

Summary
Self versus non-self discrimination is at the core of T lymphocyte recognition. To this end, αβ T-cell receptors (TCRs) ligate ‘foreign’ peptides bound to major histocompatibility complex (MHC) class I or class II molecules (pMHC) arrayed on the surface of antigen-presenting cells (APCs). Since the discovery of TCRs ~30 years ago, considerable structural and functional data have detailed the molecular basis of their extraordinary ligand specificity and sensitivity in mediating adaptive T-cell immunity. This review focuses on the structural biology of the Fab-like TCRαβ clonotypic heterodimer and its unique features in conjunction with those of the associated CD3εγ and CD3εδ heterodimeric molecules, which, along with CD3ζζ homodimer, comprise the TCR complex in a stoichiometry of 1:1:1:1. The basis of optimized TCRαβ docking geometry on the pMHC linked to TCR mechanotransduction and required for T-cell signaling as well as CD4 and CD8 co-receptor function are detailed. A model of the TCR ectodomain complex including its connecting peptides suggests how force generated during T-cell immune surveillance and at the immunological synapse results in dynamic TCR quaternary change involving its heterodimeric components. Potential insights from the structural biology relevant to immunity and immunosuppression are revealed.
Keywords: T-cell receptor, adaptive immunity, CD4, CD8, structural immunology, mechanoreceptor
Introduction
Adaptive immunity endows mammals and other jawed vertebrates with precursors of T (thymus-derived) and B (bone marrow-derived) lymphocytes able to generate a repertoire of clonotypic antigen receptors [T-cell receptors (TCRs) and B-cell receptors (BCRs)] of immense diversity from somatic rearrangements of variable gene segments (VDJ and VJ recombination). Spatio-temporally controlled differentiation and selection processes of those cells shape two complementary lineages of the immune system, offering protection with exquisite specificity, sensitivity, and long-term memory.
Key discoveries during the last 50 years have unraveled the cellular and molecular nature of adaptive immunity. In the 1960s, T and B lymphocytes were identified and their interactions shown to be essential for antibody production (1, 2). The basic paradigm of immunoglobulin (Ig) gene rearrangements that generate antibody diversity was revealed in 1976 (3). The ‘dual’ specificity of T cells for foreign peptide and self-major histocompatibility complex (MHC) inferred by functional studies was discovered and clearly noted to be distinct from the ‘single’ specificity of antibody recognition of foreign proteins (4, 5). This realization then led to an intense effort to understand the molecular puzzle represented by self versus non-self discrimination and the receptor and ancillary molecules on T cells responsible for this unusual recognition.
Initial studies suggesting the existence of an ‘I-J-specific’ suppressor factor secreted by T cells and TCR specificity achieved through Ig genes were refuted. Rather, the discovery of how to expand T cells in vitro, via IL-2-dependent T-cell cloning (6), in conjunction with monoclonal antibody (7) and flow cytometry screening (8) technologies plus in vitro functional analyses were decisive in molecular identification for the long sought-after TCR. The key breakthroughs came in the early 1980s with the identification in human of a clonotypic disulfide-linked heterodimer, the Ti αβ TCR heterodimer, which together with CD3 molecules, were essential for peptide/MHC complex recognition and cellular activation (9–14). Biochemical evidence showed that, similar to immunoglobulin (Ig) molecules, both Ti α and β chains possessed variable and constant regions (9, 10). A comparable αβ Ti was soon identified also in the mouse in 1983, with similar cognate immune recognition features (15, 16). Those murine studies supported an earlier conjecture that a tumor-specific marker on mouse T-lymphoma cells might be TCR-related (17). Within two years, cDNAs for TCRαβ subunits were obtained beginning with cloning efforts of Davis and Mak (18–20) in mouse and human, respectively, identifying the β chain as shown by protein sequence (21). Collectively, these results confirmed the clonotypic nature of the TCRαβ heterodimer first identified biochemically. These studies showed that TCR combinatorial diversity was generated by the same type of site-specific gene recombination mechanisms as with Ig genes but without somatic hypermutation and led to identification of a second type of TCR, the γδ TCR (reviewed in 3).
CD4 and CD8, surface molecules identified during the same period, were recognized as co-receptors that optimize TCR recognition and T-cell activation via interaction with monomorphic segments of MHC class II and I molecules, respectively (22, 23). A few years later, the dual recognition puzzle was solved when it was shown that MHC class I and class II proteins bound foreign and self-peptides derived from degradation of intracellular or exogenous proteins and that such complexes could be recognized by the TCR (reviewed in 24). Structures of peptides complexed with MHC molecules (pMHC) then followed (25, 26), as did structures of αβ TCR heterodimers in complex with pMHC ligands (27–30).
It is now known that the αβ TCR is a multimeric transmembrane complex composed of a disulfide-linked antigen-binding clonotypic heterodimer in non-covalent association with the signal-transducing CD3 subunits (CD3εγ, CD3εδ, and CD3ζζ) (31–33). TCR signaling via CD3 dimers evokes T-cell lineage commitment and repertoire selection during development, maintains the peripheral T-cell pool, and further differentiates naive T cells into effector or memory cell populations upon immune stimulation.
This review focuses on selected aspects of the structural biology of the TCR complex. In particular, we consider those novel structural features of the TCRαβ heterodimer and their functional importance, TCRαβ heterodimer docking onto pMHCI and pMHCII ligands, structures of the CD3 heterodimeric ectodomains, and a model of TCR complex topology. As these structural features can be rationalized in view of recent knowledge that the TCR is a mechanosensor, the evidence for TCR mechanotransduction function upon pMHC ligation is reviewed. The bidentate interaction of CD4 or CD8 co-receptors with pMHC in concert with the TCRαβ complex coordinates p56lck-mediated phosphorylation of the exposed immunoreceptor tyrosine-based activation motifs (ITAMs) of the CD3 cytoplasmic tails. Accessibility of CD3 ITAMs to tyrosine kinase appears to require their release from the inner leaflet of the plasma membrane post-TCR ligation (34–36). Downstream signaling then follows (33). Lastly, we shall discuss the implications of these structural biology insights for translational medicine.
The novel structural features of TCRαβ heterodimers and their functional significance
In the mid-1990s, the first wave of publication of TCRαβ heterodimer structures and their complexes with cognate ligand, pMHC appeared (27–29, 37). As expected from their primary sequence analysis, the ectodomains of these cell surface receptors form an Fab-like structure with each subunit consisting of one variable and one constant Ig-like domain. In our MHC class I-restricted N15 TCRαβ structure (specificity: VSV8 octapeptide bound to H-2Kb), a monoclonal antibody (mAb) H57 Fab was used to aid in the crystallization (PDB code, 1NFD) (37) (Fig. 1A). This complex provided us with an opportunity to directly compare structures of a TCRαβ heterodimeric ectodomain with that of an Fab, the antigen recognition fragment of a BCR. From that comparison, we observed four major features that make a TCRαβ heterodimer (hereafter termed TCRαβ) distinct from an Fab. These differences can be appreciated in Fig. 1 (panels B and C) (37).
Fig. 1. Structure of N15 TCRαβ ectodomains in complex with an Fab fragment of the H57 mAb.

(A) The complex structure with H57 bound to the protruding FG loop of the N15 Cβ domain (adapted from 37). (B) The structure of the N15 αβ ectodomains. The α and β subunits are represented in the same color as in (A). The inset offers the detailed internal structure of the Cβ FG loop. (C) The structure of H57 Fab fragment. Note the overall narrow shape of the Fab in comparison to the TCRαβ. The β strands and the CDR2 loop of VL domain are labeled for comparison with that of the N15 Vα domain depicted in (B).
TCRαβ has a relatively flat ligand-binding surface suitable for pMHC interaction (Fig. 1B), whereas Fabs often have a concave surface for antigen recognition (Fig. 1C). This difference stems from the fact that antibodies recognize antigens with a multitude of diverse shapes. A cavity formed by six complementary-determining region (CDR) loops can better complement the epitope shape of myriad antigens. In contrast, the binding partners of TCRs are universally linear antigenic peptides loaded onto an MHC molecule with two flanking helices exposed for interaction. This pMHC TCR-contacting face is relatively flat. Second, in general, the C module of the TCRαβ bends more than that of an Fab and has an asymmetric arrangement relative to the V module, with the Cβ domain significantly angled more acutely toward the Vα domain. This disposition makes the TCR wider than an Fab (Fig. 1). Of note, the Cα domain substantially deviates from a canonical Ig-like domain. Its outside face is even difficult to be designated as a β sheet. Instead, there is a helical F strand and a loosely packed C strand (Fig. 1B). This peculiar Cα domain was first noted by Garcia and his coworkers (27). The asymmetric Cα-Cβ module is topologically conserved among the first three crystallographically solved structures, 2C TCR (27), A6 TCR (28), and N15 TCR (37). The module was also predicted to be preserved in the pre-TCR in the form of pTα-Cβ domain-domain interaction (37). This prediction was confirmed by a recently solved structure of the pre-TCR pTα-β chain heterodimer (38). The major new discovery of the pre-TCR structure is that pTα has a more regular Ig-like fold. Our follow-up work suggests that this conserved C module topology is a critical structural feature for TCR signaling as will be described below. Third, in contrast to a typical V-set Ig-like domain that has an ABED β sheet and a C″C′CFG β sheet, the TCR Vα domain C″ β strand has translocated, switching from a canonical position to the opposite sheet and thereby creating an ABEDC″ sheet and a C′CFG β sheet (Fig. 1B). This translocation makes the Vα CDR2 C′C″ loop more parallel to the α2 helix of the MHC molecule. As a consequence, Vα CDR2 creates additional contacts with a pMHC ligand. Fourth, the Cβ domain carries a unique 12 amino acid residue insertion, forming an unusually long rigid protruding FG loop (Fig. 1B). The functional importance of this feature will be elaborated on below.
