

Abstract
Werner syndrome (WS) is a premature aging disorder caused by mutations in a RecQ-like DNA helicase. Mice lacking the helicase domain of the WRN homologue exhibit many phenotypic features of WS. Importantly, mutant WrnΔhel/Δhel mice show abnormal increases in visceral fat deposition and fasting blood triglyceride levels followed by insulin resistance and high blood glucose levels. These mice also exhibit increased heart and liver tissue reactive oxygen species concomitantly with oxidative DNA damage, indicating a pro-oxidant status. We treated mice with either ascorbate or catechin hydrate for 9 months. Vitamin C supplementation reduced oxidative stress in liver and heart tissues and reversed hypertriglyceridemia, hyperglycemia, and insulin resistance and reduced fat weight in mutant WrnΔhel/Δhel mice. Although the free scavenger catechin hydrate also reduced oxidative DNA damage in heart and liver tissues, it did not reverse any of the metabolic phenotype aspects in treated mutant mice. Finally, vitamin C and catechin hydrate did not affect the metabolic status of wild-type mice. These results indicate that vitamin C supplementation could be beneficial for WS patients.
Keywords: Werner syndrome, metabolic syndrome, vitamin C, catechin, Wrn mutant mice
Aging is defined as a progressive deterioration of physiological functions that impairs the ability of an organism to maintain its homeostasis and consequently increases susceptibility to diseases and death. Much of the recent progress in the understanding of aging has been fueled by the study of human progeroid syndromes.1 One of the most fascinating of human aging disorders is Werner syndrome (WS). WS is a human autosomal recessive disorder characterized by genomic instability and the premature onset of a number of age-related diseases, including osteoporosis, ocular cataracts, graying and loss of hair, diabetes mellitus, arteriosclerosis, and atherosclerosis.2–5 It is, to our knowledge, the human disease model closest to normal aging. The defective enzyme responsible for WS is a helicase or exonuclease involved in DNA repair, replication, transcription, and telomere maintenance.6–10 We previously generated a mouse model with a deletion in the helicase domain of the murine WRN homologue11 that recapitulates most of the WS phenotypes, including an abnormal hyaluronic acid excretion, higher reactive oxygen species (ROS) levels, and increased genomic instability and cancer incidence.12,13 In addition, WrnΔhel/Δhel mice show hallmarks of a metabolic syndrome that includes premature visceral obesity, hypertriglyceridemia, and insulin-resistant type 2 diabetes, like WS patients.13 Metabolic syndrome afflicts up to one-half of Western populations and is considered an age-related, proinflammatory lipidic disorder.14 As WS patients exhibit a pro-oxidative status in vivo,15 it is believed that an antioxidant treatment might alleviate some of the risk factors (including metabolic anomalies) associated with age-related diseases in such patients. The WrnΔhel/Δhel mouse provides a unique opportunity to test such compounds. In this study, we first tested the impact of two molecules on visceral obesity, hypertriglyceridemia, insulin-resistant diabetes, and ROS levels in specific tissues known to have antioxidant properties, namely, ascorbate (or vitamin C) and the polyphenolic flavonoid (+)-catechin hydrate [or (+)-cyanol-3; Sigma, St. Louis, MO, USA].
The drinking water of WrnΔhel/Δhel mice was supplemented daily with either 0.4% sodium L-ascorbate (w/v) as described before16 or with 0.2% catechin (w/v) at weaning. The concentration of catechin used was based on the observation that it prevented vascular endothelial dysfunction and reduced vascular ROS production in dyslipi-demic mice expressing the human apolipoprotein B-100 gene.17 We first examined ROS and DNA damage levels in the liver and heart tissues of ascorbate- and catechin-treated WrnΔhel/Δhel mice compared to untreated mutant and wild-type mice. We measured ROS and oxidative DNA damage in five males and five females of each cohort at 9 months of age as described previously.18 WrnΔhel/Δhel animals exhibited higher amounts of ROS and oxidative DNA damage in the liver and heart than wild-type animals (Fig. 1A–D). Both ascorbate and catechin treatments of WrnΔhel/Δhel mice reduced these amounts to equal or even inferior values than those observed in the untreated wild-type cohort. These results indicate that both ascorbate and catechin molecules were absorbed by mutant mice and exerted their antioxidant effects in different tissues.
Figure 1.

Impact of ascorbate and catechin on levels of ROS and oxidative DNA damage in liver and heart of WrnΔhel/Δhel mice. (A) Liver tissue ROS levels. Tissues were excised and homogenized in RIPA buffer. The homogenate was incubated with the dye 2′,7′-dichlorofluorescein for 1 h at 37°C. This dye is highly fluorescent upon oxidation. (B) Oxidative DNA damage levels in liver. Oxidative DNA damage was measured with the oxidative DNA damage kit from Kamiya Biomedical Co. (Seattle, WA). (C) Heart ROS levels. (D) Oxidative DNA damage levels in heart. Ascorbate and catechin were added in drinking water (0.4% and 0.2%, w/v, respectively) of WrnΔhel/Δhel mice at weaning. *, P < 0.05 by unpaired t-test compared to WrnΔhel/Δhel animals. Bars in the histograms represent the standard errors of the means. Nine-month-old males (n = 5) and females (n = 5) of each cohort were used for all measurements.
