

Abstract
This paper presents an industrial scale process for extraction, purification, and isolation of epiisopiloturine (EPI) (2(3H)-Furanone,dihydro-3-(hydroxyphenylmethyl)-4-[(1-methyl-1H-imidazol-4-yl)methyl]-, [3S-[3a(R*),4b]]), which is an alkaloid from jaborandi leaves (Pilocarpus microphyllus Stapf). Additionally for the first time a set of structural and spectroscopic techniques were used to characterize this alkaloid. EPI has shown schistomicidal activity against adults and young forms, as well as the reduction of the egg laying adult worms and low toxicity to mammalian cells (in vitro). At first, the extraction of EPI was done with toluene and methylene chloride to obtain a solution that was alkalinized with ammonium carbonate. The remaining solution was treated in sequence by acidification, filtration and alkalinization. These industrial procedures are necessary in order to remove impurities and subsequent application of the high performance liquid chromatography (HPLC). The HPLC was employed also to remove other alkaloids, to obtain EPI purity higher than 98%. The viability of the method was confirmed through HPLC and electrospray mass spectrometry, that yielded a pseudo molecular ion of m/z equal to 287.1 Da. EPI structure was characterized by single crystal X-ray diffraction (XRD), 1H and 13C nuclear magnetic resonance (NMR) in deuterated methanol/chloroform solution, vibrational spectroscopy and mass coupled thermal analyses. EPI molecule presents a parallel alignment of the benzene and the methyl imidazol ring separated by an interplanar spacing of 3.758 Å indicating a π-π bond interaction. The imidazole alkaloid melts at 225°C and decomposes above 230°C under air. EPI structure was used in theoretical Density Functional Theory calculations, considering the single crystal XRD data in order to simulate the NMR, infrared and Raman spectra of the molecule, and performs the signals attribution.
Introduction
Alkaloids are organic compounds found in plant kingdom, fungus, bacteria and animals. The majority of these natural substance exhibits an alkaline character related particularly to the presence of basic amino groups (frequently heterocyclic) in their chemical structure. The word alkaloid is derived from the Latin “alkali” (basic) with the suffix “-oid” (-like). In the literature, many authors claim that a true alkaloid is composed by one or more basic nitrogen atoms in a heterocyclic ring, beside the intense biological activity in the presence of living organisms [1]. Additionally, alkaloids are known to be used as therapeutical agents for anesthetics, analgesics, and psycho-stimulant, among other pharmacological activities.
Over the past one hundred and fifty years, thousands of alkaloids have been isolated [2], and several high standard techniques can now be employed to evaluate the pharmacological and toxicological activities of these substances. Thus, new applications of centenarians alkaloids have been discovered, as the case of Pilocarpine, which has been isolated in 1875 and applied for decades in the treatment of glaucoma and, recently, also used for in the treatment of xerostomy [3], [4].
Several alkaloids have been isolated from the Pilocarpus genus, but many of them are still in analysis to evaluate their therapeutic applications [5]. The jaborandi species (Pilocarpus sp.) provides several molecules of the alkaloid class such as: pilocarpine, isopilocarpine, pilocarpidine, isopilocarpidine, pilosine, isopilosine, epiisopilosine, epiisopiloturine, 13–7-noria (11)-dehydro-pilocarpine, N,N-dimethyl-5-methoxy-tryptamine, N,N-dimethyl tryptamine, plastidesmine (1H)-4-methoxy-2-quinolone and dictamine [5].
The alkaloid epiisopiloturine (EPI) in particular was identified by Voightlander et al. in 1973 [6]. This substance has shown in vitro activity against Schistosoma mansoni [7], [8], the main agent of schistosomiasis, a neglected disease of poor, rural and forgotten populations. The parasitic disease represents one of the main public health problems in more than 70 tropical and subtropical countries, especially in Africa, Asia and Latin America[9]. Approximately 240 million of people have been infected worldwide and about 700 million of people are at risk areas [10], [11].
In adults and young (schistosomula) forms of S. mansoni, the EPI alkaloid had schistomicidal activity at a concentration of 150 µg/mL and 300 µg/mL, respectively [7], [9]. Besides the death, the sub lethal dose (100 µg/mL) of this alkaloid promotes the total reduction of the egg laying in the paired adult form, differently from what has been observed when praziquantel (the drug reference standard) was used in the treatment of schistosomiasis. Furthermore, the EPI was not cytotoxic in the peritoneal macrophages, and can be obtained, in large scales, by sustainable and economical process [7], [8].
The activity against S. mansoni, no toxicity to mammalian cells and the plant extraction by sustainable form turn up the studies and characterization of EPI indispensable. The meticulous investigation of the waste from the extraction of a natural product could introduce an important strategy for the discovery and development of a new drug [7].
This work reports the industrial extraction, purification and isolation of EPI from jaborandi leaves, and also its physicochemical characterization by single crystal X-ray diffraction, 1H and 13C nuclear magnetic resonance (NMR) in deuterated methanol/chloroform solution, vibrational infrared (FT-IR) and Raman (FT-Raman) spectroscopies, and mass coupled thermal analyses. The spectroscopic characterization data are further supported by computational calculations performed in the framework of the Density Functional Theory (DFT).