Diagonal TCRαβ docking onto pMHCI offers an optimal binding geometry for adaptive recognition
It has now been well established that TCRαβ heterodimers dock onto pMHCI using a common diagonal mode such that the Vα domain contacts MHCI α2 helix and the Vβ domain overlays the α1 helix. The initial representative TCRαβ footprints onto MHC are shown in Fig. 2A,B. One interesting question is what determines the common docking mode? Garboczi (28) first noted two high points on the TCR-binding surface of MHCI molecules that force the TCRαβ to dock diagonally so that the TCR can best achieve contacts with the antigenic peptide in the MHCI groove (Fig. 2C). We superimposed the first three TCRαβ/pMHCI complex structures using the MHC molecule as a reference to demonstrate this common docking mode (Fig. 2A, B). Relative to pMHCI, we observed that different TCRs have their positions twisted, tilted, and shifted with respect to one another (29). The 2C TCR in 2C-dEV8-Kb complex, for example, appears to twist the most and hence, assumes the more diagonal orientation with respect to the antigen-binding groove of pMHCI (Fig. 2B). The A6 TCR in A6-TAX-HLA-A2 complex, on the other hand, is so tilted that its Vβ subunit can barely touch pMHCI via CDR1 and CDR2 (29) (Fig. 2A). Furthermore, we indicated that the two high points in the MHCI presentation platform helices are the result of the inherent left-handed twist of the large 8-stranded β sheet that forms the bottom of the antigen-binding groove of the MHC molecule (29) (Fig. 2C). When the two helices run across this twisted β sheet, the uneven β sheet platform disrupts the two helices at these two high points, segmenting them into two components each. Note, however, that the two helices break in relatively different positions. Whereas the α1 helix breaks near its N-terminal end, the helix α2 (or helix β1 in MHCII molecule) has a break closer to the middle (Fig. 2B). We discuss the extremely important functional significance of this difference later. Since the β sheet floor in the antigen-presenting platform is a common feature of every MHC molecule in humans and mouse, whether class I or class II, it enforces a more or less common TCR diagonal docking onto pMHC (Fig. 2C).
Fig. 2. TCRαβ dockings onto pMHCI molecules.

(A, B) Side and top views, respectively, of three TCR/pMHCII complexes (adapted from 29). For clarity, only one MHCI peptide-binding groove is shown. The N15 TCR is in white (PDB 1NFD), the 2C TCR in red (PDB 2CKB), and the A6 TCR in green (PDB 1QSE). Relative to the MHC molecule, the three TCR molecules have a similar diagonal docking with Vα and Vβ domains on α2 and α1 helices, respectively. The orientation difference can be described by parameters of twist, tilt, and shift. (C) The TCR docking constraint imposed by the MHC molecule is shown by the two arrows indicating the high points on α1 and α2 helices. The helical breaks are caused by the inherent left-handed twist of the β sheet seen at the bottom of peptide-binding groove.
A separate question needs to be addressed with regard to docking polarity. Why does the Vα domain always contact an MHC molecule’s α2 helix (or β1 helix in case of MHCII) and the Vβ domain overlay the MHCI α1 helix, rather than the other way around? There is no definitive answer at the moment. Suggested germline-encoded V docking preferences (39, 40) and/or early thymic selection may mandate such a preference (41).
It must also be noted that there are exceptional TCRαβ docking topologies, manifesting a binding mode significantly different from the conventional docking approach. The first striking example is the structure of a human autoimmune TCR from a patient with multiple sclerosis, Ob.1A12, in complex with HLA-DR loaded by a self-peptide [residues 85–99 of myelin basic protein (MBP)] (Ob.1A12 in dark blue color in Fig. 3) (42). In this complex, the TCRαβ largely shifts toward the N-terminal part of the peptide (Fig. 3A) and is also tilted against the β helix of HLA-DR (Fig. 3B). Strikingly, this TCR actually sits over the high point of the α helix of HLA-DR in an unfavorable binding mode. Overall, this orientation results in a reduced TCRαβ interaction surface with substantially smaller buried area and hence weaker binding affinity compared with a conventional TCR-pMHC complex. It is probable, as postulated by the authors, that the unique CDR3 interaction permits autoreactive T cells to escape intrathymic deletion during negative selection and thereby induce an inflammatory brain disease (42).
Fig. 3. Unconventional TCR docking orientations.
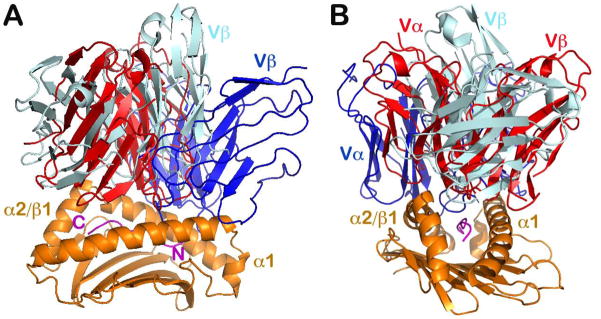
The two examples given are the human autoimmune receptor Ob.1A12 in complex with HLA-DR (PDB 1YMM) and NKT TCR in complex with MHCIb, CD1d (PDB 2PO6). (A, B) Two views rotated 90° vertically with respect to each other. The two unconventional TCR/MHC structures are superimposed together with the conventional N15/H2-Kb structure (PDB 1NFD) based on their respective MHC peptide-binding groove. For clarity only one MHC molecule and each TCR V module are shown. N15 TCR is in red, Ob.1A12 is in dark blue and NKT TCR is colored in silver. N- and C-termini of peptide are labeled in magenta in (A). The Ob.1A12 shifts more towards the peptide’s N-terminal is seen in A, whereas the NKT TCR appears more parallel with the MHC groove, as best seen in (B).
Another unusual docking is observed in the structure of a natural killer T cell (NKT) TCRαβ in complex with the MHC class Ib molecule, CD1d loaded by the lipid α-GalCer (43). In this case, the NKT TCRαβ binds to CD1d in a parallel rather than diagonal mode (NKT TCRαβ in silver color best viewed in Fig. 3B) and also shifts more to the C-terminal part of the peptide (Fig. 3A), as opposed to the autoimmune Ob.1A12 TCR. This docking mode guarantees that the highly conserved NKT TCR α-chain dominates the TCR ligand binding surface (estimated to be two thirds of total buried surface area contributed by the α-chain), and ensures its ‘preconfigured’ innate-like contribution to immunity. Notably, the NKT TCR has an even smaller overall contact area and weaker binding affinity compared to conventional adaptive αβTCRs.
Despite the very unconventional docking modes exemplified above, one key rule remains intact: the Vβ domain contacts the α1 helix and the Vα domain contacts the α2 helix (or the β1 helix in the case of MHCII) (Fig. 3B). In that sense, any ‘alternative’ binding topology can still be regarded as a variation on the conventional TCR docking mode but with a more dramatic twist and/or shift with respect to pMHC. These ‘unconventional’ TCRs may even sit on or near the high point of the helices of the MHC. For adaptive TCRαβ to recognize an antigenic foreign peptide leading to productive T-cell activation, the conventional docking mode is most suitable. In fact, more recent structural and functional analysis demonstrated that a single TCRαβ may bind with stronger affinity in solution to an MHC molecule loaded with a non-stimulatory peptide than the same MHC loaded with an agonist peptide (44). The functionally unproductive non-agonist binding engenders a more parallel docking mode in the TCR pMHC crystal structure than does the agonist in the complex structure. This paradox becomes explicable when considering the mechanotransduction function of TCRαβ described below.
The structural features of TCRαβ-pMHCII complexes
Fig. 4A shows the first TCRαβ/MHC class II structure, the single-chain D10 TCR V module in complex with the murine class II MHC molecule I-Ak loaded with a peptide derived from conalbumin (CA), dubbed scD10-CA/I-Ak (30). Class I and class II MHC molecules have evolved to facilitate T-cell detection of pathogens residing in distinct intracellular compartments (45). Accordingly, TCRs are restricted to these two major classes of MHC molecules. One might have expected to see structural distinctions among TCRs recognizing peptides bound to the two different classes of MHC molecules. However, when the structure of first class II MHC-restricted D10 TCR VαVβ was determined, it was striking to observe no significant difference with the known structure of a class I MHC restricted TCRαβ, the 2C TCR (30). Fig. 5A represents an overlay of the V module of the 2C TCRαβ onto the V module of D10 TCRαβ. The RMSD value for superimposed Vα domain’s 110 Cα atoms is 0.98Å, while that for the Vβ domain’s 107Cα is 0.72Å, respectively (30). If the two Vα domains are superimposed, then the two Vβ domains only differ by 3.7° (30). This analysis clearly demonstrates no intrinsic structural differences between the TCR recognition elements binding to pMHCI and pMHCII ligands. Likewise, Fig. 5B offers two views of superimposed MHCI and MHCII molecules. As shown, the overall configuration of peptide presenting platform is also very similar despite a substantially different chemical composition of the two MHC molecules (26).
Fig. 4. Structure of a TCR/pMHCII complex, the scD10-CA/I-Ak (PDB 1D9K).

(A) The complex structure with TCR V module in green (Vα) and cyan (Vβ), MHCII in yellow (α subunit) and orange (β subunit) and CA peptide in magenta. The N- and C- termini of the TCR contacting portion of the peptide are indicated by the P-1 and P8 labels, respectively. (B) The peptide-binding groove of I-Ak is illustrated. The side chain of conserved residues from MHC α and β subunits make extensive hydrogen bonds to the main chain of the peptide such that the 16 amino acid residue peptide adopts a fixed extended conformation on the groove.
Fig. 5. Absence of intrinsic structural differences between the two classes of TCR as well as MHC molecules among their interacting domains.
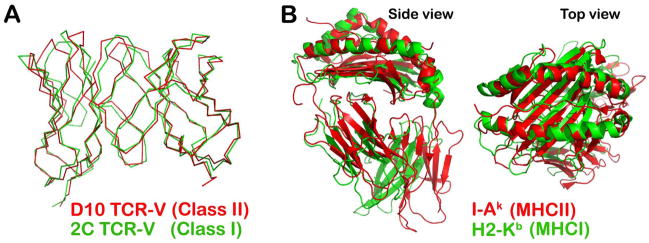
(A) The superposition of the V modules of the D10 TCR (PDB 1D9K) and 2C TCR (PDB 2CKB) in Cα is shown using a Cα atom skeleton drawing. (B) Side view (left panel) and top view (right panel) of superimposed I-Ak (PDB 1D9K) and H2-Kb (PDB 2CKB) molecules represented as ribbon drawings. The two peptide-binding grooves overlay well, despite their very different chemical compositions.
