

The zebrafish has become a significant model system for studying renal organogenesis and disease as well as for the quest for new therapeutics, thanks to the structural and functional simplicity of the embryonic kidney. Inroads to the nature and disease states of kidney related ciliopathies and acute kidney injury (AKI) have been advanced by zebrafish studies. This model organism has been instrumental in the analysis of mutant gene function for human disease with respect to ciliopathies. In addition, in the AKI field, recent work in the zebrafish has identified a bona fide adult zebrafish renal progenitor (stem) cell that is required for neo-nephrogenesis, both during the normal lifespan and in response to renal injury. Taken together, these studies solidify the zebrafish as a successful model system for studying the broad spectrum of ciliopathies and AKI that affect millions of humans worldwide, and point to a very promising future of zebrafish drug discovery. The emphasis of this review will be on the role of the zebrafish as a model for human kidney-related ciliopathies and acute kidney injury, and how our understanding of these complex pathologies is being furthered by this tiny teleost.
INTRODUCTION
The zebrafish (Danio rerio) is a freshwater tropical fish that spawns via external fertilization. The ex utero developing embryos progress rapidly through embryogenesis and are characterized by optical transparency. These features aid in vitro analysis of gene expression via in situ hybridization and immunohistochemistry as well as in vivo studies using transgenic reporter lines. Adult fish are small, allowing large numbers of animals to be maintained in a minimal amount of space. In addition, they breed frequently, yield large numbers of progeny, and have a generation time of three months. Perhaps the greatest reason the zebrafish has become a widely studied vertebrate model is that it is amenable to genetic screens. Several large-scale mutant screens have been performed that identified new genes involved in many aspects of organogenesis, including kidney development (Driever et al. 1996; Haffter and Nusslein-Volhard 1996; Drummond et al. 1998; Amsterdam et al. 1999).
In the thirty years since George Streisinger first broke new ground using zebrafish as a genetically amenable research organism at the University of Oregon (Streisinger et al. 1981; Chakrabarti et al. 1983; Walker and Streisinger 1983), it has developed from a pet store novelty to a model for studying embryogenesis and human disease pathologies. During this progression from basic to translational research, the zebrafish has been employed in a plethora of studies modeling human disease including those for congenital defects such as Fraser syndrome (Carney et al. 2010), Waardenburgh syndrome (Dutton et al. 2009) and muscular dystrophy (Thornhill et al. 2008); numerous cancers like melanoma (Patton et al. 2005; Ceol et al. 2011; White et al. 2011), epithelial tumors (Shepard et al. 2007), neuroendocrine carcinoma (Yang et al. 2004), and leukemia (Langenau et al. 2003); as well as neurodegenerative disorders including tauopathies (Bai et al. 2007; Paquet et al. 2010), Parkinson’s disease (Flinn et al. 2009), and Huntington’s disease (Williams et al. 2008). In addition, several zebrafish models of regeneration have been utilized to understand the repair potential of the limb (fin) (White et al. 1994; Akimenko et al. 1995), heart (Poss et al. 2002; Jopling et al. 2010; Kikuchi et al. 2010), retina (Cameron and Carney 2000; Vihtelic and Hyde 2000), lateral line hair cells (Harris et al. 2003; Lopez-Schier and Hudspeth 2006) and kidney (Reimschuessel 2001; Hentschel et al. 2005; Zhou et al. 2010; Diep et al. 2011).
This review will focus on the zebrafish kidney and will illustrate how kidney organogenesis studies have evolved into translational-based research. The zebrafish has become an important model system for studying renal disease thanks to the anatomical simplicity of the embryonic kidney (Drummond 2005). Although functionally similar, three types of kidneys have evolved in vertebrates: the pronephric, mesonephric, and metanephric kidneys. The metanephros, the most complex kidney, is only present in birds and mammals, but develops from the two simpler, pronephric and mesonephric, kidneys. Zebrafish have pronephric (embryonic) and mesonephric (adult) kidneys. All three types of kidney utilize a common functional substructure called the nephron. Generally, kidney nephrogenesis can be divided into four stages: (1) specification of intermediate mesoderm as nephrogenic mesenchyme, (2) growth and epithelialization of the anlagen, (3) induction and patterning of the nephron, and (4) formation of the glomerular capillary tuft from invading endothelial cells (Drummond 2003). In the zebrafish embryonic kidney, these stages occur once during organ formation to create two bilaterally paired nephrons, whereas, in the mammalian kidney, they are reiterated many times to create a million nephrons in humans. Significantly, there is a striking similarity in the molecular and segmental organization of the mammalian and zebrafish nephrons (Wingert et al. 2007; Wingert and Davidson 2008).
The simple nature of the zebrafish pronephric kidney makes it a suitable system to study the early developmental events that lay the foundation for genesis of more complex kidneys. Even though the pronephros in mammalian and avian embryos is not functional, it is important for initiation of the molecular events of kidney development (Vize 2003). Microsurgical inhibition of pronephros development in chick embryos and loss of the pronephros in mouse knockouts results in a lack of mesonephros and metanephros development (Shawlot and Behringer 1995; Mauch et al. 2000; Bouchard et al. 2002; James and Schultheiss 2003). Chick embryological experiments also revealed that initial patterning of the intermediate mesoderm is dependent upon signals from the axial/paraxial mesoderm (Mauch et al. 2000; James and Schultheiss 2003). However, the mechanism of how the specified intermediate mesoderm differentiates into nephric mesenchyme is not completely understood. Several studies have defined a set of genes that are expressed within the intermediate mesoderm of all vertebrate species, which later become restricted to the nephrogenic mesenchyme, and have therefore been implicated in kidney formation, including lhx1, pax8, pax2, c-ret, hnf-1β and components of the Notch pathway (Barnes et al. 1994; Toyama and Dawid 1997; Pfeffer et al. 1998; Mauch et al. 2000; Bouchard et al. 2002; Drummond 2002; Kopan et al. 2007; Drews et al. 2011). The emphasis of this review will be on the role of the zebrafish as models for human kidney-related ciliopathies and acute kidney injury.