We next examined visceral fat weight, blood triglyceride, fasting blood glucose, insulin levels, and the homeostatic model assessment of insulin resistance (HOMA-IR) index in treated animals. As indicated in Figure 2A, visceral fat weight was increased in WrnΔhel/Δhel mice compared to wild-type animals. Ascorbate treatment decreased visceral fat weight of WrnΔhel/Δhel mice to wild-type levels. In contrast, catechin had no effect on the accumulation of visceral fat weight in WrnΔhel/Δhel mice. Similarly, blood triglyceride levels were increased in WrnΔhel/Δhel mice compared to wild-type animals (Fig. 2B). Ascorbate treatment decreased visceral fat weight of WrnΔhel/Δhel mice to wild-type levels, but catechin had no effect.
Figure 2.
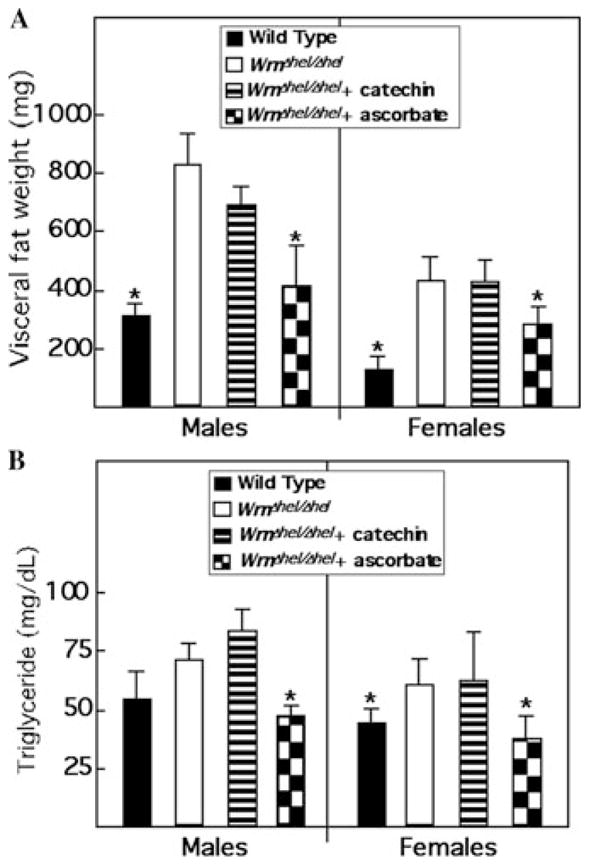
Impacts of ascorbate and catechin on visceral fat weight and blood triglyceride levels in WrnΔhel/Δhel mice. (A) Weight of retroperitoneal (visceral) white adipose tissue in males and females at 9 months of age. (B) Fasting triglyceride levels in serum of untreated wild-type and treated and untreated WrnΔhel/Δhel mice at 6 months of age. Five mice of each genotype were evaluated. *, P < 0.05 by unpaired t-test, compared to WrnΔhel/Δhel animals. WrnΔhel/Δhel mice were treated with either ascorbate or catechin as described for Figure 1.
Fasting blood glucose levels were increased in WrnΔhel/Δhel mice compared to wild-type animals. Similarly, despite this increase in glucose levels, WrnΔhel/Δhel mice also exhibited an increase in insulin levels and HOMA-IR index. Ascorbate decreased the blood glucose levels of WrnΔhel/Δhel mice to wild-type levels. The blood insulin levels were higher in ascorbate-treated WrnΔhel/Δhel mice than untreated wild-type animals (Fig. 3). Calculation of the insulin resistance index, however, indicated a significant trend toward normalization. In contrast, catechin had no impact on the increased values of fasting blood glucose, insulin, and HOMA-IR index (Fig. 3). Finally, neither ascorbate nor catechin had a significant impact on wild-type fat or glucose metabolism (data not shown).
Figure 3.