It was known from functional data that peptides presented by MHC molecules may be as long as 25 amino acid residues (46–48). Direct structural analysis of an MHCII molecule rationalized the biological data and demonstrated how the long peptide is held in an open-ended groove of an MHC class II molecule (26). In our scD10-CA/I-AK structure, the CA peptide used for crystallization is 16 residues long (designated as from position P-3 to P13). The structure of the entire peptide bound to MHCII molecule I-AK was unambiguously defined. However, the D10 TCR interaction was restricted to the nine amino acid long P-1 to P8 segment, with the other peptide residues lacking contacts with the TCR (30) (Fig. 4A). This 9-residue length peptide antigen-binding interface is the general rule for TCRαβ/pMHCII complexes. As first noted by Stern and Wiley (49) and illustrated in Fig. 4B for CA/I-AK, the peptide bound to an MHC class II molecule assumes a fixed, extended conformation, being held in the groove through a network of hydrogen bonds between the peptide’s mainchain atoms and sidechains of conserved residues of MHCII molecules, such as Asn62 and Asn69 of the α chain and Trp61 and Asn82 of the β chain. This binding contrasts with that of a peptide loaded onto an MHCI molecule, where a peptide has its N- and C-termini fixed at the same distance by amino terminal and C-terminal residue binding pockets (the distance between Cα atoms of N- and C-terminal residues is 22Å). For MHCI-bound peptides, the middle portion of the peptide bulges in a sequence dependent conformation that is impacted by length of the peptide (generally 8–11 residues but with a 13–14 residue upper limit) with longer length peptides manifesting a greater bulge (50). Whereas an MHCII-restricted TCR binding specificity is solely dependent upon the exposed peptide sidechains and the TCR footprint, by contrast, an MHCI-restricted TCR interrogates both mainchain conformation as well as sidechain residues as it docks to pMHCI. That a substantially bulged peptide bound to MHCI makes key energetic contribution to TCR binding is consistent with this view (51).
When we obtained the scD10-CA/I-AK structure, one feature of the complex was the orthogonal docking of TCRαβ onto the pMHCII, in contrast with the previously observed diagonal docking of three class I TCRαβ/pMHC structures. We attributed this difference to the expanded high point on the α1 helix of MHCII molecules (30). However, when additional TCR-pMHCII complex structures were solved, the orthogonal geometry became less clear. Currently, we have more than two dozen TCRαβ/pMHC complex structures in the database which gives us an opportunity to revisit TCR docking differences onto pMHCI vs. pMHCII. Fig. 6 is a side-by-side overlay of representative complex structures with superposition based on the MHC molecules. For clarity, only a single MHC molecule is shown, and the TCR V modules of ligated TCRαβ are displayed in the figure. Overall, the TCRαβ/pMHCII complexes tend to be more orthogonal, whereas class I complexes manifest a more variable TCRαβ docking geometry. With respect to the latter, in addition to the typical diagonal docking of 2C TCR (blue-grey in Fig. 6, left panel; PDB code 2CKB), there is also a more orthogonal example (yellow in Fig. 6, left panel; PDB code 2AK4). The structure of the one extreme outlier autoimmune TCRαβ/pMHCII (dark blue in Fig. 6, right panel; PDB code 1YMM) is also included here for comparison. We speculate that the orthogonal binding mode may be advantageous for TCR mechanotransduction upon pMHCII interaction, as described in the CD4/TCR/pMHCII orientation section below.
Fig. 6. Overlay of selected TCR/pMHCI (class I) and TCR/pMHCII (class II) complexes reveals a more conserved docking mode on pMHCII.
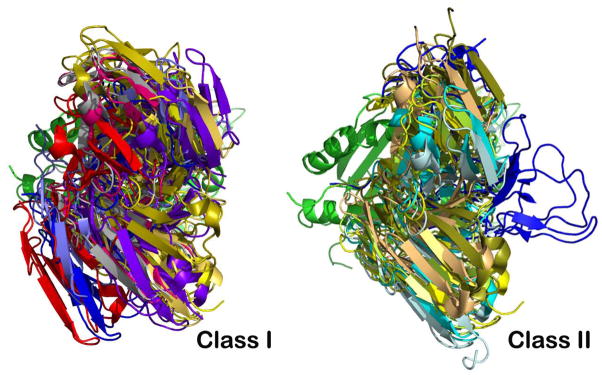
The superposition is based on the MHC molecules’ peptide-binding groove. For clarity, only TCR V modules and one MHC groove are depicted. Left panel: Selected class I TCR molecules are 2CKB in blue-grey, 1QSE in purple, 3SJV in dark blue, 1QGA in pink, 1BD2 in yellow, 2VLJ in silver, 2AK4 in gold, 3FFC in light blue and 1MT5 in red. Right panel: Selected class II TCR molecules are 1D9K in yellow, 1FYT in cyan, 1U3H in pale-green, 1YYM in dark blue, 3C5Z in sand, 3RDT in bluewhite, and 2IAM in smudge.
The functional significance of the unique FG loop of TCR Cβ domain
The αβ TCR is a complex consisting of the pMHC-binding αβ heterodimer in non-covalent association with CD3εγ, CD3εδ and CD3ζζ signaling dimers in a 1:1:1:1 stoichimetry. Structural topological detail of how the TCR complex signals is yet to be further resolved (52, 53). We noticed early on that one striking structural feature of TCR molecule is the asymmetric disposition of its C module mentioned above. The Cβ domain is about 55Å long and bends much more acutely towards the Vβ domain, compared to the 40Å-long Cα domain to Vα domain. Remarkably, a 12-residue long FG loop uniquely protrudes out of Cβ domain, as shown in Fig. 1B. This is a well-structured loop. The center is a conserved Trp residue which forms bifurcated hydrogen bonds with mainchain carbonyl oxygens from Ser229 and Pro230. These hydrogen bonds fix the sidechains of the Trp 225, which in turn makes extensive hydrophobic contacts with residues Leu219, Pro226, and Pro232 (Fig. 1B, inset). The Leu219 and Pro232 are themselves conserved residues located at strand termini to form the last hydrogen bond pair between the F and G strands. These interactions lend rigidity to the loop. This rigid FG loop forms a canopy and side-wall of a cavity created by the asymmetric disposition of TCR Cβ and Cα ectodomains. The CD and EF loops of Cα domain forms another wall (Fig. 1B). Glycans emanating from CαN121, CαN185, CβN186 and CβN236 surround the cave-like structure, as elaborated below. We proposed that this cavity has sufficient size to accommodate one non-glycosylated CD3ε ectodomain (37). Epitope mapping of an MHCI-restricted TCR suggested that one of the CD3ε subunit lies in close proximity to this Cβ FG loop (54).
Follow-up experiments using TCR transgenic mice bearing either an intact or FG loop-deleted β chain showed that the loss of the Cβ FG loop attenuates negative selection of thymocytes and affects cognate peptide-mediated activation of mature T cells, confirming the functional importance of Cβ FG loop (55). Subsequent studies using T-cell transfectants and thymic reconstitution analysis revealed that the unique Cβ FG loop appendage primarily controls αβ T-cell development through intrathymic selection processes at TCR and pre-TCR levels (56). In addition, on mature T cells, deletion of the Cβ FG loop, while maintaining TCR expression and surface copy number, dramatically reduced the functional sensitivity of TCR-mediated activation. That is, 1000 to 10,000-fold higher molar concentrations of cognate peptide were required to stimulate cytokine production from Cβ FG loop-deleted T cells versus wildtype TCRs (56). We attribute this attenuation to loss of efficient mechanotransduction from the TCRαβ to CD3εγ heterodimers, as described below.
Our proposal of the critical association of TCR Cβ FG loop with the CD3εγ gained further support from analysis of CD3 sequence divergence and TCR evolution in jawed vertebrates (Gnathostomata) (57). Distinct CD3γ and CD3δ subunits do not exist in non-mammalian species such as birds, amphibians, reptiles, and bony fish. Instead, in these species there is a single CD3γ/δ precursor (CD3P gene). The elongated FG loop with Leu219, Trp225, and Pro232 conserved in mammalian β chains is absent therein (Fig. 7). Concurrently, when the elongated Cβ FG loop feature evolved in the mammalian species, CD3P duplicated and diverged to create distinct CD3γ and CD3δ genes and their products, as shown in Fig. 7. Thus, the elongated Cβ FG loop and the distinct CD3γ and CD3δ genes appear to have co-evolved (57).
Fig. 7. Co-evolution of the elongated TCRCβ FG loop as well as CD3γ plus CD3δ genes from a single precursor (CD3P) in Gnathostamata.
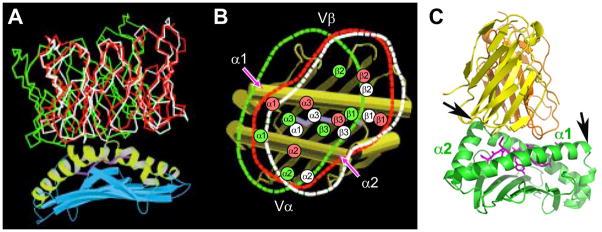
(Adapted from 57). (A) Sequence comparison of the TCRCβ FG loop regions among various species. The position of the F and G strands is defined based on the N15 TCR structure (37, 120). The bracketed region defines the elongated FG loop in mammalian species with well-conserved key residues (L219, W225, and P232) forming the hydrophobic core of the loop. The two cysteines contributing to the intra-chain and an inter-chain disulfide bonds are indicated by the filled and open circles, respectively. The conserved lysine residue in the transmembrane region of Cβ is also highlighted in yellow. (B) Schematic representation of evolutionary relationships between TCRβ and CD3 gene products. Possession of adaptive immunity with recombinatorial-based immune receptors is known for Agnathans and Gnathostomata. Gnathostomata possess a developed adaptive immune system supporting various VDJ recombinations for immunoglobulin (Ig) and TCR rearrangement, whereas Agnathans do not but contain variable lymphocyte receptors (VLR). Distinctions between mammals versus birds, amphibians, reptiles, and bony fish are described and shown schematically.
The elongated Cβ FG loop and the CD3εγ heterodimer are paired for efficient TCR assembly and signaling in mammalian species (57). Using structure-guided mutational analysis, we investigated the consequences of a striking asymmetry in CD3γ and CD3δ G-strand geometries impacting ectodomain structure (defined in the CD3 section immediately below). The uniquely kinked conformation of the CD3γ G-strand is crucial for maximizing pMHC-triggered T cell activation and TCR surface expression, offering a geometry to accommodate the juxtaposition of CD3γ and TCRβ ectodomains and to foster quaternary change that cannot be replaced by the isologous CD3δ subunits extracellular region.
Structures of CD3 heterodimers
The signal-transducing invariant CD3 subunits (CD3ε, CD3γ and CD3δ) each comprise a single extracellular Ig-like domain followed by a short stalk region, referred to as a connecting peptide (CP), a transmembrane (TM) helix, and a cytoplasmic tail. The interaction between TCRαβ and pMHC ligand initiates a cascade of downstream signaling events via the ITAMs in the cytoplasmic tail of the associated CD3 subunits (58–60). To understand CD3 function in TCR signaling, it was critical to define the CD3 structures uncovering how these subunits pair into specific heterodimers and assemble with αβ TCR onto the TCR complex.