CILIOPATHIES
Cilia are microtubule-based organelles that are anchored and extend from the apical surface of most eukaryotic cells. They can generally be classified as either motile or immotile. The importance of motile cilia has been documented for some time in a number of organisms. For example, the cilia that line the trachea or are found on the ependymal cells of brain ventricles are critical for the movement of mucus and cerebrospinal fluid, respectively (Yoder 2007; Vincensini et al. 2011). However, the immotile or primary cilium has only recently gained respect within the scientific community. Evidence suggests that the primary cilium can act as a mechanosensor and transducer of various signaling pathways which are critical for normal development and when disrupted, for disease progression (Yoder 2007; Lancaster and Gleeson 2009; Goetz and Anderson 2010).
The assembly of the primary cilium is a dynamic process that is tightly linked to the cell cycle, such that cilia are built during G1 and retracted as cells enter mitosis (Pedersen et al. 2008; Santos and Reiter 2008). Derived from the mother centriole, the basal body serves as the foundation of the cilium and is composed of nine triplet microtubules that form a barrel-like structure (Yoder 2007; Pedersen et al. 2008). The core of the cilium, the axoneme, extends from this basal body and consists of nine microtubule doublets. The presence of a central pair of microtubules (9+2 configuration) has been linked to motile cilia while this pair is normally absent in primary cilia (9+0 configuration), although exceptions to this rule are found (Yoder 2007; Vincensini et al. 2011). Construction of the ciliary axoneme requires intraflagellar transport (IFT), a bidirectional motility system that localizes between the axoneme and the membrane of the cilium (Pedersen et al. 2008). IFT particles are transported to the distal tip of the growing axoneme (anterograde) by kinesin-2 motors and return to the base of the cilium (retrograde) with the assistance of cytoplasmic dynein-2. These IFT particles carry a variety of different cargo including axonemal precursors and components of important developmental signaling pathways (Goetz and Anderson 2010).
Deciphering the roles that primary cilia play during development and adult homeostasis has been the driving force behind the work from many laboratories. While it has been appreciated for some time that motile cilia participate in fluid flow, the function of primary cilia was always less clear. However, studies from a number of groups have suggested that primary cilia function as environmental sensors, detecting changes in flow and light (Vincensini et al. 2011). They also appear to function as signaling centers since a number of important developmental pathways, including hedgehog and Wnt, converge on this organelle (Veland et al. 2009; Goetz and Anderson 2010). The number of proteins that localize to the cilium, basal body, or centrosome (centrioles with associated pericentriolar material) has increased dramatically in recent years. There are currently over 2600 entries in the cilia proteome database (www.ciliaproteome.org), highlighting the importance of this organelle in a variety of cell types and cellular processes. As a result of these localization studies, connections have been made between cilia dysfunction and human diseases/syndromes, collectively termed ciliopathies. Disruption of motile cilia often leads to early embryonic death due to respiratory dysfunction, hydrocephalus, and/or the failure of embryonic turning (Badano et al. 2006; Cardenas-Rodriguez and Badano 2009; Tobin and Beales 2009). In less severe cases, defects in left-right asymmetry are apparent, resulting in the randomization of visceral organs or complete situs inversus. Compromising the function of primary cilia results in a range of syndromes, with the development of kidney cysts being a common denominator. Other common phenotypes include liver disease, polydactyly, retinal degeneration, mental retardation, and neural tube defects.
Model organisms have been instrumental in the analysis of gene function, and have been used extensively in the study of human disease, particularly with respect to ciliopathies. While researchers often look to the mouse for answers, the zebrafish is emerging as an excellent system in which to analyze ciliary function. In the zebrafish, apical cilia are present in Kupffer’s vesicle (KV) (the equivalent of the mouse node), the spinal cord, and the pronephric kidney (Kramer-Zucker et al. 2005). The cilia in all of these organs are motile but have a slightly different architecture, namely, the cilia in the KV and kidney are 9+2 while those in the central canal are 9+0. Interestingly, knockdown of the zebrafish IFT88 ortholog (polaris/oval) results in phenotypes similar to those observed when IFT88 function is compromised in the mouse (Pazour et al. 2000; Kramer-Zucker et al. 2005). Not only does this data suggest that the function of this IFT component is conserved within vertebrates but also that studies in the fish can be used to tease out the intricacies within the field of cilia biology.
Polycystic kidney disease
Polycystic kidney disease (PKD), which can be subdivided into autosomal dominant (ADPKD) and autosomal recessive (ARPKD), is characterized primarily by the formation of renal cysts (Torres and Harris 2009). ADPKD is one of the most common monogenic disorders, affecting between 1/400 and 1/1000 individuals, and has been linked to mutations in two genes, PKD1 (polycystin-1) and PKD2 (polycystin-2). Polycystin-1 contains a large extracellular N-terminus, 11 transmembrane domains, and a shorter cytoplasmic C-terminus while polycystin-2 is a much smaller protein, spanning the membrane only 6 times with intracellular N- and C- termini (Chapin and Caplan 2010). Data suggest that the polycystins form a complex that regulates the level of intracellular Ca+2, and this interaction is facilitated by their colocalization, especially to the primary cilium.