Materials and Methods
Industrial extraction, pre-purification and isolation of alkaloid from jaborandi leaves
The initial step is based on the alkalinization of 1,500 kg of jaborandi leaves with a solution of potassium carbonate, followed by the extraction of all alkaloids (solid-liquid extraction) with toluene and methylene chloride solvents ( Figure 1 ). The organic phase was submitted to liquid-liquid extraction with an aqueous solution of sulfuric acid. Hereafter, 250 L of the aqueous solution with a mean content of 2% (m/v) of EPI in the sulfate salt form was cooled, alkalinized with ammonium hydroxide solution and treated with activated carbon and diatomaceous sand.
Figure 1. Scheme of all necessary steps in obtaining Epiisopiloturine with >98% purity from Jaborandi leaves.
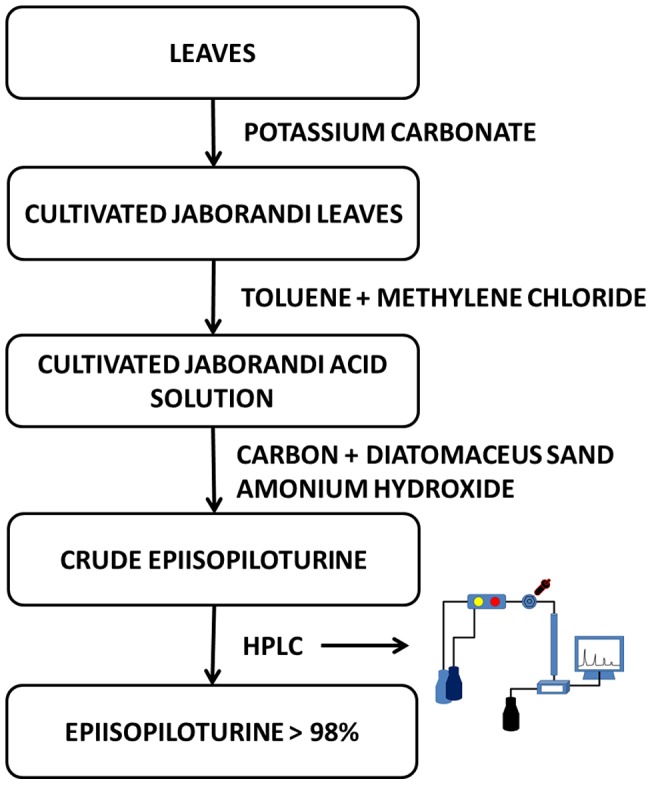
After treatment with carbon and diatomaceous sand, the impure EPI was dissolved in an aqueous solution containing hydrochloric acid and filtered on a pressure lentil filter (filter medium: cloth/two filter papers/cloth) under reduced pressure. The filtrate containing EPI hydrochloride was alkalinized with ammonium hydroxide solution to precipitate the EPI neutral form, and then the solution was filtered under reduced pressure [8]. The aim of the previous step was to remove the impurities such as carbon and diatomaceous sand in order to submit EPI to further purification process by high performance liquid chromatography ( Figure 1 ) (all details from this steps is in patently process – 000121-INPI, Brazil).
The crude EPI ( Figure 1 ) at a concentration of 10 mg/mL was dissolved in the mobile phase with potassium phosphate 5% (v/v), filtered by a membrane of 0.45 µm and set to the preparative high performance liquid chromatography – HPLC (SHIMADZU Prominence, AUTOSAMPLER SIL-10AF, CTO-20A, DGU-20A5, LC-6AD, CBM-20A, SPD-20A, Tokyo, Japan).
The preparative chromatographic conditions set were performed in a column of LiChrospher 60 RP Select B (250×25 mm, 5 µm). The mobile phase was 367.59 mM potassium phosphate adjusted at pH 2.5 with a flow rate of 10 mL/min for a time run of 90 min. The detection was done using a UV detector at a wavelength of 216 nm and the column oven was set to 50°C.
The injection volume was 1000 µL and 500 mL fractions were collected with a concentration of 100 mg/L of crude EPI. The solution obtained after preparative HPLC was alkalinized between pH 9 to 9.5 and subjected to liquid-liquid extraction with industrial chloroform. The organic phase was evaporated with a vacuum controller V-850, a water bath – B – 491 and an evaporator – R – 215 (BUCHI, Switzerland).
For fine analyses an analytical HPLC was employed to verify each process step, with the LiChrospher 60 RP Select B (250×4.6 mm, 5 µm), using external standard (EPI standard at 20 µg/mL and pilocarpine standard at 50 µg/mL, Merck, Darmstadt, Germany). The mobile phase was 367.59 mM potassium phosphate adjusted at pH 2.5. The flow rate was 1 mL/min, the column oven was set to 50°C for a time run of 50 min with UV detection at a wavelength of 216 nm.