The determination of solution NMR structures of CD3εγ (61) and CD3εδ (52) as well as the crystal structures of related antibody in complex with CD3εδ (62) and CD3εγ (63) offered the remaining pieces of the TCR ectodomain architecture. In NMR studies of CD3εγ and CD3εδ ectodomains, a single-chain strategy was employed to create constructs for E coli expression. For CD3εγ, the murine CD3γ Ig-like domain was linked to CD3ε Ig-like domain through a 26-residue peptide linker (61). This approach facilitated protein expression, proper heterodimer pairing, and yielded protein that was stable at neutral pH. A similar method was used for CD3εδ° except that sheep CD3δ was linked to the C-terminus of the murine CD3ε through a 33-residue linker. Sheep CD3δ was chosen since this orthologue has fewer surface exposed hydrophobic residues and manifests improved refolding and solubility characteristics (52).
Fig. 8 illustrates the NMR structures of CD3εγ and CD3εδ. The major structural observation can be summarized as follows. First, all three CD3 subunit ectodomains adopt a C-type Ig-like fold. Whereas CD3ε and CD3γ belong to the C2-set, CD3δ falls into the C1-set; CD3δ has its C′ edge β strand translocated from one β sheet to the other, thereby becoming a D strand (52). This edge strand translocation is a common phenomena for an Ig-like domain (64). From Fig. 8B it can be seen that neither the edge strand C′ in CD3ε nor the strand D in CD3δ is a regular β strand. Another distinct aspect of CD3δ is an extremely short BC loop (Fig. 8B), contributing to the relatively small domain size. Notably, the CD3ε domain in structures of CD3εγ and CD3εδ are virtually identical, suggesting its robustness. Perhaps the most interesting structural feature of the two CD3 heterodimers is how their extracellular domains associate with each other in a parallel fashion. Their respective G strands pair with extensive mainchain hydrogen bonds such that one ectodomain’s CFG sheet merges with another ectodomain’s GFC sheet to form a ‘super’ β sheet. This, in turn, brings hydrophobic residues into the interface in an interdigitating fashion, thereby consolidating heterodimer formation. The view in Fig. 8B illustrates the most obvious parallel G-strand pairing in CD3εδ.
Fig. 8. NMR solution structures of CD3 heterodimers.
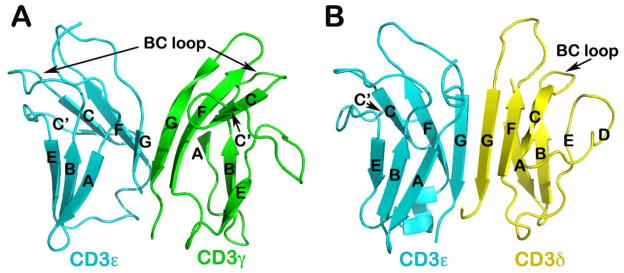
(A) Structure of CD3εγ (PDB 1JBJ): Note the cleft or notch at the top portion of the dimer, compared to CD3εδ shown in (B). (B) Structure of CD3εδ (PDB 1XMW): The BC loop of the CD3δ subunits significantly shorter compared to that of CD3ε and CD3γ subunits (52, 61). The linker to make the single-chain constructs is omitted in these two figures.
This conjoined β-sheet domain-domain amalgamation creates a solid but squat ectodomain unit vertically arrayed on the cell membrane, well-suited to function in signal transduction as described below. One obvious difference in the geometry of the two CD3 heterodimers is a pronounced cleft between the CD3 ectodomain tops in CD3εγ (Fig. 8A). A similar comparative difference was identified in the antibody bound crystal structures of human CD3εγ (63) and CD3εδ (62), confirming the importance of their surface topological distinction across species. As noted above, we have experimentally demonstrated that this uniquely kinked conformation of the CD3γ G strand is crucial for maximizing antigen-triggered TCR activation and surface TCR expression (57). Moreover, this CD3εγ geometry accommodates the TCR β subunit’s juxtaposition as described (57).
The αβ TCR complex and mechanotransduction
These rigidified CD3 heterodimers in turn are associated with the TCR αβ heterodimer whose own rigid structure is reinforced by the FG loop of the β chain constant domain (37). Thus, unsurprisingly, comparison of unligated and pMHC ligated TCRαβ structures does not show major conformational changes (reviewed in 32). A model of the TCR complex of αβ, CD3εγ° and CD3εδ ectodomains (52) defines a plausible topology and emphasizes its glycan richness (Fig. 9B). The multiple N-linked glycan adducts of the TCR complex (Fig. 9B, top panel) help guide pMHC ligands to the TCR recognition surface, reducing entropic penalties by directing binding to the exposed, glycan-free CDR loops. Given that CD3ζ has only a 9 amino acid long ectosegment, it is omitted from Fig. 9B as are the CPs. This rendering incorporates the consequences of several known TCR characteristics: (i) putative transmembrane charge pairs involving TCR subunit chain association (Fig. 9A) with CD3ε-CD3δ-TCRα-CD3ζ-CD3ζ as one cluster and CD3ε-CD3γ-TCRβ as a second cluster (65, 66), (ii) extracellular domain associations involving other in vitro chain association data (67, 68), TCR chemical crosslinking results (69, 70), and (iii) proximity of one CD3ε subunit to the TCR Cβ FG loop revealed by quantitative T-cell surface immunofluorescent antibody binding analysis (54). In addition, structural insights from crystallographic data on the glycosylated N15 TCRαβ heterodimer ectodomain in complex with the Cβ-FG loop-binding H57 Fab discussed above and the likely position of glycans in both CD3εγ and CD3εδ (37) are considered. Evident in Fig 9B (bottom panel) is the central position of the TCRαβ heterodimer with a vertical dimension of 80Å projecting from the cell membrane, flanked on either side by the shorter (40Å) CD3 heterodimers, CD3εδ on the ‘left’ TCRα side and CD3εγ on the ‘right’ TCRβ side. Note that the width of the CD3εδ and CD3εγ components, 50Å and 55Å, respectively, are comparable in size to that of the TCRαβ heterodimer (58Å), and together (excluding glycans) span ~160Å. These flanking CD3 ectodomain components will likely impede lateral movement of the TCRαβ heterodimer upon pMHC binding.
Fig. 9. TCR complex interaction with pMHC.

(A) TCR components (ectodomains, stalk CP, TM, and cytoplasmic tails) are labeled and shown in distinct colors. The pMHC on the APC and the interacting CD8αβ heterodimer are not colorized. The relative positions of the positively charged residues in TCR α and β TMs are shown and putative interactions with CD3 acidic residues indicated (boxed insert). In the bottom right insert, arrows denote direction of mAbs binding to CD3εγ relative to the T cell membrane. The view shown is a 90° Y-axis clockwise rotation relative to (A) above. (B) Ectodomain structure in ribbon form based upon PDB code IFND, 1XMW and 1JBJ are shown from the perspective of pMHC (top) and side (bottom) views with the position of T-cell membrane shown in the latter. In the top view, adducted glycans in CPK visualization are colored according to subunit whereas all sugars are denoted in beige in the side view. The FG loop is labeled by an * in each panel.
The 5–10 amino acid squat and rigid CD3 CP segments (71) contrast sharply with the long (19–26aa) and flexible TCR α and β CP linking their respective constant domains to the transmembrane segments (Fig. 9A). The functional importance of this contrasting arrangement was revealed through analysis of interactions of activating (i.e. 2C11 or 500A2) and non-activating (17A2) anti-CD3ε mAbs, which bind to the CD3εγ ectodomains with virtually identical affinity on T cells (72). Activating antibodies footprint to the membrane distal CD3ε lobe which they approach diagonally (Fig. 9A, bottom), adjacent to the lever-like Cβ FG loop noted to facilitate pMHC-triggered activation (56). In contrast, the non-activating mAb (17A2) binds to the cleft between CD3ε and γ in a mode perpendicular to the T-cell membrane (72). Polystyrene bead-bound 17A2 mAb became stimulatory, however, upon application of ~50 pN of external tangential force to the bead (Fig. 10B). Importantly, specific bead-bound pMHC (but not irrelevant peptide bound to the same MHC) also activates a T cell upon application of a similar tangential mechanical force (MF) via optical tweezers to initiate intracellular calcium flux (Fig. 10D). These findings imply that the TCR is a mechanosensor, converting mechanical energy into a biochemical signal upon specific pMHC ligation that occurs as a T cell moves over antigen-presenting cells during the course of immune surveillance (Fig. 10A). As shown in Fig. 10C, the pMHC on the APC is first ligated by a specific TCR. Then as the T cell continues to move, prior to a stop movement signal mediated through inside-out integrin affinity upregulation, pMHC functions as a force transducing handle to pull on the TCRαβ heterodimer. This force is amplified and exerted on CD3εγ by the lever arm where the TCRβ TM acts as a fulcrum. For activation, force must be applied to the TCR complex tangentially and not perpendicular to the plane of the T-cell membrane, showing that the TCR is an anisotropic mechanosensor (i.e. direction matters)(72). The rupture force and bond lifetime under load between pMHC and TCRαβ heterodimer are potentially important parameters which can determine the potency of pMHC stimulation as can the angle of TCR-pMHC interaction (44, 73). The lateral pull from pMHC most probably causes the Cβ FG loop to push on the upper outer lobe of CD3ε. A common TCR quaternary change rather than conformational alterations can better facilitate structural signal initiation, given the vast array of TCRs and their pMHC ligands. During this force driven quaternary change, TCR-decorating glycans can serve as steric and spring-like barriers that require force to overcome in order to deliver signaling to CD3 subunits. Since our report (72), other groups now have provided evidence that physical force applied to TCR components activates T cells (74–77).
Fig. 10. TCR activation by mechanical force.
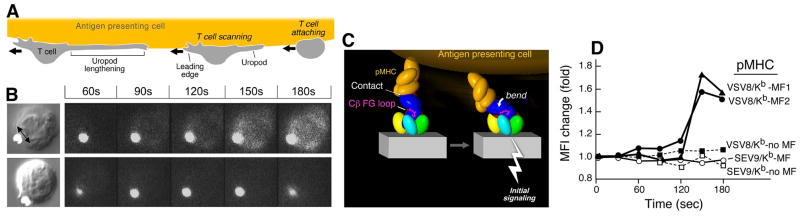
(A) Cartoon showing tangential T-cell scanning of an APC surface. (B) A 17A2 anti-CD3 mAb coated bead is approximated to a T cell using an optical trap. A tangential force is applied on the cell as shown by the double-headed arrow. Fluorescence image reporting of intracellular calcium dynamics show signaling in the T cells upon application of mechanical force to the bead but not in the absence of mechanical force (top and bottom rows, respectively). (C) Torque on TCR-pMHC interaction initiates signaling action (pMHC, orange; Cβ FG loop, magenta; and TCR complex, other colors). (D) Signaling (fluorescence increase) in TCR transgenic N15 T cells [specific for vesicular stomatitus virus octapeptide (VSV-8) bound to MHC Kb] occurs only in the presence of tangential mechanical force (MF) and only for stimulatory VSV8/Kb pMHC but not irrelevant SEV9/Kb pMHC molecules (72). Stimulation is achieved at 10 relevant pMHC copies per bead.