In particular, the zebrafish has served as an excellent model system for analyzing the role of pkd2 during development. Initial studies showed that pkd2 was expressed in multiple ciliated-tissues, including Kupffer’s vesicle (KV), the pronephric duct, floorplate, and brain (Bisgrove et al. 2005). Knockdown of pkd2 gene function resulted in axis curvature, kidney cysts, pericardial edema, and hydrocephalus as well as defects in left-right asymmetry, consistent with phenotypes observed in Pkd2−/− mice (Wu et al. 1998; Wu et al. 2000; Pennekamp et al. 2002; Sun et al. 2004; Bisgrove et al. 2005). It was later shown that the Pkd2 protein localizes to cilia and the basolateral surface of epithelial cells in the anterior pronephros (Obara et al. 2006), and this localization is required for proper function of Pkd2 within the kidney (Obara et al. 2006; Streets et al. 2006; Fu et al. 2008).
Since both PKD1 and PKD2 localize to the cilium, it is reasonable to suggest that proper function of this organelle is required to prevent cyst formation. In support of this idea, two independent screens in the zebrafish identified a large number of genes that, when mutated, gave rise to cystic kidneys (Drummond et al. 1998; Sun et al. 2004). Two such genes, curly and moe, were identified as the zebrafish orthologs of ift57 and ift172, respectively (Sun et al. 2004; Cao et al. 2010). Analysis of pronephric cilia in ift57hi3417 and ift172hi2211 mutants demonstrated that a subset of ciliated cells was affected. There are both single-ciliated (SCC) and multi-ciliated (MCC) cells present in the zebrafish kidney at 48 hours post-fertilization (hpf) (Liu et al. 2007; Ma and Jiang 2007). While the SSCs were unaffected, there was a disorganization of the MCCs due to disorganized basal bodies in the tubule epithelial cells (Cao et al. 2010). This phenotype was also observed upon knockdown of prickle1 (pk1), a core component of the planar cell polarity (PCP) pathway. Interestingly, both ift57 and ift172 genetically interact with pk1, suggesting that alterations in the PCP pathway are responsible for pronephric cyst formation in these ift mutants.
Understanding the mechanism of cyst formation is crucial for designing possible treatment options for PKD. Consequently, studies in zebrafish have focused on characterizing this process within the pronephros. Using mutations in switch hitter (swt), the zebrafish ortholog of LRRC50, researchers have found that defects in the motility of pronephric cilia contribute to cyst formation (Sullivan-Brown et al. 2008; van Rooijen et al. 2008). Interestingly, these defects were identified as early as 26 hpf and suggest that disruption of normal ciliary function is the initial cause of pronephric cysts in this mutant. By 48 hpf, cysts within the proximal tubule segments (PCT and PST (Wingert et al. 2007)) develop, followed shortly by glomerular dilation. There is also an increase in proliferation and a mislocalization of Na+/K+ATPase around and within the tubules, respectively (Sullivan-Brown et al. 2008; van Rooijen et al. 2008). However, these are only thought to contribute to cyst formation because they occur after the identification of cysts (Sullivan-Brown et al. 2008). These phenotypes differ slightly from those observed in pkd2 morphants (Bisgrove et al. 2005; Obara et al. 2006; Schottenfeld et al. 2007; Sullivan-Brown et al. 2008). While pronephric cysts develop, they occur without defects in ciliary motility and appear localized to the glomerulus. These studies highlight the heterogeneity that exists within cystic kidney disease as well as the importance for normal ciliary function in the prevention of kidney cyst formation.
Nephronopthisis
While nephronopthisis (NPHP) is a cystic kidney disease, the kidney does not become enlarged due to cyst formation throughout the organ (Hildebrandt and Zhou 2007; Hildebrandt et al. 2009). Rather, the cysts that develop are localized to the corticomedullary region, resulting in little change in the overall size of the kidney. This disease is also characterized by tubular basement membrane disruption and tubulointerstitial fibrosis. NPHP is an autosomal recessive disorder and represents the most common genetic cause of end-stage renal disease during the first 3 decades of life. To date, mutations in ten genes have been shown to cause NPHP (Hildebrandt et al. 2009). Since these proteins generally localize to primary cilia, basal bodies, and centrosomes, a role for the cilium in the pathogenesis of NPHP has been suggested.
Similar to phenotypes seen in humans and mice (Gagnadoux et al. 1989; Mochizuki et al. 1998; Morgan et al. 1998), knockdown of inversin (invs) (nphp2) in zebrafish results in ventral axis curvature, severe pronephric cyst development, and randomization of heart looping (Otto et al. 2003). Interestingly, both the axis curvature and cyst development can be rescued by injection of the wildtype mouse Invs mRNA, suggesting a conserved function for NPHP2/inversin within the vertebrate lineage. This model of cystogenesis, generated by knockdown of invs in zebrafish, was utilized to test the role of canonical Wnt signaling in renal development (Simons et al. 2005). Injection of diversin, a molecule structurally related to inversin and capable of inhibiting the canonical Wnt pathway, prevented renal cyst formation in invs morphants suggesting that proper regulation of Wnt signaling is important for renal development. Knockdown of other NPHP genes in zebrafish using morpholinos produced similar phenotypes to those observed in invs morphants (Schafer et al. 2008; Zhou et al. 2010). Both nphp3 and nphp5 morphants displayed body axis curvature, pronephric cyst development, and hydrocephaly. In addition to these phenotypes, left-right patterning defects were also seen upon knockdown of nphp3 (Schafer et al. 2008; Zhou et al. 2010). These phenotypes suggest a conserved role for both proteins in proper ciliary function.