The molecular mass confirmation was performed by mass spectrometry (AmaZon SL, Bruker Daltonics, Bremen – Germany), which was used in positive electrospray ionization mode. The capillary voltage was – 1,800 V, temperature at 250°C, and the mass spectra were acquired in mass range of m/z 160 – 300 Da. MS/MS was carried out in manual mode with fragmentation of the precursor ion by collision induced dissociation (CID) using helium (He) as the collision gas. Precursor ions were selected within an isolation width of 2 u and scans were accumulated with variable RF signal amplitudes. The m/z scale of the mass spectrum was calibrated using the external calibration standard G2421A electrospray ‘tuning mix’ from Agilent Technologies (Santa Rosa, USA).
Physical measurements
Nuclear Magnetic Resonance (NMR)
10 mg of EPI was dissolved in 2 mL of CD3OD/CDCl3 1∶1 mixture. Afterward 600 µL of this solution was transferred to a 5 mm NMR sample tube. For the measurements of NMR data, standard parameter sets created for the Bruker CMC-se (complete molecular confidence-structure elucidation) program were uniformly employed. Gradient COSY (Correlation Spectroscopy) (2 scans per increment) and 1H -13C HMBC (Heteronuclear Multiple-Bond Correlation spectroscopy) (8 scans per increment) were acquired using 4 k complex data points in F2 and 512 points in F1 dimension. A 1H-15N HMBC was acquired with 32 scans per increment with a time domain of 256 in F1 and 2 k points in F2. The multiplicity edited gradient HSQC (Heteronuclear Single Quantum Correlation) (2 scans per increment) was acquired with 2k data points in F2 and 400 points in the F1 dimension. The instrument used was an AVANCE III 600 MHz NMR spectrometer equipped with a 5 mm TXI probe head (Bruker Biospin, Rheinstetten, Germany). The NMR data acquired were processed according to the general experimental procedures.
The Bruker structure elucidation package CMC-se (Topspin 3.1) was used to get the peak and multiplet lists in a fully automated way. The multiplet lists are collected into a correlation table. The automatic step is usually followed by short visual inspection of the results. The SELU module contains combined display linking the correlation table and the spectra display. This allows fast inspection and correction of generated HSQC, HMBC and COSY multiplet lists. After the correlation table is populated and inspected, the structure generator calculates structures which are consistent with the NMR data acquired. Finally an independent 13C chemical shift prediction method is used to validate the result.
Single crystal X-ray diffraction (XRD)
The X-ray diffraction data were collected at room temperature using a KAPPA-CCD Diffratometer with MoKα radiation (λ = 0.71073 Å). The cell refinements were performed using the software Collect and Scalepack [12]. The final unit cell parameters were obtained on all reflections. Data reduction was carried out using the software Denzo-SMN and Scalepack. The structure was solved by Direct Methods and anisotropically refined with full-matrix least-squares on F2 using SHELXL97 [13]. The hydrogen atoms bonded to C and N atoms were positioned geometrically and refined with riding constraints with distance restraints of N-H = 0.86 Å, aromatic C-H = 0.93 Å and with Uiso(H) = 1.2 Ueq(N,C). The crystallographic data were deposited at the Cambridge Crystallographic Data Center under the numbers CCDC 915132. Copies of the data can be obtained, free of charge, via www.ccdc.cam.ac.uk.
Vibrational FT-IR and FT- Raman
FT-IR spectrum of EPI sample diluted in KBr was recorded in the 4000–400 cm−1 range on a Bomen spectrophotometer, model MB-102, with a coupled diffuse reflectance accessory (Pike Technologies, Inc.). FT-Raman spectrum was recorded in a FT-Raman Bruker FRS-100/S spectrometer using 1064 nm exciting radiation (Nd:YAG laser Coherent Compass 1064–500 N) and a Ge detector.
Mass coupled thermal analyses (TGA-DSC-MS)
The thermal analyses were recorded on a Netzsch thermoanalyser model TGA/DSC 490 PC Luxx coupled to an Aëolos 403 C mass spectrometer, using a heating rate of 10°°C/min and under synthetic air flow of 50 mL/min.
Computational analysis
The Density Functional Theory (DFT) [14] in the Kohn-Sham (KS) scheme [15] was used to investigate the electronic structure, vibrational properties and NMR 13C and 1H isotropic chemical shifts for the isolated EPI alkaloid molecule. The B3LYP [16] exchange correlation functional and the 6–311++G** basis set were used as implemented in the Gaussian 09 computational package [17]. Gauge Including Atomic Orbitals (GIAO) method [18]–[20] and diffuse functions in the basis set were applied for the calculation of the C and H NMR spectra. All simulations were carried out in vacuum conditions and T = 0 K.
Results and Discussion
The results, here presented, describe a methodology for extraction, purification and isolation of EPI from jaborandi leaves. The HPLC ( Figure 2 ) and LC/MS ESI+/Ion Trap ( Figure 3 ) techniques have demonstrated the purity and initial characterization of each process step. For industrial scale, 1,500 kg of jaborandi leaves have been used through all steps described in Figure 1 , what lead to obtain around 2 kg of pure EPI, used in the structural and spectroscopic characterization described in this work.
Figure 2. Analytical HPLC used LiChrospher 60 RP column and eluted with potassium phosphate.