Several theories on T-cell triggering have been proposed to explain how recognition of pMHC by a weakly interacting (~1–100 μM Kd) clonotypic αβ heterodimer on the T-cell surface evokes intracellular signaling via the adjacent CD3 components with minimal, if any, stable αβ ectodomain interface contacts (78–81). These include conformational change, aggregation, and segregation models. The rigidity of αβ (37) and CD3εγ and CD3εδ heterodimeric ectodomains (52, 61, 79) noted above imply that conformational change within TCR subunits cannot initiate signaling, thereby eliminating earlier conformational models (80). We suggest that mobility of the αβ heterodimeric ectodomains relative to the fixed CD3 heterodimers allows for transient ectodomain interactions, quaternary TCR complex change, and pMHC ligation to be sensed. Force must then be transduced via the individual TCR complex TM segments, potentially modifying individual transmembrane helices, TM-TM interactions, and/or the lipid environment and cytoplasmic tail exposure/configuration. In the future, determining the structures of these segments separately and as relevant oligomers, their adjacent peptide connecting extensions, and cytoplasmic tails will be the key first step in elucidating the molecular basis of signal transfer from outside the cell to inside the cell.
A singularly vertical or piston-like motion previously suggested as a basis for T-cell signaling seems excluded by optical trap analysis (72). The notions of permissive geometry involving dimers of TCRs and pMHC (82) or pseudodimers (83) are difficult to reconcile with the extensive glycosylation of the TCR complex (Fig. 9B). Microclustering of TCR complexes in the absence of TCR and/or co-receptor protein ectodomain oligomerization can be mediated by intracellular domain interactions with scaffold and/or cytoplasmic protein (84–86). Likewise, kinetic segregation involving a partitioning of inhibitory phosphatases (CD45) away from the activation machinery (p56lck) in the T cell-APC contact zone referred to as the immunological synapse can be operative in conjunction with mechanotransduction (87). The accelerated kinetics of TCR/pMHC interaction, including off-rate in situ dependence upon the actin cytoskeleton, will impact and contribute to sustained mechanotransduction at the immunological synapse (88). Studies suggesting that pMHC applies an external force to push on or ‘deform’ the TCR (61, 89) are consistent with the optical trap results.
That the TCR is a mechanosensor activated by direction-specific physical force (72) has immediate implications. First, since the total force applied to the T-cell surface is essentially defined by movement of the T-cell membrane relative to that of the APC, ligation of several TCRs by several cognate pMHCs on the opposing APC will exert a greater physical force per individual TCR than multiple TCR ligations through a large number of TCR-pMHC interactions on the same T cell. Hence, sensitivity is built into TCR mechanosensor function. Second, in principle, shear forces can form catch bonds at the TCR-pMHC interface to enhance binding and/or confer additional ligand specificity. These bonds, which are strengthened by tensile force, have been described for cell adhesion molecules (90). Third, a recent study by Adams et al. (44) suggesting that docking geometry impacts 2D binding and T-cell activation is entirely consistent with the notion of TCR mechanotransduction; the pMHC-TCR lever arm length and force vector parameters determining torque will be modified via such differential docking, independent of 3D solution affinity for pMHC ligand (73). Precedent for mechanoreceptors in the hematopoietic system is the von Willebrand factor (VWF) receptor on platelets where tensile stress on bonds between the GPIbα subunit and the VWFA1 domain under fluid dynamic conditions triggers integrin αIIbβIII activation to support platelet adhesion (91).
CD4 and CD8 co-receptor structures, bidentate attachment to MHC, and delivery of p56lck kinase to the TCR-pMHC complex
As early as 1980, it was clear that two major T-cell subsets, cytotoxic T lymphocytes and helper T lymphocytes, could be distinguished by their surface expression of CD8 and CD4, respectively (12, 22, 23, 92). Using T-cell clones and monoclonal antibody blocking studies, it was shown that MHC class II-restricted T-cell recognition was mediated by CD4+ T cells, whereas MHC class I-restricted T-cell recognition was performed by CD8+ T cells. Given that CD4 and CD8 were invariant structures, we speculated that they interacted with conserved regions of polymorphic MHC molecules as co-receptors, unlike the clonotypic TCRαβ heterodimer whose recognition was dependent on specific peptide and polymorphic MHC segments (23). From structural studies accomplished in the last two decades (93–97), we learned that whereas the TCR interacts with the antigenic peptide bound to the MHC molecule’s membrane-distal helical surface and β-sheet floor (i.e. the antigen presenting platform), co-receptors contact a conserved membrane-proximal region of their MHC molecular target (vide infra).
Co-receptor biology
The CD8 transmembrane co-receptor is encoded by two distinct genes: CD8α and CD8β. Each consists of a single Ig-like domain followed by a lengthy stalk region of 30–50 residues with multiple O-glycosylated adducts, a TM helix, and a short cytoplasmic tail (reviewed in 98). The CD8α but not CD8β tail binds to p56lck, essential for T-cell signaling. While CD8αα homodimers and CD8αβ heterodimers are found on the surface of lymphocytes, CD8ββ homodimers are absent. The CD8αβ heterodimer is the dominant isoform expressed on CTLs (99). The CD8αα isoform is expressed on γδ T cells, some NK cells, and a subset of intraepithelial lymphocytes (100). By contrast to CD8, CD4 comprises four Ig-like domains in tandem with a short stalk region and TM helix, but its cytoplasmic tail also binds p56lck. In fact, a zinc clasp tethers p56lck to the cytoplasmic tail of both CD4 and CD8α (101).
The major function of the co-receptor in T cell-mediated adaptive responses is not to facilitate adhesion and/or binding per se but rather to deliver p56lck into the area of TCR-pMHC interaction so that exposed ITAM(s) on CD3 tails can be phosphorylated on tyrosine residues therein to allow Zap-70 recruitment and the remainder of downstream signaling apparatus to assemble (102). This selective co-receptor delivery of an essential TCR kinase to the MHC during TCR recognition skews αβ TCR repertoire selection toward a pMHC ligand specification. As a result, αβ TCRs in mice lacking co-receptors and MHC do not have a bias for pMHC ligands, instead possessing antibody-like receptor specificities (103).
The affinity of CD4 for pMHCI is extremely weak (200 μM or higher)(104) and that of CD8αβ for pMHCII only slightly (several fold) stronger (105, 106). By contrast, ~1 μM affinities of TCR-pMHC interactions are not uncommon (107). The half-life of TCR-pMHC is ~1000 times longer than that of CD4-pMHC; thus, little binding contribution for pMHC is contributed by the co-receptor ectodomain. Likely, CD8αβ offers more pMHC interaction energy than CD4. More importantly, however, for both co-receptors, the ability to recruit p56lck to the TCR interaction site, although transient, overcomes p56lck diffusion thereby affording targeted kinase delivery to the TCR-pMHC complex. A formal mathematical modeling of this process has recently been performed, consistent with this view (108). Given that only a fraction of p56lck is catalytically active in unstimulated T cells and that amount does not increase after TCR and co-receptor engagement (109), rapid entry and exit of co-receptors into the TCR-pMHC site is critical. If a co-receptor were loaded with inactive p56lck and bound tightly to pMHC, it could function as a dominant negative inhibitor of TCR activation. A fast on and off rate of co-receptor-pMHC will likely bring an active kinase to an accessible CD3 tail ITAM.
The bidentate interaction of TCR and CD8αβ with a single agonist pMHC has been elegantly studied by a micropipet adhesion assay (110). Kinetic analysis reveals a two stage cooperative process with the first stage representing TCR dominant binding to pMHC. The second stage binding, delayed by one second, is Src-tyrosine kinase-dependent (i.e. presumably p56lck) resulting in the CD8αβ co-receptor binding to the TCR-engaged pMHC molecule. This ordered and cooperative trimeric interaction favors agonist ligands and synergistically augments the bidentate binding to pMHC in turn linked to T-cell signaling.
Aside from their binding specificities, CD8αβ and CD4 differ with respect to their ectodomain flexibility and tunable biology (98, 111). The combined presence of O-glycans and prolines in the stalk CD8 subunits indicates that the stalks most likely adopt extended and somewhat stiff conformations, similar to that observed for leukosialin and mucins. When mucins are heavily O-glycosylated, they exhibit a threefold expansion in chain dimension compared to their unglycosylated counterpart (112, 113). Notwithstanding, there remains some CD8 stalk flexibility in counter-distinction to the rigidity of the four concatamerized CD4 Ig-like domains. This CD8αβ co-receptor flexibility perhaps compensates for and accommodates to the variability of TCRαβ docking onto pMHCI. In addition, developmentally regulated glycosylation of the CD8αβ stalk modulates pMHC binding (98). Immature CD4+CD8+ double positive (DP) thymocytes bind MHCI more avidly than mature CD8 single positive (SP) thymocytes. This differential binding is governed by developmentally programmed O-glycan modification of several CD8 stalk threonine residues proximal to the CD8β headpiece and controlled by ST3Gal-I sialyltransferase (98, 111). ST3Gal-I specifically localizes to the medulla of the thymus where SP thymocytes reside. ST3Gal-I induction and attendant core 1 sialic acid addition to CD8β on mature thymocytes decreases CD8αβ-MHCI avidity by altering CD8αβ domain-domain association and/or orientation. Hence, glycans on the CD8β stalk appear to modulate the ability of the distal binding surface of the dimeric CD8 globular head domains to clamp MHCI. The DP stage facilitates efficient elimination by negative selection of autoreactive TCR specificities through this enhanced co-receptor function working in tandem with highly specific TCR-pMHC triggered apoptosis (114). Once a thymocyte has differentiated to the CD8 SP stage, however, CD8αβ O-glycan sialylation reduces the strength of the CD8αβ co-receptor interaction with pMHC, mandating a greater requirement for TCR-pMHC interaction to achieve a subsequent activation threshold in mature T-lineage cells (98).
Co-receptor structures
Structures of human and murine CD8αα homodimers and CD8αβ heterodimer in complex with pMHCI molecules (93–95) as well as the N-terminal two Ig-like domains of CD4 complexed to pMHCII (96) are available. Fig. 11 depicts our structures of the murine CD8αα homodimer in complex with H-2Kb (Panel A) and the human CD4 N-terminal two-domain construct bound to the I-Ak pMHCII molecule (Panel B).
Fig. 11. Structure of TCR co-receptors.