In individuals with NPHP, there is often involvement of multiple organs outside of the kidney. When NPHP is combined with cerebellar vermis aplasia, patients are diagnosed with a separate, rare autosomal recessive disorder called Joubert syndrome (JBTS) (Hildebrandt and Zhou 2007). JBTS occurs in between 1/80,000 and 1/100,000 live births and is primarily diagnosed by the presence of the “molar tooth sign,” a midbrain-hindbrain malformation visible on brain imaging (Brancati et al. 2010). Abnormal eye movements, breathing difficulties, hypotonia, and developmental delay/mental retardation also characterize this syndrome (Harris 2007; Brancati et al. 2010). Currently, there are ten causative genes that have been identified and they may explain the pleiotropy associated with this syndrome (Brancati et al. 2010).
Interestingly, the NPHP6 gene has been implicated in both NPHP and JBTS. Morpholino knockdown of cep290 (nphp6) results in pronephric cyst development, ectopic tissue in the fourth ventricle, and a reduction in retinal tissue (Sayer et al. 2006). These phenotypes are reminiscent of the meningoencephalocele and retinal degeneration seen in some patients with JBTS. Further characterization of the molecular function of NPHP6 has also been performed in the zebrafish. Nphp6 was shown to genetically interact with nphp5 (Schafer et al. 2008) as well as with cc2d2a/JBTS9, another causative gene linked to JBTS (Gorden et al. 2008). Injection of the cep290 morpholino into sentinal (snl) (cc2d2a) mutants resulted in larger and more prevalent pronephric cyst formation, suggesting a functional synergism between these two genes (Gorden et al. 2008).
Similar to CEP290, RPGRIP1L has also been linked to both NPHP and JBTS. Retinal degeneration is a common but not universal phenotype in many ciliopathies. Resequencing of known genes involved in ciliopathies identified a mutation in RPGRIP1L (A229T) that was enriched in individuals with retinal defects (Khanna et al. 2009). To test the functional significance of this mutation, a splice-blocking morpholino was designed to target the zebrafish ortholog, rpgrip1l. Knockdown of this gene resulted is gastrulation defects as well as tail curvature. These phenotypes could be rescued by the wildtype human mRNA but could not be rescued to the same degree by a human mRNA encoding the mutant Thr229. Thus, studies in zebrafish have enabled researchers to identify genes that can serve as phenotypic modifiers, and this may explain, at least in the case of CEP290 and RPGRIP1L, why they have been implicated in multiple ciliopathies.
Many of the studies examined so far have utilized zebrafish to test the functionality of a gene once it has been linked to an established disease. However, one of the strengths of zebrafish as a model organism is the ability to conduct forward genetic screens. As the result of one such screen, the first analysis of a gene associated with JBTS was performed. Scorpion (sco)/arl13b mutants displayed a curved body axis as well as kidney cysts (Sun et al. 2004). Because of the phenotypic similarity to other mutants identified in the screen, later determined to be zebrafish orthologs of IFT components, cilia formation was analyzed in sco mutants. Based on the absence of acetylated tubulin staining in the kidney (pronephros), it was concluded that sco was required for cilia formation. Further analysis showed that arl13b was expressed in ciliated tissues and was localized to the cilium in the pronephric duct, neural tube, olfactory placode, and KV (Duldulao et al. 2009).
Genome mapping and sequencing of families affected by Joubert syndrome and related disorders (JSRD) later identified ARL13B (JBTS8), a member of the Ras GTPase family, as a causative gene (Cantagrel et al. 2008). Interestingly, this group utilized the zebrafish to test the functionality of the human protein containing the mutations that were identified in individuals with JSRD. Injection of the wildtype human ARL13B mRNA was able to rescue the curved axis and kidney cysts observed in zebrafish sco mutants, suggesting a conservation of function across vertebrate lineages for this protein. However, two separate human mRNAs containing the missense mutations identified in JSRD families could not show the same degree of rescue. Taken together, the above studies suggest that Joubert syndrome should be considered a ciliopathy. To provide further evidence for this classification, the cilia in sco mutants and morphants were analyzed in more detail (Duldulao et al. 2009). In scohi459 mutants, closer examination of cilia in the pronephric duct demonstrated that the number of cilia was significantly reduced and their motility was compromised. Deletion analysis suggested that both the small GTPase domain as well as the coiled-coil domain are required for localization of Arl13b to the cilium, and that this localization is required for function. These studies not only demonstrate the conserved function of ARL13B in vertebrates but also highlight the utility of zebrafish in determining the functionality of a protein associated with human disease.
Bardet-Biedl syndrome
It has been suggested that Bardet-Biedl syndrome (BBS) is a model ciliopathy because the characteristic features of BBS are shared with other disorders (Zaghloul and Katsanis 2009). This overlap can be seen at the level of phenotype, involvement of common signaling pathways, and protein co-localization. While BBS is normally inherited in an autosomal recessive fashion, the genetic heterogeneity and presence of second-site modifiers makes this syndrome a representative of the ciliopathy disease spectrum (Zaghloul et al. 2010). Currently, mutations in 14 loci have been shown to cause BBS (Table 1), which is characterized by retinal degeneration, obesity, hypogonadism, polydactyly, renal dysfunction, and mental retardation (Zaghloul and Katsanis 2009). Through the use of the zebrafish, significant progress has been made with respect to understanding not only the in vivo functions of the BBS genes but also the variants that contribute to this and related syndromes.
Table 1.