. (A) Standard EPI (20 µg/mL), (B) Standard pilocarpine (50 µg/mL), (C) “cultivated jaborandi leaves” solution, resulted from first extraction step, (D) “cultivated jaborandi acid” solution, obtained EPI under salt form, (E) Solution of “crude EPI” with some impurities as pilocarpine and other alkaloids, (F) last step of isolation showing EPI >98% purity.
Figure 3. Mass spectrum obtained from ESI+/Ion Trap.

(A) free EPI with a pseudo molecular ion m/z 287.1 Da [M+H]+, (B) MS2 with characteristic fragment at m/z 269.1 Da [M – H2O + H]+, (C) MS3 with fragments at m/z 251.0 Da [M – 2H2O + H+] and 168.06 Da with proposed chemical structure.
The MS/MS analysis showed a pseudomolecular ion with m/z 287.1 Da [M + H]+ and a MS2 fragment with m/z 269.1 Da [M – H2O + H]+. This “molecular fingerprint” of EPI has been previously reported in the literature[6]. Structural and spectroscopic characterizations were also performed to assure the integrity of the EPI molecule.
The EPI molecular structure is shown in Figure 4 , created by the ORTEP [21] software. An interesting feature of this molecule is the parallel alignment of the benzene and the methyl imidazole ring separated by an interplanar spacing of 3.758 Å, indicating a π-π bond interaction. Another feature of the crystalline packing is the presence of a hydrogen bond between hydroxyl H group, from one molecule, and the N1 of the methyl imidazole ring of another, forming in this way a continuous chain of hydrogen bonded molecules, as can be seen in Figure 5 .
Figure 4. Isolated Epiisopiloturine molecular structure.

Figure 5. Epiisopiloturine Crystalline form with the molecules represented in stick format.
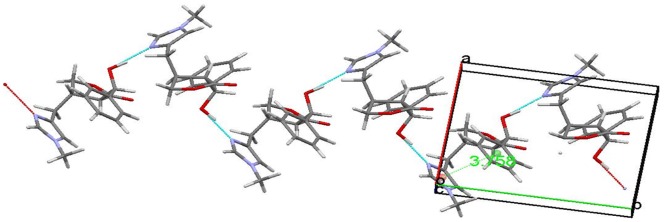
Color code: carbon (gray), hydrogen (white), nitrogen (blue) and oxygen (red). Cyan lines are only guide lines to illustrate hydrogen bonds between the hydroxyl group and the imidazole ring of neighboring molecules in the solid.
Table 1 shows the differences between experimental X-ray diffraction and structural relaxation results obtained through DFT calculations, where we can notice that the largest absolute differences are related with hydrogen atoms. It can be seen, from the bond angle results shown in Table 2 , that the maximum distortion difference is 5.64° in the C3-C2-H2 angle.
Table 1. EPI bond distances obtained through x-ray diffraction (Experimental) and DFT results (Calculated).
| Experimental (Å) | Calculated (Å) | Difference (Experimental -Calculated) (Å) | |
| O1–C9 | 1.419 (3) | 1.429 | −0.010 |
| O1–H1 | 0.82 | 0.963 | −0.143 |
| C1–N1 | 1.314 (3) | 1.312 | 0.002 |
| C1–N2 | 1.341 (3) | 1.364 | −0.023 |
| C1–H1A | 0.93 | 1.080 | −0.150 |
| N1–C3 | 1.379 (3) | 1.379 | 0.000 |
| C3–C2 | 1.359 (3) | 1.374 | −0.015 |
| C3–C4 | 1.481 (3) | 1.496 | −0.015 |
| N2–C2 | 1.369 (3) | 1.381 | −0.012 |
| N2–C16 | 1.452 (3) | 1.453 | −0.001 |
| – | 1.511 (3) | 1.520 | −0.009 |
| C9–C8 | 1.536 (3) | 1.543 | −0.007 |
| C9–H9 | 0.98 | 1.098 | −0.118 |
| C5–C4 | 1.531 (3) | 1.546 | −0.015 |
| C5–C6 | 1.532 (3) | 1.539 | −0.007 |
| C5–C8 | 1.537 (3) | 1.539 | −0.002 |
| C5–H5 | 0.98 | 1.090 | −0.110 |
| C2–H2 | 0.93 | 1.078 | −0.148 |
| C10–C11 | 1.383 (3) | 1.396 | −0.013 |
| C10–C15 | 1.390 (3) | 1.398 | −0.008 |
| C4–H4A | 0.97 | 1.095 | −0.125 |
| C4–H4B | 0.970 | 1.095 | −0.125 |
| C8–C7 | 1.515 (3) | 1.533 | −0.018 |
| C8–H8 | 0.980 | 1.092 | −0.112 |
| O3–C7 | 1.345 (3) | 1.353 | −0.008 |
| O3–C6 | 1.446 (3) | 1.448 | −0.002 |
| C16–H16A | 0.960 | 1.092 | −0.132 |
| C16–H16B | 0.960 | 1.089 | −0.129 |
| C16–H16C | 0.960 | 1.092 | −0.132 |
| O2–C7 | 1.207 (3) | 1.199 | 0.008 |
| C11–C12 | 1.381 (3) | 1.394 | −0.013 |
| C11–H11 | 0.930 | 1.083 | −0.153 |
| C15–C14 | 1.382 (3) | 1.392 | −0.010 |
| C15–H15 | 0.930 | 1.086 | −0.156 |
| C6–H6A | 0.970 | 1.090 | −0.120 |
| C6–H6B | 0.970 | 1.090 | −0.120 |
| C13–C12 | 1.371 (4) | 1.392 | −0.021 |
| C13–C14 | 1.376 (4) | 1.394 | −0.018 |
| C13–H13 | 0.930 | 1.084 | −0.154 |
| C12–H12 | 0.930 | 1.084 | −0.154 |
| C14–H14 | 0.930 | 1.085 | −0.155 |
Atom labels accordingly to Figure 4.