(A) Crystal structure of murine H2-Kb in complex with CD8αα homodimer (PDB 1BQH). For the H2-Kb, the heavy chain is in green and β2 microglobulin is in cyan, whereas the two subunits of CD8αα are in yellow and pink, respectively. The peptide is shown as a stick model within the groove. Note that the CD loop of H2-Kb is clamped by CDR loops of CD8αα homodimer. (B) Crystal structure of murine I-Ak in complex with the N-terminal two domains of CD4 (PDB 1JL4). The α and β subunits of I-Ak are in cyan and green, respectively, whereas the CD4 is in yellow. The peptide is in stick model representation lying in the groove. Note how the CD4 domain 1 wedges between α2 and β2 domain of I-Ak with hotspot residue, F43 poking into the MHCII hydrophobic pocket. (C) Specific interaction between CD8αα and H2-Kb’s CD loop. Conserved residues involved in these hydrogen bondings are labeled. (D) Specific interaction between CD4 and I-Ak. The two main chain hydrogen bonds between the edge β strands of CD4 and β2 domain of I-Ak docks the CD4 onto I-Ak. This brings F43 of CD4 into hydrophobic pocket formed between α2 and β2 domains whose key contacts are shown.
It is remarkable how the two co-receptors employ divergent strategies to achieve interaction with the membrane proximal region of their respective MHC molecules. The murine CD8αα homodimer binds primarily to the CD loop of the MHCI α3 domain in a fashion similar to an antibody binding to antigen (94), as does the human analogue in binding to HLA-A*0201 (93). The two Ig-like CD8 homodimer ectodomains are comparable to an Fv module of an antibody, using the six CDR-like loops to clamp the CD loop of the MHC α3 domain. There are conserved residues engaging in a specific hydrogen bond network between CD8αα and the CD loop of MHCI molecule residues Glu222, Gln226 and Asp227 (Fig. 11C). The CD loop itself is well structured through an internal hydrogen bonding network. Therefore, CD8-binding does not require detectable conformational change. A very similar binding mode has been observed as well for the CD8αα homodimer interaction with a non-classical MHCIb molecule, TL (115), suggesting the robustness of the protruding CD loop of MHC near the plasma membrane for CD8-binding. Based on the relatively shorter stalk length of the CD8β subunit compared to that of CD8α, we predicted that the CD8β ectodomain would occupy the APC membrane distal position in the CD8αβ heterodimer when complexed with pMHCI (94). This hypothesis was confirmed by the recent structure of CD8αβ/MHCI complex revealing how the CDR loops of CD8αβ Ig-like domains also clamp the CD loop (95).
Since CD4 is a single subunit co-receptor, unlike CD8, an antibody-like MHC-binding mode is less likely. The structure of the CD4/pMHCII complex unexpectedly showed that the CD4 binding to MHC involves wedging of the CD4 N-terminal domain D1 between the two membrane-proximal α2 and β2 domains of MHCII (Fig. 11B). The most protruding C″ β strand of CD4 D1 makes a mini-antiparallel β structure with the outermost strand of the MHCII β2 domain. A pair of mainchain hydrogen bonds illustrated in Fig. 11D mediates this interaction. This docking topology brings the key hotspot binding residue of CD4 D1, Phe43, into the hydrophobic environment created by conserved MHCII residues, including Phe92 and Trp178 from the α2 domain as well as Ile148 and Leu158 from the β2 domain (Fig. 11D).
The CD4/MHCII structure is of biological relevance in two important respects. First, it allowed us to build a ternary complex of TCR/MHCII/CD4 by superimposing the known structures of TCR/MHCII (PDB code 1D9K and 1FYT) and the entire ectodomain of CD4 (D1–D4) (PDB code 1WIO) onto our CD4/MHCII complex (96). This yielded a V-shaped ternary complex model clearly implying that there was no direct contact between a TCR and its CD4 co-receptor from the same plasma membrane as shown below. More interestingly, perhaps, the ternary association geometry suggests how a TCR from a CD4+ T cell must scrutinize an APC surface and trigger its signaling as elaborated below. Very recently, using yeast display technology, the Mariuzza group has been able to enhance the binding affinity between CD4 and MHCII (116) to the point where they can successfully co-crystallize a ternary complex of TCR/pMHCII/CD4 (117). Their ternary structure confirms the complex model we proposed.
Second, our CD4/MHCII complex structure provided a molecular explanation for how CD4 is subverted by HIV-1 as the virus’s portal of entry into the human cell. By comparing the CD4/MHCII structure with the previously published structure of CD4/HIV-gp120 (118), it is obvious that the virus mimics MHCII binding to CD4. HIV surface glycoprotein gp120, like MHCII, also uses its edge β-strand to pair with the C″-strand of CD4 D1, in turn bringing the key Phe43 residue into a hydrophobic pocket of gp120. Since gp120 effectively makes contacts with additional CD4 surface area as well, the CD4/gp120 binding affinity is ~1000 times stronger than that of CD4/MHCII binding (119). In this way, the HIV envelope protein successfully out-competes the physiological ligand MHCII for CD4-binding. In so doing, the effectiveness of the immune response is diminished.
Overall topology of the CD4/TCR/pMHCII ternary complex and its immunological significance
When Bjorkman, Strominger, and Wiley first published their landmark structure of HLA (25), the view was that the MHCI molecule vertically presented a peptide at a distance from the APC membrane, well-exposed for TCR binding from an opposing T cell (Fig. 12A). The same ‘classic’ orientation was assumed for MHCII (Fig. 12B). However, given that MHCI has a single TM linked to the heavy chain in turn noncovalently associated with β2M, whereas the MHCII has two TM-containing subunits of similar size, this view requires modification. The C-terminal stalk regions of α2 and β2 domains of MHCII molecules are both roughly 10 residues in length. The stalks and TM segments constrain the position of the α and β ectodomains. Thus, the pMHCII cannot be oriented as shown in Fig. 12B akin to the MHCI (Fig. 12A). Rather, pMHCII must be oriented in a fashion more equivalent to the orientation provided in Fig. 12C. There the C-terminal residues (in dark blue stick model) of the two ectodomains are placed at equivalent distance above the plane of the membrane below. In this orientation, the peptide-binding groove appears tilted such that the β1 helix projects out significantly more above the membrane than the α1 helix.
Fig. 12. Overall topology of the CD4/TCR/pMHCII ternary complex.

(A) A ‘classic’ view of the MHCI molecule demonstrating how an MHC molecule on an APC presents the antigenic peptide vertically toward an opposing T cell for TCR recognition. The light chain, β2 microglobulin (yellow) is non-covalently attached to the heavy chain. (B) A similar ‘classic’ view of MHCII molecule as that of MHCI in (A). In this view, the α subunit in yellow has its C-terminal α2 domain residue (dark blue stick model) high above the plasma membrane relative to the C-terminal residue (dark blue stick model) of the β2 domain in orange. (C) Revised MHCII orientation. Given that the stalks of both the α and β subunits are ~10-residue long, the stalk and TM should impose constraints on the two subunits such that the MHCII molecule must be oriented in a manner more tilted compared to the view shown in (B). As a consequence, the two MHC terminal residues are at about the same height on the membrane. The helical region of the β1 domain would significantly project upward in comparison to that of the α1 domain. (D) The overall orientation of the ternary complex (coordinates from PDB 3T0E). This representation takes into account the V-shaped CD4-MHCII architecture as well as the MHCII topology on the APC membrane given in (C) above. It becomes obvious how the MHC class II-restricted TCR must contact the MHCII molecule tangentially from the right in the figure. The TCRαβ will ‘bump’ up against the ‘wall’ or barrier of the helical region of the β1 domain to provide a mechanical force, supporting T-cell mechanotransduction. Note that although the CPs are not shown from the T-cell side, CD4 has a shorter stalk than that of the TCRαβ heterodimer subunits.
Once this constraint is applied to the V-shaped ternary complex, the overall CD4/TCR/pMHCII orientation would appear as illustrated in Fig. 12D. Here, the central break in the β1 helix serves not only as an interaction site boundary but creates a wall to impact TCR movement. If this notion is correct, kinetic energy may then be converted into a torque to pivot the TCRαβ heterodimer about its TM segments, pushing on the CD3 subunit close to the membrane as described in the prior section on mechanotransduction. How this geometry relates to that of CD8-dependent TCR recognition remains to be determined. Given that MHCI uses a single TM for presentation and is associated with a co-receptor with some stalk flexibility, differences in ternary complex recognition geometry cannot be excluded.
Summary/implications
Here we have reviewed a wealth of structural information gleaned by many laboratories on the nature of the TCRαβ-pMHC interaction. We described the relatively flat VαVβ module recognition surface adapted via thymic selection processes and malleable germline interaction preferences for recognition of linear peptides bound to the groove of MHC molecules. We analyzed the TCRαβ docking geometries onto pMHCI and pMHCII. Whereas a general diagonal footprint with considerable variability in tilt, twist, and shift accurately accounts for TCRαβ-pMHCI interaction, the TCRαβ approach onto pMHCII is more limited to the orthogonal footprint. Although there are no apparent intrinsic structural distinctions between TCR V modules of the CD4+ and CD8+ T-cell subsets nor the antigen-presenting platforms of MHCI and MHCII, differential projection of MHCI and MHCII from the APC surface, at least in part, may foster binding footprint distinction. The CD8αβ and CD4 co-receptors use different strategies to ligate pMHCI and pMHCII, respectively, via weak affinities that similarly function to deliver p56lck into the site of TCRαβ-pMHC interaction.
The current understanding of TCR complex mechanotransduction is reviewed, affording important insights into the rigidity of the TCRαβ, CD3εγ° and CD3εδ heterodimeric ectodomains, the unusual Cβ FG loop adaptation and TCR quaternary change that is mechanical force dependent. Mammalian development with co-evolution of the Cβ FG loop and CD3γ and CD3δ molecular speciation enhances the sensitivity of cognate TCRαβ recognition to the level of endowing one or several pMHC per APC surface with the capability of triggering T-cell activation. We present data suggesting how mechanotransduction is operative, both during immune surveillance and within the immunological synapse.
At a practical level, there are implications of the present work for translational medicine. T-cell-based vaccines or immunotherapies eliciting effector T cells with specificity for epitopes on target cells need not focus on pMHC ligands exclusively present at high copy number, given the sensitivity of TCR mechanotransduction and its mode of action. Thus, the number of suitable immune targets for T-cell-based preventive and therapeutic approaches is greater than previously envisioned. Future single molecule analysis of TCR force transduction will provide additional insights into early T-cell signaling events and, in conjunction with structural elucidation of TCR subunit CP, TM and cytoplasmic tail segments, potentially afford new approaches towards development of immunosuppressive compounds based on disruption of TCR mechanotransduction.