Causative Loci in Multiple Ciliopathies and their zebrafish orthologs
| Causative Loci in Humans | Zebrafish Ortholog | Phenotype | References |
|---|---|---|---|
| Polycystic Kidney Disease (PKD) | |||
| Pkd1 (Polycystin-1) | pkd1a/pkd1b | Dorsal axis curvature, pronephric kidney cysts, hydrocephalus, jaw defects | (Mangos et al. 2010) |
| Pkd2 (Polycystin-2) | pkd2/curly up | Dorsal axis curvature, pronephric kidney cysts, pericardial edema, hydrocephalus, laterality defects | (Bisgrove et al. 2005; Obara et al. 2006; Schottenfeld et al. 2007; Fu et al. 2008; Sullivan-Brown et al. 2008) |
| Nephronopthisis (NPHP) | |||
| NPHP1 | N.D. | ||
| NPHP2/inversin | inversin | Ventral axis curvature, pronephric kidney cysts, pericardial edema, laterality defects | (Otto et al. 2003; Simons et al. 2005) |
| NPHP3 | nphp3 | Ventral axis curvature, pronephric kidney cysts, hydrocephalus, laterality defects, CE defects, fewer and shorter cilia in KV | (Zhou et al. 2010) |
| NPHP4 | N.D. | ||
| NPHP5 | nphp5/iqcb1 | Axis curvature, pronephric kidney cysts, hydrocephalus | (Schafer et al. 2008) |
| NPHP6/CEP290 | cep290 | Ventral axis curvature, pronephric kidney cysts, hydrocephalus, CE defects, deficient retinal tissue | (Sayer et al. 2006; Leitch et al. 2008; Schafer et al. 2008) |
| NPHP7/GLIS2 | N.D. | ||
| NPHP8/RPGRIP1L | rpgrip1l | Dorsal axis curvature, CE defects | (Khanna et al. 2009) |
| NPHP9/NEK8 | nek8 | Kidney cysts | (Liu et al. 2002) |
| NPHP11/MKS3 | mks3 | CE defects | (Leitch et al. 2008) |
| Joubert Syndrome (JBTS) | |||
| JBTS1/INPP5E | inpp5e | N.D. | |
| JBTS2/TMEM216 | tmem216 | Ventral axis curvature, pericardial edema, hydrocephaly, CE defects | (Valente et al. 2010) |
| JBTS3/AHI1 | ahi1 | N.D. | |
| JBTS4/NPHP1 | See NPHP1 | ||
| JBTS5/CEP290 | See NPHP6 | ||
| JBTS6/TMEM67/MKS3 | tmem67 | CE defects | (Leitch et al. 2008; Valente et al. 2010) |
| JBTS7/RPGRIP1L | See NPHP8 | ||
| JBTS8/ARL13B | arl13b/scorpion | Ventral axis curvature; pronephric kidney cysts; laterality defects, CE defects, reduced number of cilia in pronephros and KV | (Sun et al. 2004; Cantagrel et al. 2008; Duldulao et al. 2009) |
| JBTS9/CC2D2A | cc2d2a/sentinal | Dorsal axis curvature, pronephric kidney cysts | (Gorden et al. 2008) |
| JBTS10/OFD1 | ofd1 | Axis curvature, pericardial edema, hydrocephalus, laterality defects, CE defects | (Ferrante et al. 2009) |
| Bardet-Biedl Syndrome (BBS) | |||
| BBS1 | bbs1 | CE defects, KV defects, delay in melasome transport, expansion of Shh expression in fin bud | (Gerdes et al. 2007; Tayeh et al. 2008; Zaghloul et al. 2010) |
| BBS2 | bbs2 | KV defect, degenerate cila in KV, delay in melanosome transport | (Yen et al. 2006) |
| BBS3 | arl6/bbs3 | KV defects, laterality defects, delay in melanosome transport | (Tayeh et al. 2008) |
| BBS4 | bbs4 | CE defects, KV defect, degenerate cilia in KV, laterality defects, delay in melanosome transport | (Yen et al. 2006; Gerdes et al. 2007; Zaghloul et al. 2010) |
| BBS5 | bbs5 | KV defects, laterality defects, delay in melanosome transport | (Yen et al. 2006) |
| BBS6/MKKS | mkks/bbs6 | Pronephric kidney cysts, CE defects, KV defect, degenerate cilia in KV, laterality defects, delay in melanosome transport | (Yen et al. 2006; Gerdes et al. 2007; Zaghloul et al. 2010) |
| BBS7 | bbs7 | KV defect, degenerate cila in KV, laterality defects, delay in melanosome transport, expansion of Shh expression in fin bud | (Yen et al. 2006; Tayeh et al. 2008) |
| BBS8 | ttc8/bbs8 | Ventral axis curvature, pronephric kidney cysts, KV defect; laterality defects, delay in melanosome transport | (Yen et al. 2006; Tobin and Beales 2008) |
| BBS9 | bbs9 | CE defects | (Zaghloul et al. 2010) |
| BBS10 | bbs10 | CE defects | (Zaghloul et al. 2010) |
| BBS11 | trim32/bbs11 | KV defect, delay in melanosome transport | (Chiang et al. 2006) |
| BBS12 | bbs12 | CE defects | (Stoetzel et al. 2007; Zaghloul et al. 2010) |
| BBS13/MKS1 | mks1 | Ventral axis curvature, pronephric kidney cysts, CE defects | (Leitch et al. 2008; Tobin and Beales 2008) |
| BBS14/CEP290 | See NPHP6 | ||
Abbreviations: N.D. (Not Determined); CE (convergent extension); KV (Kupffer’s Vesicle)
Although the proteins encoded by the BBS genes have all been shown to localize to the basal body, centrosome, or cilia (Cardenas-Rodriguez and Badano 2009), their structures do not place them in similar functional categories (Tayeh et al. 2008). To determine the roles of these genes during development, a number of groups have utilized morpholinos to knockdown the BBS genes individually or in combination. Individual knockdown of bbs1-8 resulted in defects in KV formation (Yen et al. 2006; Tayeh et al. 2008). In particular, cilia in the KV appeared to degenerate following injection of morpholinos targeting bbs2, bbs4, bbs6, and bbs7, which lead to defects in both heart and gut looping (Yen et al. 2006). It was also shown that knockdown of bbs1-8 lead to defects in retrograde melanosome transport, a process that requires the minus-end directed motor dynein (Yen et al. 2006; Tayeh et al. 2008). These results suggest that BBS genes are important for ciliary maintenance and intracellular transport.
