, persistence reflects the duration of time from initiation to discontinuation of therapy, and can be measured at the regimen or patient level. In developed countries, improved regimen persistence, or durability, has been observed with regimens dosed once daily and containing fewer pills.47 In the ECHO and THRIVE trials,24,25 adherence to treatment was one of the most important factors associated with virological response. In the pooled analysis in patients with baseline HIV-RNA levels ≤100,000 copies/mL at 48 weeks, response rates were ≈20% higher in patients with >95% adherence than in those with ≤95% adherence in both the RPV/FTC/TDF and EFV/FTC/TDF groups;30 similar results were derived from pooled 96-week data.29 The RPV/FTC/TDF STR, as well as having a convenient once-daily administration schedule as a single tablet, is well tolerated, which may also improve adherence and patients’ quality of life.
Health care system perspective
The RPV/FTC/TDF STR has not yet been thoroughly evaluated in terms of cost-effectiveness; however, the price of the STR in Europe makes it one of the cheapest treatment options for naïve patients. As an example from the Italian market, starting a patient on the RPV/FTC/TDF STR compared to the use of the same NNRTI backbone plus any boosted protease inhibitor ensures a monthly saving of about €150. More generally, recent studies have shown that the HAART regimens based on only one tablet a day (STR) are associated with significantly better adherence and a lower hospitalization risk in comparison to the administration of three or more tablets a day.9 In this regard, STRs present potential benefits for patients and, in the end, for the health care system.48
According to Markov models, the economic value of the improved response by patients to the STR has been quantified in terms of quality of life and in terms of cost per gained quality-adjusted life year. Owing to the better quality of life perceived by patients, the incremental cost-effectiveness ratio is more favorable and often dominating when STRs are used compared to more complex, although highly active, regimens.44,49–51
Place in therapy
Although clinical trials of the STR are lacking, RPV/FTC/TDF was an effective and generally well-tolerated regimen in two clinical trials24,25 in treatment-naïve patients with HIV infection.52
Generally speaking, within the approved indications,1 RPV/FTC/TDF STR may prove an advantageous choice for first-line therapy in HIV-positive patients. Certain patient populations, in particular, could benefit from this regimen, including:
Women of child-bearing potential. There are no clinically significant drug interactions between RPV/FTC/TDF and oral contraceptives. Moreover, RPV has a US Food and Drug Administration use-in-pregnancy rating of category B (no evidence of harm to the fetus in animal studies, but no adequate and well-controlled studies in pregnant women).10
Patients with an increased risk of central nervous system events in which other specific drugs (eg, efavirenz) may be contraindicated.
Patients with high cardiovascular risk, as the RPV/FTC/TDF combination has shown an excellent lipid profile.
Patients on methadone substitutive therapy, because there is no need to adjust the dosage of methadone, as there are no clinically significant drug interactions.
Patients with mild or moderate hepatic impairment, because there is no need of dosage adjustment, although caution is required in patients with moderate hepatic impairment.
Conclusion
The once-daily STR of FTC, TDF, and RPV (Complera™/Eviplera™) provides a convenient option for antiretroviral therapy-naïve patients with HIV infection.
Footnotes
Disclosure
FM has served as a consultant on advisory boards for Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Merck Sharp and Dome, Roche, Tibotec; has received lecture fees from Abbott, Bayer, Bristol-Myers Squibb, Gilead GlaxoSmithKline, Merck Sharp and Dome, Pfizer, and Roche; and has received research and educational grants from Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Jansen-Cilag, and Roche. The authors report no other conflicts of interest in this work.