Table 2. EPI bond angles obtained through x-ray diffraction (Experimental) and DFT results (Calculated).
| Experimental (°) | Calculated (°) | Difference(Experimental – Calculated) (°) | |
| C9–O1–H1 | 109.50 | 108.21 | 1.29 |
| N1–C1–N2 | 112.37 (19) | 112.32 | 0.05 |
| N1–C1–H1A | 123.80 | 125.88 | −2.08 |
| N2–C1–H1A | 123.80 | 121.80 | 2.00 |
| C1–N1–C3 | 105.62 (17) | 105.54 | 0.08 |
| C2–C3–N1 | 108.60 (18) | 109.82 | −1.22 |
| C2–C3–C4 | 130.68 (19) | 129.22 | 1.46 |
| N1–C3–C4 | 120.60 (18) | 120.95 | −0.35 |
| C1–N2–C2 | 106.14 (17) | 106.27 | −0.13 |
| C1–N2–C16 | 126.60 (20) | 126.76 | −0.16 |
| C2–N2–C16 | 127.24 (19) | 126.96 | 0.28 |
| O1–C9–C10 | 112.88 (16) | 112.61 | 0.27 |
| O1–C9–C8 | 107.18 (16) | 106.45 | 0.73 |
| C10–C9–C8 | 111.68 (16) | 113.75 | −2.07 |
| O1–C9–H9 | 108.30 | 109.79 | −1.49 |
| C10–C9–H9 | 108.30 | 108.25 | 0.05 |
| C7–C8–C5 | 103.81 (17) | 103.80 | 0.01 |
| C9–C8–C5 | 114.85 (17) | 116.96 | −2.11 |
| C7–C8–H8 | 109.20 | 107.96 | 1.24 |
| C9–C8–H8 | 109.20 | 107.18 | 2.02 |
| C5–C8–H8 | 109.20 | 111.91 | −2.71 |
| C7–O3–C6 | 109.86 (17) | 110.48 | −0.62 |
| N2–C16–H16A | 109.50 | 110.89 | −1.39 |
| N2–C16–H16B | 109.50 | 110.70 | −1.2 |
| H16A–C16–H16B | 109.50 | 108.64 | 0.86 |
| N2–C16–H16C | 109.50 | 108.87 | 0.63 |
| H16A–C16–H16C | 109.50 | 109.10 | 0.4 |
| H16B–C16–H16C | 109.50 | 108.57 | 0.93 |
| O2–C7–O3 | 121.60 (20) | 122.96 | −1.36 |
| O2–C7–C8 | 126.90 (20) | 127.01 | −0.11 |
| O3–C7–C8 | 111.48 (18) | 110.03 | 1.45 |
| C12–C11–C10 | 121.60 (20) | 120.53 | 1.07 |
| C8–C9–H9 | 108.30 | 105.75 | 2.55 |
| C4–C5–C6 | 113.10 (17) | 111.44 | 1.66 |
| C4–C5–C8 | 112.45 (18) | 112.38 | 0.07 |
| C6–C5–C8 | 102.70 (17) | 101.97 | 0.73 |
| C4–C5–H5 | 109.50 | 108.19 | 1.31 |
| C6–C5–H5 | 109.50 | 110.75 | −1.25 |
| C8–C5–H5 | 109.50 | 112.07 | −2.57 |
| C3–C2–N2 | 107.27 (17) | 106.04 | 1.23 |
| C3–C2–H2 | 126.40 | 132.04 | −5.64 |
| N2–C2–H2 | 126.40 | 121.80 | 4.6 |
| C11–C10–C15 | 117.60 (20) | 118.84 | −1.24 |
| C11–C10–C9 | 122.62 (18) | 121.49 | 1.13 |
| C15–C10–C9 | 119.79 (18) | 119.67 | 0.12 |
| C3–C4–C5 | 112.38 (16) | 113.42 | −1.04 |
| C3–C4–H4A | 109.10 | 108.57 | 0.53 |
| C5–C4–H4A | 109.10 | 109.84 | −0.74 |
| C3–C4–H4B | 109.10 | 109.96 | −0.86 |
| C5–C4–H4B | 109.10 | 108.00 | 1.10 |
| H4A–C4–H4B | 107.90 | 106.80 | 1.10 |
| C7–C8–C9 | 110.25 (17) | 108.68 | 1.57 |
| C12–C11–H11 | 119.20 | 120.25 | −1.05 |
| C10–C11–H11 | 119.20 | 119.22 | −0.02 |
| C14–C15–C10 | 121.0 (2) | 120.05 | 0.95 |
| C14–C15–H15 | 119.50 | 120.09 | −0.59 |
| C10–C15–H15 | 119.50 | 119.85 | −0.35 |
| O3–C6–C5 | 107.00 (18) | 106.26 | 0.74 |
| O3–C6–H6A | 110.30 | 107.92 | 2.38 |
| C5–C6–H6A | 110.30 | 111.82 | −1.52 |
| O3–C6–H6B | 110.30 | 107.06 | 3.24 |
| C5–C6–H6B | 110.30 | 113.48 | −3.18 |
| H6A–C6–H6B | 108.60 | 109.98 | −1.38 |
| C12–C13–C14 | 119.80 (20) | 119.60 | 0.2 |
| C12–C13–H13 | 120.10 | 120.22 | −0.12 |
| C14–C13–H13 | 120.10 | 120.18 | −0.08 |
| C13–C12–C11 | 119.9 (2) | 120.26 | −0.36 |
| C13–C12–H12 | 120.00 | 120.09 | −0.09 |
| C11–C12–H12 | 120.00 | 119.65 | 0.35 |
| C13–C14–C15 | 120.10 (20) | 120.05 | 0.05 |
| C13–C14–H14 | 120.00 | 120.09 | −0.09 |
| C15–C14–H14 | 120.00 | 119.85 | 0.15 |
Atom labels accordingly to Figure 4.