Data also suggest that BBS proteins play an important role in modulating the non-canonical Wnt pathway and PCP. Disruption of PCP in zebrafish results in convergent extension defects, which include a shortened and widened body axis (Keller 2002). This phenotype was observed along with widening of the somites and a kinked notochord when bbs4 or bbs6 were knocked down (Badano et al. 2006). Both of these genes genetically interact with vangl2, a core PCP gene (Ross et al. 2005), as well as Wnt5a and Wnt11 (Gerdes et al. 2007). Interestingly, while non-canonical Wnt signaling is compromised in bbs4 and bbs6 morphants, the level of canonical Wnt signaling is increased suggesting that BBS proteins and the cilium are important for regulating the balance between these two signaling pathways (Gerdes et al. 2007).
The PCP phenotype that was first observed with knockdown of either bbs4 or bbs6 has since been used to identify modifiers of BBS and to characterize the spectrum of this disease. Meckel syndrome (MKS), a lethal autosomal recessive disorder, is characterized by cystic kidneys, liver fibrosis, polydactyly, and encephalocoele (Tobin and Beales 2009). Since there is significant phenotypic overlap between MKS and BBS, with the primary exception of encephalocoele, it is possible that MKS represents a more severe form of BBS. To this end, mutations in MKS genes could contribute to or modify BBS mutations. The MKS1 gene was sequenced from over 150 families affected by BBS and a number of mutations were identified (Leitch et al. 2008). Knockdown of mks1 in zebrafish phenocopied bbs morphants and could be rescued with the wildtype MKS1 human mRNA. These morphants, however, could not be rescued to the same degree by constructs containing the identified human mutations. Depending on the degree of rescue, these mutations were classified as benign, hypomorphic, or null. Mks1 was also shown to genetically interact with bbs1, bbs2, and bbs4, supporting the hypothesis that MKS genes can modify BBS phenotypes, and that the penetrance of BBS is dependent on the number of mutations affecting overall ciliary function.
This method of analysis was extended to all of the known BBS genes to test the functionality of 125 identified alleles (Zaghloul et al. 2010). Morpholinos were designed against bbs1-12 and knockdown resulted in the typical PCP phenotypes previously observed, namely a shortened body axis, broadened somites, and a widened/kinked notochord. Based on the ability of human constructs harboring the identified mutations to rescue the morphant phenotypes, an allelic series was created. A number of dominant negative alleles were isolated and at first, seemed inconsistent with a disease that is inherited in a recessive fashion. However, these alleles were mapped to epistatic loci (rather than primary) and function as dominant modifiers. Taken together, these elegant studies in the zebrafish have begun to unravel some of the complexity associated with this syndrome.
ACUTE KIDNEY INJURY
Acute kidney injury (AKI) is a multi-factorial disorder associated with significantly high mortality rates that have remained unchanged for the last twenty years. AKI commonly results from ischemic injury, the use of nephrotoxic agents, and/or sepsis. It occurs in approximately 7% of in-patient hospital admissions (Lameire et al. 2006), and is an independent predictor of in-hospital mortality (Yalavarthy and Edelstein 2008). Severe AKI requiring renal replacement therapy occurs in 4% of critically ill patients and has a 50% in-patient mortality (Goldberg and Dennen 2008). Despite the high mortality rates, at present, renal replacement and dialysis are the only therapies that have proven to be beneficial in humans. Thus, there is an increasing interest in the development of treatments able to prevent or limit kidney injury, and/or enhance regeneration of normal kidney epithelium following injury. Recently, regeneration in the zebrafish kidney has received considerable attention given the potential for the development of new therapeutic strategies.
Mammalian AKI
The vertebrate kidney possesses a remarkable potential to regenerate after an ischemic or nephrotoxic insult (Duffield et al. 2005; Romagnani and Kalluri 2009). Mammalian renal epithelium has the capacity to self renew by symmetric division of differentiated cells that are indistinguishable from their progeny (Vogetseder et al. 2007; Vogetseder et al. 2008). However, the generation of new nephrons has never been observed in mammals, and repair mechanisms often lead to the formation of fibrotic non-functional tissue (Romagnani and Kalluri 2009). Following AKI, the injured cells undergo a transient process of dedifferentiation, characterized by cytoskeletal disruption that results in changes in cell polarity, redistribution of the basolateral Na+/K+ATPase pump to the apical membrane, loss of brush border, and re-expression of mesenchymal markers and markers typical of early nephron development (Sharfuddin and Molitoris 2011). This damage results in epithelial cell loss and denudation of areas of basement membrane. In experimental models of toxic and ischemia reperfusion (IR) kidney injury in rodents, epithelial cell death is largely restricted to the S3 segment of the proximal tubule and occurs within the first 12–24 hours of injury. Over this time period, there is loss of cell-cell junctions between surviving renal tubular epithelial cells along with with cellular flattening and expression of mesenchymal markers, such as vimentin (Witzgall et al. 1994; Belleri et al. 2005; Verghese et al. 2008). Depending on the severity of the insult, regeneration of injured renal tubular epithelial cells occurs over a one to three week period as rapidly-dividing, vimentin-positive cells undergo a mesenchymal-to-epithelial transition and, once again, form functional, polarized epithelia. Recent lineage tracing studies indicate that the bulk of regenerating renal tubular epithelial cells are derived from intrinsic renal epithelium following IR injury (Humphreys et al. 2008). This process of dedifferentiation is believed to promote cell migration into the area where cell death occurred, and the proliferation of these cells reconstitutes the tubular epithelium (Bonventre 2003; Hader et al. 2010). The identification of renal stem cells and mesenchymal stem cells in the repair process has received considerable attention, given the potential for identifying new therapeutic strategies for treatment of kidney disease (Little 2006). However, the contribution of adult stem cells, either of intrarenal origin or from the bone marrow, during the repair phase after AKI is still controversial (Cantley 2005; Benigni et al. 2010).