References
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents Department of Health and Human Services; [updated February 12, 2013]. Available from: http://aidsinfo.nih.gov/contentfles/lvguidelines/adultandadolescentgl.pdfAccessed February 25, 2013 [Google Scholar]
- 2.Maltêz F, Doroana M, Branco T, Valente C. Recent advances in antiretroviral treatment and prevention in HIV-infected patients. Curr Opin HIV AIDS. 2011;6(Suppl 1):S21–S30. doi: 10.1097/01.COH.0000410238.80894.81. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MS, Chen YQ, McCauley M, et al. HPTN 052 Study Team Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011;365(6):493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson MA, Aberg JA, Hoy JF, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA. 2012;308(4):387–402. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 5.Hasse B, Ledergerber B, Furrer H, et al. Swiss HIV Cohort Study Morbidity and aging in HIV-infected persons: the Swiss HIV cohort study. Clin Infect Dis. 2011;53(11):1130–1139. doi: 10.1093/cid/cir626. [DOI] [PubMed] [Google Scholar]
- 6.Krentz HB, Cosman I, Lee K, Ming JM, Gill MJ. Pill burden in HIV infection: 20 years of experience. Antivir Ther. 2012;17(5):833–840. doi: 10.3851/IMP2076. [DOI] [PubMed] [Google Scholar]
- 7.Marzolini C, Back D, Weber R, et al. Swiss HIV Cohort Study Members Ageing with HIV: medication use and risk for potential drug-drug interactions. J Antimicrob Chemother. 2011;66(9):2107–2111. doi: 10.1093/jac/dkr248. [DOI] [PubMed] [Google Scholar]
- 8.Llibre JM, Arribas JR, Domingo P, et al. Spanish Group for FDAC Evaluation Clinical implications of fixed-dose coformulations of antiretrovirals on the outcome of HIV-1 therapy. AIDS. 2011;25(14):1683–1690. doi: 10.1097/QAD.0b013e3283499cd9. [DOI] [PubMed] [Google Scholar]
- 9.Sax PE, Meyers JL, Mugavero M, Davis KL. Adherence to antiretroviral treatment and correlation with risk of hospitalization among commercially insured HIV patients in the United States. PLoS One. 2012;7(2):e31591. doi: 10.1371/journal.pone.0031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tibotec Pharmaceuticals . Edurant™ (rilpivirine) tablets [prescribing information] Raritan, NJ: 2011. [Google Scholar]
- 11.Ghosn J, Chaix ML, Delaugerre C. HIV-1 resistance to first- and second-generation non-nucleoside reverse transcriptase inhibitors. AIDS Rev. 2009;11(3):165–173. [PubMed] [Google Scholar]
- 12.Deeks ED, Keating GM. Etravirine. Drugs. 2008;68(16):2357–2372. doi: 10.2165/0003495-200868160-00007. [DOI] [PubMed] [Google Scholar]
- 13.Azijn H, Tirry I, Vingerhoets J, et al. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob Agents Chemother. 2010;54(2):718–727. doi: 10.1128/AAC.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov. 2003;2(10):812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 15.Goebel F, Yakovlev A, Pozniak AL, et al. Short-term antiviral activity of TMC278 – a novel NNRTI, in treatment-naive HIV-1-infected subjects. AIDS. 2006;20(13):1721–1726. doi: 10.1097/01.aids.0000242818.65215.bd. [DOI] [PubMed] [Google Scholar]
- 16.European Medicines Agency Edurant® 25 mg Film-Coated Tablets: Summary of Product Characteristics Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002264/WC500118874.pdfAccessed December 9, 2011
- 17.Lachau-Durand S, Mamidi RNVS, Cuyckens F, et al. Absorption, metabolism and excretion of TMC278, a next- generation non-nucleoside reverse transcriptase inhibitor (NNRTI), after a single oral dose of 150 mg in healthy male volunteers; 12th European AIDS Conference; November 11–14, 2009; Cologne, Germany. Abstract no PE7.1/3. [Google Scholar]
- 18.European Medicines Agency Eviplera ™ 200 mg/25 mg/245 mg film coated tablets: Summary of product characteristics Available from http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002312//WC500118802.pdfAccessed February 25, 2013
- 19.Gilead Sciences, Inc Complera™: US Prescribing Information Available from: www.gilead.com/medicines#hivaidsAccessed February 25, 2013
- 20.Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of renal disease among people living with HIV: a systematic review and meta-analysis. BMC Public Health. 2012;12(1):234. doi: 10.1186/1471-2458-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young J, Schäfer J, Fux CA, et al. Swiss HIV Cohort Study Renal function in patients with HIV starting therapy with tenofovir and either efavirenz, lopinavir or atazanavir. AIDS. 2012;26(5):567–575. doi: 10.1097/QAD.0b013e32834f337c. [DOI] [PubMed] [Google Scholar]