The EPI was further characterized through 1H and 13C NMR spectroscopy. The standard way of chemical shift assignments [22] was compared with theoretical DFT calculations for the isolated molecule and can be seen in Supporting Information S1. The 1H NMR data obtained in this work are in conformity with seminal results reported by Voigtlander et al [6].
Measured and calculated FT-IR and FT-Raman EPI molecule spectra are shown in Figure 6 and Figure 7 . The vibrational band assignments, presented in Table 3 , were proposed based on general literature about organic molecules [22]–[25] and a study reported about an isomer of EPI named epiisopilosine [26]. The vibrational assignments were supported by DFT calculations performed in this work as can be seen in Table 3 . The very strong band at 1769 cm−1 is assigned to C = O stretching of the lactone ring, in the FT-IR spectrum shown in Figure 6 , and corresponds to the medium intensity band at 1758 cm−1 in the FT-Raman spectrum, shown in Figure 7 . The FT-Raman spectrum reveals three strong bands of EPI around 1602–1568 cm−1 ( Figure 7 ) characteristic of the C-C stretching vibrational mode of benzene and imidazole rings; these bands are weak in the FT-IR spectrum. On the other hand, the bands around 1524–1494 cm−1, attributed to modes related to imidazole and lactone groups (see Table 3 ) are present in the FT-IR spectrum but practically absent in the FT-Raman spectrum. Well-defined band at 1385 cm−1 assigned to the scissoring C9-O1-H vibrational mode of the secondary alcohol group attached to the organic molecule is observed in both spectra. The strong band at 1004 cm−1 (FT-Raman) is attributed to the C-C-C trigonal bending of benzene, while the bands around 990–930 cm−1 are assigned to the bending of the lactone ring, in both FT-IR and FT-Raman spectra. Several bands attributed to the vibration of C-H group of benzene and imidazole rings can be observed in the 900–730 cm−1 range, as listed in Table 3 . The low frequency region is dominated by bands associated to vibrations involving C-C-C of benzene, imidazole (600–500 cm−1) and also to all the EPI structure bending modes (430–400 cm−1).
Figure 6. Epiisopiloturine FT-IR spectra: A) experimental and B) calculated.

Figure 7. Epiisopiloturine FT-Raman spectra: A) experimental and B) calculated.

Table 3. Infrared (IR) and Raman wavenumbers (cm−1) of solid state EPI.