In addition to the expression of mesenchyme markers, there is evidence that regenerating renal tubular epithelial cells express genetic markers normally associated with the early embryonic renal epithelia (in the pre-tubular aggregates and renal vesicles) including Pax2, Wnt4, Lhx1 and components of the Notch signaling pathway (Terada et al. 2003; Lin et al. 2005; Villanueva et al. 2006; Kobayashi et al. 2008). These markers appear within the first 24 hours following injury and are lost as the cells undergo epithelial differentiation. Since these embryonic markers are required for specification of diverse tubular epithelial cell types during embryonic development of the kidney it has been proposed that expression of these markers is required to specify a number of different cell types within the regenerating renal tubular epithelium (Kopan et al. 2007). However, the mechanisms regulating embryonic gene expression following AKI are unknown. Interestingly, recent studies have shown that renal ischemia is associated with a reversible reduction in histone deacetylase (HDAC) activity and increased histone acetylation (K9 acetyl-histone H3) in renal tubular epithelial cells (Marumo et al. 2008). As epigenetic regulation of gene expression during development is regulated, in part, by histone acetylation (Haberland et al. 2009), these findings suggest that alterations in HDAC activity may mediate epigenetic reprogramming of regenerating renal tubular epithelial cells to a more primitive embryonic epithelial state, and that this may be required for the kidney to undergo complete cellular and functional recovery following injury.
Zebrafish AKI
The zebrafish pronephric kidney has been established as a valid vertebrate model system for AKI studies (Hentschel et al. 2005; Zhou et al. 2010; Diep et al. 2011). Using gentamicin and cisplatin as nephrotoxicants, Hentshel et al., demonstrated nephrotoxic effects in zebrafish larvae similar to the aminoglycoside toxicity observed in mammalian AKI models. In particular, gentamicin mediated damage resulted in pronephric tubule flattening of the brush border, tubular and glomerular distension, lysosomal phospholipidosis, formation of debris in the tubular lumen and accumulation of leukocytes. Similarly, cisplatin treatment caused cellular vacuolization, flattening and loss of brush borders of the tubular cells, and distension of the tubular lumen. These changes resulted in a decline in renal function, due to an inability to maintain water balance, and were associated with the development of pericardial edema. Similar results for a gentamicin-mediated AKI model were described in a detailed methodology for inducing AKI in zebrafish larvae (Cianciolo Cosentino et al. 2010). In addition, the loss of tubular cell polarity, one of the first hallmarks of sub-lethal injury in AKI, was clearly demonstrated by the redistribution of the basolateral Na+/K+ATPase pump to the apical membrane (Fig. 1).
Figure 1. Gentamicin-mediated AKI in zebrafish.

(A,B) Zebrafish larvae at 5 days post fertilization (dpf) injected with (A) vehicle or (B) 7.5 ng gentamicin. Arrow indicates pericardial edema reflecting kidney damage. (C–D) Haematoxylin and eosin (H&E) staining of kidney sections 72 hours post-treatment in (C) vehicle and (D) gentamicin-injected larvae. Inset shows the pronephric tubules, with tubular distension and loss of brush border (compare D with C, arrowheads). (E–F) Immunofluorescence for Na+/K+ATPase 48 hours post-treatment in (E) vehicle and (F) gentamicin-injected larvae. White arrowhead (F) indicates loss of basolateral polarity in proximal tubules. Scale bar is 20 μm.
As opposed to mammals, in which development of new nephrons stops before birth (Kuure et al. 2000), fish can generate new nephrons throughout their lifespan (Reimschuessel and Williams 1995; Reimschuessel 2001). This, perhaps, serves as the largest advantage in using zebrafish as an AKI model. Interestingly, neo-nephrogenesis in fish is greatly enhanced after kidney injury. This was first demonstrated in the goldfish, Carassius auratus, following exposure to hexaclorobutadiene (Reimschuessel et al. 1990). This same process of neo-nephrogenesis has subsequently been found in other fish species with different nephrotoxicants (Reimschuessel 2001). The new nephrons appear as basophilic clusters of cells, called nephrogenic bodies, that are either spread throughout the renal mesenchymal tissue (Watanabe et al. 2009; Diep et al. 2011) or, like in the skate, Leucoraja erinacea, are restricted to a specific nephrogenic zone, containing mesenchymal cells and immature nephrons which resemble the embryonic kidney (Elger et al. 2003). Interestingly, following partial nephrectomy in skates, the formation of new nephrons and the rate of proliferation in the nephrogenic zone were greatly enhanced in both the operated and the intact contralateral kidney.