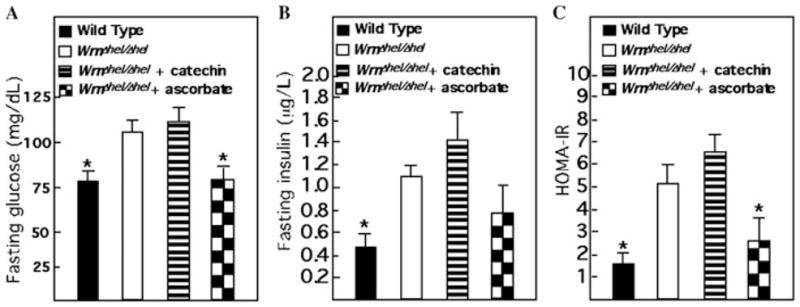
Impact of ascorbate and catechin on fasting blood glucose, insulin, and HOMA-IR index in WrnΔhel/Δhel mice. (A) Fasting blood glucose levels. (B) Fasting insulin levels. (C) Index for the HOMA-IR for fasting mice. *, P < 0.05 by unpaired t-test compared to WrnΔhel/Δhel animals. Bars in the histograms represent the standard errors of the means. Nine-month-old males (n = 5) and females (n = 5) of each cohort were used for all measurements. WrnΔhel/Δhel mice were treated with either ascorbate or catechin as described for Figure 1.
The above data indicate that ascorbate can reverse the increased ROS and oxidative DNA damage in liver and cardiac tissues as well as the increased fat, blood triglyceride, glucose, and insulin levels observed in WrnΔhel/Δhel mice. These results suggest that treatment of WS patients with high doses of ascorbate may be beneficial for such individuals. Similar experiments with Wrn null mice lacking a functional telomerase will be required. Such double-knockout mice exhibit a more severe premature aging phenotype than WrnΔhel/Δhel mice.19 Notably, high doses of ascorbate do not ameliorate the mean life span of wild-type animals.20 This suggests that ascorbate may have a beneficial effect only on individuals or animals with specific metabolic abnormalities. Other animal models exhibiting genomic instability and premature aging will need to be tested21 to resolve this question.
Although catechin is as efficient as ascorbate in reducing overall ROS and oxidative DNA damage, it did not reverse the fat and glucose metabolic anomalies observed in WrnΔhel/Δhel mice. Since the concentration of catechin used did not exacerbate the health effects in these animals when administered for up to 9 months, it is possible that a higher concentration of catechin would be required to have a positive effect on the metabolism of these mice. Alternatively, the antioxidant effect of catechin alone was not sufficient to improve the metabolism of WrnΔhel/Δhel mice. In this regard, ascorbate is not only an antioxidant but it can also affect the expression of many genes in vivo by an unknown mechanism.20 We will need to test several different antioxidants to determine whether regulating tissue redox status is sufficient to improve the metabolism of WrnΔhel/Δhel mice. Finally, chemicals known to affect directly metabolic enzymes involved in fat or glucose metabolism will be tested in our mouse model to determine their effects on overall health and longevity.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research. M.L. is a Senior Scholar of the Fonds de la Recherche en Santé du Québec.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Kipling D, et al. What can progeroid syndromes tell us about human aging? Science. 2004;305:1426–1431. doi: 10.1126/science.1102587. [DOI] [PubMed] [Google Scholar]
- 2.Epstein CJ, et al. Werner’s syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine. 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Salk D. Werner’s syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum Genet. 1982;62:1–5. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- 4.Yu C, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 5.Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Balajee AS, et al. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper MP, et al. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;4:907–912. [PMC free article] [PubMed] [Google Scholar]
- 8.Shen JC, Loeb LA. The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet. 2000;16:213–220. doi: 10.1016/s0168-9525(99)01970-8. [DOI] [PubMed] [Google Scholar]
- 9.Saintigny Y, et al. Homologous recombination resolution defect in werner syndrome. Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabbe L, et al. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 11.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebel M, et al. Genetic cooperation between the Werner syndrome protein and poly(ADP ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol. 2003;162:1559–1569. doi: 10.1016/S0002-9440(10)64290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massip L, et al. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the Werner syndrome gene homologue. Exp Gerontol. 2006;41:157–168. doi: 10.1016/j.exger.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 15.Pagano G, et al. In vivo prooxidant state in Werner syndrome (WS): results from three WS patients and two WS heterozygotes. Free Radic Res. 2005;39:529–533. doi: 10.1080/10715760500092683. [DOI] [PubMed] [Google Scholar]
- 16.Maeda N, et al. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci USA. 2000;97:841–846. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gendron M-E, Thorin E. A change in the redox environment and thromboxane A2 production precede endothelial dysfunction in mice. Am J Physiol. 2007;293:H2508–H2515. doi: 10.1152/ajpheart.00352.2007. [DOI] [PubMed] [Google Scholar]
- 18.Deschênes F, et al. In vivo misregulation of genes involved in apoptosis, development and oxidative stress in mice lacking both functional Werner syndrome protein and poly(ADP-ribose) polymerase-1. Hum Mol Genet. 2005;14:3293–3308. doi: 10.1093/hmg/ddi362. [DOI] [PubMed] [Google Scholar]
- 19.Chang S, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 20.Massip L, et al. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 2009;24:158–172. doi: 10.1096/fj.09-137133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher B, et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008;4:e1000161. doi: 10.1371/journal.pgen.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]