| Epiisopiloturine | Assignment | ||
| Experimental | Calculated | ||
| IR | Raman | ||
| 412 | 407 | δ (all structure) | |
| 433 | 438 | δ (all structure) | |
| 498 | 499 | 501 | C-C-C out of plane bending (benzene) |
| 526 | 525 | 528 | C-C-C out of plane bending (benzene) |
| 567 | 568 | 575 | C-C-C in-phase bending (benzene) |
| 621 | 620 | 628 | ring puckering (imidazole) |
| 634 | C-C-C in-phase bending (benzene) | ||
| 644 | 657 | sc (O3-C7-C8) | |
| 663 | 664 | 670 | C-C-C in-phase bending (benzene) |
| 712 | 711 | 713 | C-C-C in-phase puckering (benzene) |
| 729 | 717 | C-H out-of-plane in-phase (benzene) | |
| 758 | 730 | 740 | C-H out of plane bend (imidazole) |
| 775 | 783 | C-H out-of-plane in-phase (benzene) | |
| 800 sh | 796 | 804 | ring puckering (imidazole) |
| 831 | 821 | C-H out of plane bend (imidazole) | |
| 846 | 854 | r CH2 (C4) | |
| 893 | 894 | 900 | r CH2 (C4), r CH2 (C6) |
| 910 | 911 | 923 | C-H out of plane (benzene), r CH2 (C4) |
| 932 | 932 | 944 | r CH2 (C4), β (lactone) |
| 984 | 993 sh | 983 | β (lactone) |
| 1004 | 1016 | C-C-C trigonal bending | |
| 1022 | 1025 | 1038 | β (lactone), r CH2 (C4) |
| 1046 | 1048 | C-H in plane bending (benzene) | |
| 1063 | 1073 | 1076 | w CH3 (C16) |
| 1088 | 1078 | w CH3 (C16), C-H in plane bending (benzene) | |
| 1105 | 1107 | 1116 | t CH2 (C4), γ (lactone) |
| 1148 | 1148 | 1145 | w CH3 (C16) |
| 1169 | 1156 | 1165 | t CH2 (C4), γ (lactone) |
| 1184 | 1172 | 1182 | β (lactone) |
| 1207 | 1198 | C-H in plane bending (benzene), t CH2 (C4) | |
| 1197 | 1210 | C-H in plane bending (benzene) | |
| 1236 | 1220 | t CH2 (C4), t CH2 (C6) | |
| 1254 | 1235 | 1244 | β (lactone) |
| 1254 | 1256 | t CH2 (C6), C-H in plane bending (imidazole) | |
| 1263 | 1264 | 1266 | t CH2 (C4), t CH2 (C6), C-H in plane bending (imidazole) |
| 1286 | 1286 | 1292 | w CH2 (C4) |
| 1312 | 1312 | 1315 | C-C stretching (benzene) |
| 1330 | 1318 | C-N stretching (imidazole) w CH2 (C4) | |
| 1348 | 1349 | C-C stretching (benzene) | |
| 1362 | 1354 | w CH2 (C4) | |
| 1385 | 1387 | 1376 | sc(C9-O1-H), νas(C8-C9-C10) |
| 1423 | 1426 | 1418 | νs(N1-C1-N2), w CH3 (C16), νas(C16-N2-C1) |
| 1450 | 1445 | 1455 | w CH3 (C16) |
| 1472 | 1472 | 1484 | C-C stretching (benzene) |
| 1494 | 1486 | sc CH3 (C16) | |
| 1508 | 1520 | sc CH2 (lactone), w CH3 | |
| 1524 | 1535 | νas N-C-N (imidazole) | |
| 1568 | 1568 | 1592 | ν C-C (imidazole) |
| 1587 | 1586 | 1627 | ν C-C (benzene) |
| 1602 | 1647 | ν C-C (benzene) | |
| 1769 | 1758 | 1849 | ν(C = O lactone) |
Calculated vibrational wavenumbers (cm-1) for the isolated EPI molecule. A tentative assignment of the observed vibrational modes is also shown. See text for theoretical details. ν = stretching, δ = bending, β = bending in plane, γ = bending out of plane, r = rocking, τ = twist, sc = scissoring, ω = wagging, νs = symmetric stretching, νa = antisymmetric stretching, sh = shoulder.
The TGA-DSC-MS curves of the isolated EPI are shown in Figure 8 , where three events can be observed. The first event (an endothermic process) occurs at 225°C, and can be attributed to the melting process of the imidazole alkaloid ( Figure 8A ). Partly according to Voigtlander et al [6], the melting point of EPI is about 218–219°C when measured using a copper block. In line with the DTG-MS curves ( Figure 8B ), EPI is decomposed around 230–350°C (ca. 87 wt. %), in air atmosphere, producing water (m/z = 18), carbon monoxide (m/z = 28) and carbon dioxide (m/z = 44) molecules. The third event observed in the temperature range between 360–695°C (12 wt.%) is related to the decomposition of the remaining organic molecule, producing H2O and CO2. Under the experimental conditions used in this work, no other released gases were identified. A residual product of 0.5 wt.% can be attributed to some impurities from the raw material and/or introduced during the process of EPI isolation.
Figure 8. Epiisopiloturine TGA-DSC (A) and DTG-MS (B) curves under air atmosphere.

Conclusion
This work describes, for the first time, an industrial process to obtain EPI in high purity. The treatment with low-polarity solvents combined with HPLC technique allowed the isolation of the EPI alkaloid from jaborandi leaves. The technique of ESI/Ion Trap allowed attesting that its purity is higher than 98%, besides fragment characteristics of imidazole alkaloids produced by MSn. Single crystal X-ray diffraction data has shown the structure of the EPI molecule as well as its arrangement in solid state. The 1H and 13C NMR, IR and Raman spectroscopy data were supported by DFT simulations. Each assay had their contribution to characterize the EPI, allowing the interpretation of the experimental data which shows the integrity of the molecule isolated by the procedures of extraction and purification presented in this paper. According to TGA-DSC data, EPI melts at 225°C, and undergoes decomposition mainly in the 230–350°C range under air atmosphere.
The results presented in this work contribute to the advance of the isolation of EPI and provide a set of structural, spectroscopic and thermal properties of the alkaloid molecule. This study supports efforts to develop EPI as a new antiparasitic agent.
Supporting Information
Experimental (black) and theoretical (red) 1H NMR EPI spectra.
(TIF)
Experimental (black) and theoretical (red) 13C NMR EPI spectra.
(TIF)
Chemical Shift Assignments.