- 22.Scherzer R, Estrella M, Li Y, et al. Association of tenofovir exposure with kidney disease risk in HIV infection. AIDS. 2012;26(7):867–875. doi: 10.1097/QAD.0b013e328351f68f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pozniak AL, Morales-Ramirez J, Katabira E, et al. TMC278-C204 Study Group Efficacy and safety of TMC278 in antiretroviral-naive HIV-1 patients: week 96 results of a phase IIb randomized trial. AIDS. 2010;24(1):55–65. doi: 10.1097/QAD.0b013e32833032ed. [DOI] [PubMed] [Google Scholar]
- 24.Molina JM, Cahn P. Grinsztejn B, et al; ECHO study group. Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment-naive adults infected with HIV-1 (ECHO): a phase 3 randomised double-blind active-controlled trial. Lancet. 2011;378(9787):238–246. doi: 10.1016/S0140-6736(11)60936-7. [DOI] [PubMed] [Google Scholar]
- 25.Cohen CJ, Andrade-Villanueva J, Clotet B, et al. THRIVE study group Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet. 2011;378(9787):229–237. doi: 10.1016/S0140-6736(11)60983-5. [DOI] [PubMed] [Google Scholar]
- 26.Rimsky L, Vingerhoets J, Van Eygen V, et al. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J Acquir Immune Defic Syndr. 2012;59(1):39–46. doi: 10.1097/QAI.0b013e31823df4da. [DOI] [PubMed] [Google Scholar]
- 27.de Mendoza C, Anta L, García F, et al. HIV-1 genotypic drug resistance interpretation rules – 2009 Spanish guidelines. AIDS Rev. 2009;11(1):39–51. [PubMed] [Google Scholar]
- 28.Brochot A, Vis P, Corbett C, et al. Generalized additive modelling of virologic response to the NNRTIs rilpivirine (RPV, TMC278) and efavirenz (EFV) in treatment-naive HIV-infected patients: pooled week 48 data from ECHO and THRIVE; 13th European AIDS Conference; October 12–15, 2011; Belgrade, Serbia. Abstract no PS12/7. [Google Scholar]
- 29.Nelson M, Behrens G, Cohen C, et al. Sustained efficacy with low and similar rates of virologic failures in second year observed with rilpivirine (RPV) versus efavirenz (EFV) plus emtricitabine/tenofovir DF (FTC/TDF) in treatment-naive, HIV-1 infected adults: pooled 96-week ECHO and THRIVE analysis; 13th European AIDS Conference (EACS); October 12–15, 2011; Belgrade, Serbia. Abstract no LBPE7.3/7. [Google Scholar]
- 30.Behrens G, Rijnders B, Nelson M, et al. Efficacy and safety outcomes for rilpivirine (RPV) versus efavirenz (EFV) plus emtricitabine/tenofovir DF (FTC/TDF) in treatment-naïve, HIV-1-positive adults with baseline viral load ≤ 100,000 copies/mL-pooled 48-week ECHO and THRIVE analysis; AIDS 2012: XIX International AIDS conference; July 22–27, 2012; Washington, DC. Abstract no TUPE023. [Google Scholar]
- 31.White K, Van Eygen V, Vingerhoets J, et al. Week 96 resistance analysis of the pooled ECHO and THRIVE Truvada subset in treatment-naıïve HIV-infected adults with ≤100,000 c/mL baseline viral load; 18th Annual Conference of the British HIV Association; April 18–20, 2012; Birmingham, UK. Abstract no P187. [Google Scholar]
- 32.Mathias A, Menning M, Wei X, et al. Bioequivalence of the co-formulation of emtricitabine/rilpivirine/tenofovir DF; 18th International AIDS Conference; July 18–23, 2010; Vienna, Austria. Abstract no LBPE17. [Google Scholar]
- 33.Cohen CJ, Wohl D, Arribas J, et al. STAR study: single-tablet regimen emtricitabine/rilpivirine/tenofovir DF is non-inferior to efavirenz/emtricitabine/tenofovir DF in ART-naive adults; 11th International Congress on Drug Therapy in HIV infection; November 11–15, 2012; Glasgow, UK. Abstract no O425. [Google Scholar]
- 34.Fisher M, Palella F, Tebas P, et al. SPIRIT study: switching boosted PI to Rilpivirine In combination with Truvada as an STR adults; 11th International Congress on Drug Therapy in HIV infection; November 11–15, 2012; Glasgow, UK. Abstract no P285. [Google Scholar]
- 35.Mills A, Cohen C, DeJesus E, et al. Virologic suppression is maintained in virologically suppressed HIV-1 infected subjects switching from efavirenz/emtricitabine/tenofovir (EFV/FTC/TDF) single-tablet regimen (STR) to emtricitabine/rilpivirine/tenofovir (FTC/RPV/TDF) STR: week-24 results of GS-111; 18th Annual Conference of the British HIV Association (BHIVA); April 18–20, 2012; Birmingham, UK. Abstract no P186. [Google Scholar]
- 36.Cohen CJ, Molina JM, Cahn P, et al. ECHO Study Group. THRIVE Study Group Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. J Acquir Immune Defic Syndr. 2012;60:33–42. doi: 10.1097/QAI.0b013e31824d006e. [DOI] [PubMed] [Google Scholar]