Acknowledgments
This work was supported by research grants from US NIH to ELR and J-HW.
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Miller JFAP, Mitchell GF. Cell to cell interaction in the immune response. I. Hemolysin-forming cells in neonatally thymectomized mice reconstituted with thymus or thoracic duct lymphocytes. J Exp Med. 1968;128:801–820. doi: 10.1084/jem.128.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell GF, Miller JFAP. Cell to cell interaction in the immune response. II The source of hemolysin-forming cells in irradiated mice given bone marrow and thymus or thoracic duct lymphocytes. J Exp Med. 1968;128:821–837. doi: 10.1084/jem.128.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonegawa S. Somatic generation of immune diversity. In: Frångsmyr T, Lindsten J, editors. Nobel Lectures: Physiology or Medicine 1981–1990. Singapore: World Scientific Publishing; 1993. pp. 381–405. [Google Scholar]
- 4.Zinkernagel RM. The nobel lectures in immunology. The Nobel prize for physiology or medicine, 1996; Cellular immune recognition and the biological role of major transplantation antigens. Scand J Immunol. 1997;46:421–436. [PubMed] [Google Scholar]
- 5.Doherty PC. The nobel lectures in immunology. The Nobel prize for physiology or medicine, 1996; Cell mediated immunity in virus infections. Scand J Immunol. 1996;46:527–540. doi: 10.1046/j.1365-3083.1997.d01-170.x. [DOI] [PubMed] [Google Scholar]
- 6.Baker PE, Gillis S, Smith KA. Monoclonal cytolytic T-cell lines. J Exp Med. 1979;149:273–278. doi: 10.1084/jem.149.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milstein C. From the structure of antibodies to the diversification of the immune response. In: Frängsmyr T, Lindsten J, editors. Nobel Lectures, Physiology or Medicine 1981–1990. Singapore: World Scientific Publishing; 1993. pp. 248–70. [Google Scholar]
- 8.Julius MH, Masuda T, Herzenberg LA. Demonstration that antigen-binding cells are precursors of antibody-producing cells after purification with a fluorescence-activated cell sorter. Proc Natl Acad Sci USA. 1972;69:1934–1938. doi: 10.1073/pnas.69.7.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Acuto O, et al. The human T cell receptor: appearance in ontogeny and biochemical relationship of alpha and beta subunits on IL-2 dependent clones and T cell tumors. Cell. 1983;34:717–726. doi: 10.1016/0092-8674(83)90528-7. [DOI] [PubMed] [Google Scholar]
- 10.Acuto O, Meuer SC, Hodgdon JC, Schlossman SF, Reinherz EL. Peptide variability exists within alpha and beta subunits of the T cell receptor for antigen. J Exp Med. 1983;158:1368–1373. doi: 10.1084/jem.158.4.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meuer SC, et al. Evidence for the T3-associated 90K heterodimer as the T-cell antigen receptor. Nature. 1983;303:808–810. doi: 10.1038/303808a0. [DOI] [PubMed] [Google Scholar]
- 12.Meuer SC, Fitzgerald KA, Hussey RE, Hodgdon JC, Schlossman SF, Reinherz EL. Clonotypic structures involved in antigen-specific human T cell function. Relationship to the T3 molecular complex. J Exp Med. 1983;157:705–719. doi: 10.1084/jem.157.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinherz EL, Meuer S, Fitzgerald KA, Hussey RE, Levine H, Schlossman SF. Antigen recognition by human T lymphocytes is linked to surface expression of the T3 molecular complex. Cell. 1982;30:735–743. doi: 10.1016/0092-8674(82)90278-1. [DOI] [PubMed] [Google Scholar]
- 14.Clevers H, Alarcon B, Wileman T, Terhorst C. The T cell receptor/CD3 complex: a dynamic protein ensemble. Annu Rev Immunol. 1988;6:629–662. doi: 10.1146/annurev.iy.06.040188.003213. [DOI] [PubMed] [Google Scholar]
- 15.Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J Exp Med. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kappler J, et al. The major histocompatibility complex-restricted antigen receptor on T cells in mouse and man: identification of constant and variable peptides. Cell. 1983;35:295–302. doi: 10.1016/0092-8674(83)90232-5. [DOI] [PubMed] [Google Scholar]
- 17.Allison JP, McIntyre BW, Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J Immunol. 2005;174:1144–1151. [PubMed] [Google Scholar]
- 18.Hedrick SM, Cohen DI, Nielsen EA, Davis MM. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308:149–153. doi: 10.1038/308149a0. [DOI] [PubMed] [Google Scholar]
- 19.Hedrick SM, Nielsen EA, Kavaler J, Cohen DI, Davis MM. Sequence relationships between putative T-cell receptor polypeptides and immunoglobulins. Nature. 1984;308:153–158. doi: 10.1038/308153a0. [DOI] [PubMed] [Google Scholar]
- 20.Yanagi Y, Yoshikai Y, Leggett K, Clark SP, Aleksander I, Mak TW. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature. 1984;308:145–149. doi: 10.1038/308145a0. [DOI] [PubMed] [Google Scholar]
- 21.Acuto O, et al. Purification and NH2-terminal amino acid sequencing of the beta subunit of a human T-cell antigen receptor. Proc Natl Acad Sci USA. 1984;81:3851–3855. doi: 10.1073/pnas.81.12.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meuer SC, Schlossman SF, Reinherz EL. Clonal analysis of human cytotoxic T lymphocytes: T4+ and T8+ effector T cells recognize products of different major histocompatibility complex regions. Proc Natl Acad Sci USA. 1982;79:4395–4399. doi: 10.1073/pnas.79.14.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinherz EL, Meuer SC, Schlossman SF. The delineation of antigen receptors on human T lymphocytes. Immunol Today. 1983;4:5–8. doi: 10.1016/0167-5699(83)90094-4. [DOI] [PubMed] [Google Scholar]
- 24.Unanue ER. From antigen processing to peptide-MHC binding. Nat Immunol. 2006;7:1277–1279. doi: 10.1038/ni1206-1277. [DOI] [PubMed] [Google Scholar]
- 25.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:506–512. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 26.Brown JH, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 27.Garcia KC, et al. An alphabeta T cell receptor structure at 2. 5 A and its orientation in the TCR-MHC complex. Science. 1996;274:209–219. [PubMed] [Google Scholar]
- 28.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 29.Teng MK, et al. Identification of a common docking topology with substantial variation among different TCR-peptide-MHC complexes. Curr Biol. 1998;8:409–412. doi: 10.1016/s0960-9822(98)70160-5. [DOI] [PubMed] [Google Scholar]
- 30.Reinherz EL, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 31.Kim ST, et al. TCR mechanobiology: Torques and tunable structures linked to early T cell signaling. Front Immunol. 2012;3:76. doi: 10.3389/fimmu.2012.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 33.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 35.Deford-Watts LM, et al. The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J Immunol. 2009;183:1055–1064. doi: 10.4049/jimmunol.0900404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu C, et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J-H, et al. Atomic structure of an alphabeta T cell receptor (TCR) heterodimer in complex with an anti-TCR fab fragment derived from a mitogenic antibody. EMBO J. 1998;17:10–26. doi: 10.1093/emboj/17.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang SS, et al. The structural basis for autonomous dimerization of the pre-T-cell antigen receptor. Nature. 2010;467:844–848. doi: 10.1038/nature09448. [DOI] [PubMed] [Google Scholar]
- 39.Dai S, et al. Crossreactive T cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 41.Zhou B, et al. A conserved hydrophobic patch on Vβ domains revealed by TCRβ chain crystal structures: Implications for pre-TCR dimerization. Front Immunol. 2011;2:5. doi: 10.3389/fimmu.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borg NA, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 44.Adams JJ, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–693. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 46.Rudensky A, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA., Jr Sequence analysis of peptides bound to MHC class II molecules. Nature. 1991;353:622–627. doi: 10.1038/353622a0. [DOI] [PubMed] [Google Scholar]
- 47.Hunt DF, et al. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science. 1992;256:1817–1820. doi: 10.1126/science.1319610. [DOI] [PubMed] [Google Scholar]
- 48.Chicz RM, et al. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature. 1992;358:764–768. doi: 10.1038/358764a0. [DOI] [PubMed] [Google Scholar]
- 49.Stern LJ, Wiley DC. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure. 1994;2:245–251. doi: 10.1016/s0969-2126(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 50.Guo HC, Jardetzky TS, Garrett TP, Lane WS, Strominger JL, Wiley DC. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature. 1992;360:364–366. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- 51.Liu YC, et al. The energetic basis underpinning T-cell receptor recognition of a super-bulged peptide bound to a major histocompatibility complex class I molecule. J Biol Chem. 2012;287:12267–12276. doi: 10.1074/jbc.M112.344689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun ZY, Kim ST, Kim IC, Fahmy A, Reinherz EL, Wagner G. Solution structure of the CD3epsilondelta ectodomain and comparison with CD3epsilongamma as a basis for modeling T cell receptor topology and signaling. Proc Natl Acad Sci USA. 2004;101:16867–16872. doi: 10.1073/pnas.0407576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuhns MS, et al. Evidence for a functional sidedness to the alphabetaTCR. Proc Natl Acad Sci USA. 2010;107:5094–5099. doi: 10.1073/pnas.1000925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghendler Y, Smolyar A, Chang HC, Reinherz EL. One of the CD3epsilon subunits within a T cell receptor complex lies in close proximity to the Cbeta FG loop. J Exp Med. 1998;187:1529–1536. doi: 10.1084/jem.187.9.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sasada T, Touma M, Chang HC, Clayton LK, Wang JH, Reinherz EL. Involvement of the TCR Cbeta FG loop in thymic selection and T cell function. J Exp Med. 2002;195:1419–1431. doi: 10.1084/jem.20020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Touma M, Chang HC, Sasada T, Handley M, Clayton LK, Reinherz EL. The TCR C beta FG loop regulates alpha beta T cell development. J Immunol. 2006;176:6812–6623. doi: 10.4049/jimmunol.176.11.6812. [DOI] [PubMed] [Google Scholar]
- 57.Kim ST, et al. Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of alpha beta T cell receptors. J Immunol. 2010;185:2951–2959. doi: 10.4049/jimmunol.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. [PubMed] [Google Scholar]
- 59.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 60.Letourneur F, Klausner RD. Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3 epsilon. Science. 1992;255:79–82. doi: 10.1126/science.1532456. [DOI] [PubMed] [Google Scholar]
- 61.Sun ZJ, Kim KS, Wagner G, Reinherz EL. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell. 2001;105:913–923. doi: 10.1016/s0092-8674(01)00395-6. [DOI] [PubMed] [Google Scholar]