(DOCX)
Funding Statement
The authors would like to thank Anidro do Brasil Extrações S.A. and Phytobios LTDA for grants and supporting in access the process, waste and all industrial way. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dewick PM (2008) Medicinal Natural Products – A Biosynthetic Approach. New Phytologist. Chichester: John Wiley & Sons, Vol. 140: 763–772. [Google Scholar]
- 2. Barbosa-Filho JM, Piuvezam MR, Moura MD, Silva MS, Lima KVB, et al. (2006) Anti-inflammatory activity of alkaloids: a twenty-century review. Rev Bras Farmacogn 16: 109–139. [Google Scholar]
- 3. Nusair S, Rubinow A (1999) The use of oral pilocarpine in xerostomiaand sjögren's syndrome. Semin Arthritis Rheum 28: 360–367. [DOI] [PubMed] [Google Scholar]
- 4. Hendrickson RG, Morocco AP, Greenberg MI (2004) Pilocarpine toxicity and the treatment of xerostomia. Int J Emerg Med 26: 429–432. [DOI] [PubMed] [Google Scholar]
- 5. Santos AP, Moreno PRH (2004) Pilocarpus spp.: A survey of its chemical constituents and biological activities. Braz J Pharm Sci 40: 115–137. [Google Scholar]
- 6. Voigtländer H-W, Balsam G, Engelhardt M, Pohl L (1978) Epiisopiloturin, ein neues Pilocarpus-Alkaloid. Arch Pharm (Weinheim) 311: 927–935. [Google Scholar]
- 7. Veras L, Guimaraes M, Campelo Y, Vieira M, Nascimento C, et al. (2012) Activity of Epiisopiloturine Against Schistosoma mansoni. Curr Med Chem 19: 2051–2058. [DOI] [PubMed] [Google Scholar]
- 8.Leite JRSA, Miura LMCV, Lima DF, Carneiro SMP, Carvalho FAA, et al.. (2011) Processo de obtenção da epiisopiloturina e sua aplicação no combate à infecções parasitárias. BR PI0904110–9.
- 9. Steinmann P, Keiser J, Bos R, Tanner M;, Utzinger J (2006) Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis 6: 411–425. [DOI] [PubMed] [Google Scholar]
- 10. Gryseels B, Polman K, Clerinx J, Kestens L (2006) Human schistosomiasis. Lancet 368: 1106–1118. [DOI] [PubMed] [Google Scholar]
- 11.Schistosomiasis Report website. Available: http://www.who.int/schistosomiasis/en/index.html. Accessed 21 May 2013.
- 12. Otwinowski Z, Minor WJ, Carter C, Sweet R (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- 13. Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr, A, Found Crystallogr 64: 112–122. [DOI] [PubMed] [Google Scholar]
- 14. Hohenberg P, Kohn W (1964) Inhomogeneous Electron Gas. Phys Rev 136: B864–B871. [Google Scholar]
- 15. Kohn W, Sham L (1965) Self-Consistent Equations Including Exchange and Correlation Effects. Phys Rev 140: A1133–A1138. [Google Scholar]
- 16. Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37: 785–789. [DOI] [PubMed] [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2009) Gaussian 09, Rev A.1. Wallingford CT: Gaussian, Inc. Available: http://www.gaussian.com/. Accessed 21 May 2013.
- 18. Wolinski K, Hinton JF, Pulay P (1990) Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc 112: 8251–8260. [Google Scholar]
- 19. Cheeseman JR, Trucks GW, Keith TA, Frisch MJ (1996) A comparison of models for calculating nuclear magnetic resonance shielding tensors. J Chem Phys 104: 5497–5509. [Google Scholar]
- 20. Casabianca LB, De Dios AC (2008) Ab initio calculations of NMR chemical shifts. J Chem Phys 128: 052201–10. [DOI] [PubMed] [Google Scholar]
- 21.Johnson CK, Burnett MN (1996) ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations. Oak Ridge National Laboratory Report ORNL-6895. Available: http://www.ornl.gov/sci/ortep/. Accessed 21 May 2013.
- 22.Silverstein R, Webster F, Kiemle D (2005) Spectrometric Identification of Organic Compounds New York: Wiley. 204–244.
- 23.Varsanyi G, Szoke S (1969) Vibrational Spectra of Benzene Derivatives. New York and London: Academic Press. 430 p.
- 24.Dollish FR, Fateley WG, Bentley FF (1974) Characteristic Raman Frequencies of Organic Compounds. 1st ed. New York: John Wiley & Sons. 443 p.
- 25. Markham LM, Mayne LC, Hudson BS, Zgierski MZ (1993) Resonance Raman studies of imidazole, imidazolium, and their derivatives: the effect of deuterium substitution. J Phys Chem 97: 10319–10325. [Google Scholar]
- 26. Bento RRF, Silva LE da, Faria JLB, Freire PTC, De Oliveira MCF, et al. (2010) Comparative vibrational spectra of pilosine and epiisopilosine crystals. Braz J Phys 40: 217–223. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental (black) and theoretical (red) 1H NMR EPI spectra.
(TIF)
Experimental (black) and theoretical (red) 13C NMR EPI spectra.
(TIF)
Chemical Shift Assignments.
(DOCX)