- 37.Mills A, Antinori A, Clotet B, et al. Neurologic and psychiatric safety profile of TMC278 compared with EFV in treatment-naive HIV-1+ patients: ECHO and THRIVE trials at 48 weeks; 18th Conference on Retroviruses and Opportunistic Infections; February 27–March 2, 2011; Boston, MA. Abstract no 420. [Google Scholar]
- 38.Rashbaum B, Girard P, Rachlis A, et al. Rilpivirine (RPV, TMC278) tolerability over the first 12 weeks of treatment in the phase 3 ECHO and THRIVE studies; 51st Interscience Conference on Antimicrobial Agents and Chemotherapy; September 17–20, 2011; Chicago, IL. Abstract no H2-805. [Google Scholar]
- 39.Vanveggel S, Buelens A, Crauwels H, et al. TMC27825 mg/qd has no effect on corrected QT (QTC) interval in HIV-negative volunteers; 47th Annual Meeting of the Infectious Diseases Society of America; October 29–November 1, 2009; Philadelphia, PA. Abstract no 285. [Google Scholar]
- 40.Wohl D, Doroana M, Orkin C, et al. Change in vitamin D levels smaller and risk of development of severe vitamin D deficiency lower among HIV-1-infected, treatment-naive adults receiving TMC278 compared with efavirenz: 48-week results from the phase III ECHO trial 18th Conference on Retroviruses and Opportunistic Infections;February 27–March 2, 2011Boston, MAAbstract no O1014 [Google Scholar]
- 41.Arribas J, Andrade-Villanueva J, Bellos N, et al. Lipid profiles of TMC278 and efavirenz in treatment-naïve, HIV-1-infected patients: pooled Week 48 data from the randomized, double-blind, Phase III ECHO and THRIVE trials; 18th Conference on Retroviruses and Opportunistic Infections; February 27–March 2, 2011; Boston, MA. Abstract no 0304. [Google Scholar]
- 42.Cohen CJ, Molina JM, Cassetti I, et al. Pooled week 96 efficacy, resistance and safety results from the double- blind, randomised, phase III trials comparing rilpivirine (RPV) versus efavirenz (EFV) in treatment-naive, HIV-1-infected adults; 6th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; July 17–20, 2011; Rome, Italy. Abstract no TULBPE032. [Google Scholar]
- 43.Parienti JJ, Bangsberg DR, Verdon R, Gardner EM. Better adherence with once-daily antiretroviral regimens: a meta-analysis. Clin Infect Dis. 2009;48(4):484–488. doi: 10.1086/596482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nachega JB, Mugavero MJ, Zeier M, Vitória M, Gallant JE. Treatment simplification in HIV-infected adults as a strategy to prevent toxicity, improve adherence, quality of life and decrease healthcare costs. Patient Prefer Adherence. 2011;5:357–367. doi: 10.2147/PPA.S22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Airoldi M, Zaccarelli M, Bisi L, et al. One-pill once-a-day HAART: a simplification strategy that improves adherence and quality of life of HIV-infected subjects. Patient Prefer Adherence. 2010;4:115–125. doi: 10.2147/ppa.s10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bae JW, Guyer W, Grimm K, Altice FL. Medication persistence in the treatment of HIV infection: a review of the literature and implications for future clinical care and research. AIDS. 2011;25(3):279–290. doi: 10.1097/QAD.0b013e328340feb0. [DOI] [PubMed] [Google Scholar]
- 47.Willig JH, Abroms S, Westfall AO, et al. Increased regimen durability in the era of once-daily fixed-dose combination antiretroviral therapy. AIDS. 2008;22(15):1951–1960. doi: 10.1097/QAD.0b013e32830efd79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colombo GL, Colangeli V, Di Biagio A, Di Matteo S, Viscoli C, Viale P. Cost-effectiveness analysis of initial HIV treatment under Italian guidelines. Clinicoecon Outcomes Res. 2011;3:197–205. doi: 10.2147/CEOR.S24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colombo GL, Di Matteo S, Maggiolo F. Antiretroviral therapy in HIV-infected patients: a proposal to assess the economic value of single tablet regimen. Clinicoecon Outcomes Res. 2013;5:59–68. doi: 10.2147/CEOR.S38977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rizzardini G, Restelli U, Bonfanti P, et al. Cost of human immunodeficiency virus infection in Italy, 2007–2009: effective and expensive, are the new drugs worthwhile. Clinicoecon Outcomes Res. 2012;4:245–252. doi: 10.2147/CEOR.S35194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maggiolo F, Di Matteo S, Masini G, et al. Cost-effectiveness analysis of first line HAART; 11th International Congress on Drug Therapy in HIV infection; November 11–15, 2012; Glasgow, UK. Abstract no P096. [Google Scholar]
- 52.Lyseng-Williamson KA, Scott LJ. Emtricitabine/rilpivirine/tenofovir disoproxil fumarate single-tablet regimen: a guide to its use in HIV-1 infection. Clin Drug Investig. 2012;32(10):715–722. doi: 10.1007/BF03261925. [DOI] [PubMed] [Google Scholar]