- 62.Arnett KL, Harrison SC, Wiley DC. Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc Natl Acad Sci USA. 2004;101:16268–16273. doi: 10.1073/pnas.0407359101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kjer-Nielsen L, et al. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc Natl Acad Sci USA. 2004;101:7675–7680. doi: 10.1073/pnas.0402295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J-H, Springer TA. Structural specializations of immunoglobulin superfamily members for adhesion to integrins and viruses. Immunol Rev. 1998;163:197–215. doi: 10.1111/j.1600-065x.1998.tb01198.x. [DOI] [PubMed] [Google Scholar]
- 65.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Call ME, Pyrdol J, Wucherpfennig KW. Stoichiometry of the T-cell receptor-CD3 complex and key intermediates assembled in the endoplasmic reticulum. EMBO J. 2004;23:2348–57. doi: 10.1038/sj.emboj.7600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manolios N, Kemp O, Li ZG. The T cell antigen receptor alpha and beta chains interact via distinct regions with CD3 chains. Eur J Immunol. 1994;24:84–92. doi: 10.1002/eji.1830240114. [DOI] [PubMed] [Google Scholar]
- 68.Manolios N, Letourneur F, Bonifacino JS, Klausner RD. Pairwise, cooperative and inhibitory interactions describe the assembly and probable structure of the T-cell antigen receptor. EMBO J. 1991;10:1643–1651. doi: 10.1002/j.1460-2075.1991.tb07687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brenner MB, Trowbridge IS, Strominger JL. Cross-linking of human T cell receptor proteins: association between the T cell idiotype beta subunit and the T3 glycoprotein heavy subunit. Cell. 1985;40:183–190. doi: 10.1016/0092-8674(85)90321-6. [DOI] [PubMed] [Google Scholar]
- 70.Koning F, Maloy WL, Coligan JE. The implications of subunit interactions for the structure of the T cell receptor-CD3 complex. Eur J Immunol. 1990;20:299–305. doi: 10.1002/eji.1830200211. [DOI] [PubMed] [Google Scholar]
- 71.Touma M, et al. Importance of the CD3gamma ectodomain terminal beta-strand and membrane proximal stalk in thymic development and receptor assembly. J Immunol. 2007;178:3668–3679. doi: 10.4049/jimmunol.178.6.3668. [DOI] [PubMed] [Google Scholar]
- 72.Kim ST, et al. The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang J-H, Reinherz EL. A new angle on TCR activation. Immunity. 2011;35:658–660. doi: 10.1016/j.immuni.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Husson J, Chemin K, Bohineust A, Hivroz C, Henry N. Force generation upon T cell receptor engagement. PLos One. 2011;6:19680. doi: 10.1371/journal.pone.0019680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T lymphocyte activation. Biophys J. 2012;102:L05–L07. doi: 10.1016/j.bpj.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li YC, et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol. 2010;184:5959–5963. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- 77.Ma Z, Finkel TH. T cell receptor triggering by force. Trends Immunol. 2010;31:1–6. doi: 10.1016/j.it.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dustin ML, Zhu C. T cells like a firm molecular handshake. Proc Natl Acad Sci USA. 2006;103:4335–4336. doi: 10.1073/pnas.0600899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandes RA, et al. The T-cell receptor is a structure capable of initiating signalling in the absence of large conformational rearrangements. J Biol Chem. 2012;287:13324–13335. doi: 10.1074/jbc.M111.332783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Janeway CAJ. Ligands for the T cell receptor: hard times for avidity models. Immunol Today. 1995;16:223–225. doi: 10.1016/0167-5699(95)80163-4. [DOI] [PubMed] [Google Scholar]
- 81.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 82.Minguet S, Schamdel WW. A permissive geometry model for TCR-CD3 activation. Trends Biochem Sci. 2008;33:51–57. doi: 10.1016/j.tibs.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 83.Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 84.Hashimoto-Tane A, et al. Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation. Immunity. 2011;34:919–931. doi: 10.1016/j.immuni.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 85.Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. doi: 10.1146/annurev-biophys-042910-155238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 87.Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. 2006;7:803–809. doi: 10.1038/ni1369. [DOI] [PubMed] [Google Scholar]
- 88.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma Z, Janmey PA, Finkel TH. The receptor deformation model of TCR triggering. FASEB J. 2008;22:1002–1008. doi: 10.1096/fj.07-9331hyp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 91.Ruggeri ZM. Von Willebrand factor: looking back and looking forward. Thromb Haemost. 2007;98:55–62. [PubMed] [Google Scholar]
- 92.Reinherz EL, Schlossman SF. The differentiation and function of human T lymphocytes. Cell. 1980;19:821–827. doi: 10.1016/0092-8674(80)90072-0. [DOI] [PubMed] [Google Scholar]
- 93.Gao GF, et al. Crystal structure of the complex between human CD8alpha(alpha) and HLA- A2. Nature. 1997;387:630–634. doi: 10.1038/42523. [DOI] [PubMed] [Google Scholar]
- 94.Kern PS, et al. Structural basis of CD8 co-receptor function revealed by crystallographic analysis of a nurine CD8aa ectodomain fragment in complex with H-2Kb. Immunity. 1998;9:519–530. doi: 10.1016/s1074-7613(00)80635-4. [DOI] [PubMed] [Google Scholar]
- 95.Wang R, Natarajan K, Margulies DH. Structural basis of the CD8 alpha beta/MHC class I interaction: focused recognition orients CD8 beta to a T cell proximal position. J Immunol. 2009;183:2554–2564. doi: 10.4049/jimmunol.0901276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J-H, et al. Crystal structure of the human CD4 N-terminal two domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci. 2001;98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chang HC, et al. Structural and mutational analyses of a CD8alphabeta heterodimer and comparison with the CD8alphaalpha homodimer. Immunity. 2005;23:661–671. doi: 10.1016/j.immuni.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 98.Moody AM, Chui D, Reche PA, Priatel JJ, Marth JD, Reinherz EL. Developmentally regulated glycosylation of the CD8alphabeta coreceptor stalk modulates ligand binding. Cell. 2001;107:501–512. doi: 10.1016/s0092-8674(01)00577-3. [DOI] [PubMed] [Google Scholar]
- 99.Moebius U, Kober G, Griscelli AL, Hercend T, Meuer SC. Expression of different CD8 isoforms on distinct human lymphocyte subpopulations. Eur J Immunol. 1991;21:1793–1800. doi: 10.1002/eji.1830210803. [DOI] [PubMed] [Google Scholar]
- 100.Poussier P, Julius M. Thymus independent T cell development and selection in the intestinal epithelium. Annu Rev Immunol. 1994;12:521–553. doi: 10.1146/annurev.iy.12.040194.002513. [DOI] [PubMed] [Google Scholar]
- 101.Kim PW, Sun ZY, Blacklow SC, Wagner G, Eck MJ. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science. 2003;301:1725–1728. doi: 10.1126/science.1085643. [DOI] [PubMed] [Google Scholar]
- 102.Weiss A. T cell antigen receptor signal transduction: A tale of tails and cytoplasmic protein-tyrosine kinases. Cell. 1993;73:209–212. doi: 10.1016/0092-8674(93)90221-b. [DOI] [PubMed] [Google Scholar]
- 103.Tikhonova AN, et al. αβ T cell receptors that do not undergo major histocompatibility complex-specific thymic selection possess antibody-like recognition specificities. Immunity. 2012;36:79–91. doi: 10.1016/j.immuni.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiong Y, Kern P, Chang H, Reinherz E. T cell receptor binding to a pMHCII ligand Is kinetically distinct from and Independent of CD4. J Biol Chem. 2001;276:5659–5667. doi: 10.1074/jbc.M009580200. [DOI] [PubMed] [Google Scholar]
- 105.Gao GF, Rao Z, Bell JI. Molecular coordination of alphabeta T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol. 2002;23:408–413. doi: 10.1016/s1471-4906(02)02282-2. [DOI] [PubMed] [Google Scholar]
- 106.Kern P, Hussey RE, Spoerl R, Reinherz EL, Chang HC. Expression, purification, and functional analysis of murine ectodomain fragments of CD8alphaalpha and CD8alphabeta dimers. J Biol Chem. 1999;274:27237–27243. doi: 10.1074/jbc.274.38.27237. [DOI] [PubMed] [Google Scholar]
- 107.Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009;126:165–176. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc Natl Acad Sci USA. 2010;107:16916–16921. doi: 10.1073/pnas.1010568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nika K, et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity. 2010;32:66–77. doi: 10.1016/j.immuni.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang N, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity. 2011;34:13–23. doi: 10.1016/j.immuni.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moody AM, et al. Sialic acid capping of CD8beta core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–7246. doi: 10.1074/jbc.M210468200. [DOI] [PubMed] [Google Scholar]
- 112.Classon BJ, et al. The hinge region of the CD8 alpha chain: structure, antigenicity, and utility in expression of immunoglobulin superfamily domains. Int Immunol. 1992;4:215–225. doi: 10.1093/intimm/4.2.215. [DOI] [PubMed] [Google Scholar]
- 113.Rudd PM, et al. Roles for glycosylation of cell surface receptors involved in cellular immune recognition. J Mol Biol. 1999;293:351–366. doi: 10.1006/jmbi.1999.3104. [DOI] [PubMed] [Google Scholar]
- 114.Ghendler Y, et al. Differential thymic selection outcomes stimulated by focal structural alteration in peptide/major histocompatibility complex ligands. Proc Natl Acad Sci USA. 1998;95:10061–10066. doi: 10.1073/pnas.95.17.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Y, et al. The crystal structure of a TL/CD8alphaalpha complex at 2. 1 A resolution: implications for modulation of T cell activation and memory. Immunity. 2003;18:205–215. doi: 10.1016/s1074-7613(03)00027-x. [DOI] [PubMed] [Google Scholar]
- 116.Wang XX, et al. Affinity maturation of human CD4 by yeast surface display and crystal structure of a CD4-HLA-DR1 complex. Proc Natl Acad Sci USA. 2011;108:15960–15965. doi: 10.1073/pnas.1109438108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yin Y, Wang XX, Mariuzza RA. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc Natl Acad Sci USA. 2012;109:5405–5410. doi: 10.1073/pnas.1118801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang J. Protein recognition by cell surface receptors: physiological receptors versus virus interactions. Trends Biochem Sci. 2002;27:122–126. doi: 10.1016/s0968-0004(01)02038-2. [DOI] [PubMed] [Google Scholar]
- 120.Wang J-H, et al. Atomic structure of a fragment of human CD4 containing two immunoglobulin-like domains. Nature. 1990;348:411–418. doi: 10.1038/348411a0. [DOI] [PubMed] [Google Scholar]