The signals triggering neo-nephrogenesis following injury are unknown, but may be linked to the accumulation of metabolic waste or a salt imbalance (Davidson 2011). In higher vertebrates, it is still unclear why new nephrons cannot be generated. An intriguing hypothesis suggests that the evolution of higher blood pressures have allowed mammals to respond to an increase in renal demand through incremental increases in glomerular filtration pressure. However, in organisms with low blood pressure, such as the fish, the ability to generate new nephrons has been maintained so that the capacity of the kidney can be expanded (Davidson 2011).
Recently, two groups, using a gentamicin-mediated AKI model, published in vivo studies of neo-nephrogenesis using transgenic zebrafish lines (Zhou et al. 2010; Diep et al. 2011),. Zhou et al., followed zebrafish neo-nephogenesis using two transgenic zebrafish lines: wt1b:GFP, expressed in the glomeruli and proximal tubules, and pod:NTR-mCherry, expressed in the glomeruli. Similar to other fish species, zebrafish continued to generate nephrons throughout adulthood. This was demonstrated by an increase in wt1b:GFP-positive cell aggregates, which constituted the new nephrons. Interestingly, this response was enhanced following renal injury. A similar observation was made in medaka where expression of wt1 was seen in basophilic aggregates following kidney injury (Watanabe et al. 2009).
A more detailed analysis of zebrafish neo-nephrogenesis was conducted, following gentamicin-mediated AKI, and the formation of nephrogenic aggregates in post-AKI animals was observed (Diep et al. 2011). The newly developing nephrons recapitulated mesonephric neo-nephrogenesis, which consisted of small aggregates of lhx1a positive cells (Fig. 2) that would later enlarge and express additional renal genes, such as wt1b, pax2, pax8 and cdh17. In an elegant series of transplantation experiments, it was demonstrated that the lhx1a-positive aggregates possessed the ability to generate new nephrons. Interestingly, the ability to form new nephrons was maintained after serial transplantation assays, suggesting that this aggregate of cells, termed renal progenitor cells, possessed stem cell-like properties. Based on these results, it was proposed that lhx1a represents the earliest marker of renal progenitor cells. This is a significant result as it is the first indication that a cell-based therapy could be used to treat AKI in the future. It also establishes the zebrafish as the first and only model where adult renal progenitor cells can be studied undergoing successful engraftment into a functioning kidney.
Figure 2. Nascent nephrons develop from lhx1a:EGFP-positive cells.
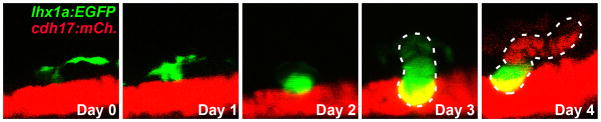
Time course of a Tg(lhx1a:EGFP;cdh17:mCherry) larva demonstrating that multiple lhx1a:EGFP-positive cells coalesce to form a progenitor aggregate, which then differentiates into a nascent nephron.
THE ZEBRAFISH KIDNEY AND CHEMICAL GENETICS
The work described in this review supports the use of zebrafish for modeling human ciliopathies and to study the pathophysiology of AKI. However, the success of any animal model also lies in the ability to use that model for the identification of new therapies for a given disease. One promising area for the continued study of ciliopathies and AKI resides in the possibility of identifying small molecules with therapeutic potential to improve the etiology of human kidney diseases.
Small molecule screens represent a novel approach to identify water-soluble chemicals that can alter a biological process and can be a complement to genetic approaches (Stein 2003; Ding and Schultz 2004). The pharmaceutical industry has focused on such screens with assays designed to uncover therapeutics with the potential to alleviate disease. However, in academic centers, historically a lack of robotics and automated equipment prevented the exploitation of the newest developments in combinatorial chemistry and rapid target validation. The zebrafish embryo has become an attractive tool for small molecule screens because it is a vertebrate that is small in size (1–2 mm), optically transparent, and can be kept alive in 96 well plates for multiple days without the need for additional nutrients (Peterson et al. 2000). Many research groups have used zebrafish to investigate the effects of small molecules (Stern and Zon 2003; Peterson et al. 2004; Burns et al. 2005; Murphey et al. 2006; Sachidanandan et al. 2008; Yu et al. 2008; Molina et al. 2009; de Groh et al. 2010), and a wide variety of assay tools have been successfully developed for analysis of zebrafish screens (Pichler et al. 2003; Tran et al. 2007; Vogt et al. 2009).
Fluorescent transgenic reporter zebrafish lines make it possible to discover specific molecules that can alter differentiation events during organogenesis (Molina et al. 2007). For example, Chan et al., identified molecules that could alter blood vessel formation in the zebrafish embryo, further elucidating the signaling pathways that are employed for vessel formation and regeneration (Chan et al. 2002; Bayliss et al. 2006). Not only did these studies yield insight into vessel formation and the process of angiogenesis in zebrafish, but they may also contribute to the development of assays to screen for anti-angiogenic molecules. In another study, Peterson et al., identified two structurally similar molecules that could rescue the zebrafish genetic mutant, gridlock, which results in the disruption of aortic blood flow and exhibits similar physiological features to aortic coarctation found in humans (Peterson et al. 2004). The implication of these results is that there is now a basis from which to develop molecules that can alleviate aortic coarctation, based on the structures of these two molecules.
Two groups have taken advantage of the zebrafish models for PKD and other ciliopathies to test the ability of several drugs or compounds to ameliorate the presence of kidney cysts. Knockdown of bbs4, bbs6, bbs8, ift80, nphp2, nphp5, nphp6, mks1, mks3, or ofd1 results in the formation of cystic kidneys along with edema (Tobin and Beales 2008). Treatment of each morphant with rapamycin, an inhibitor of the mTOR pathway, significantly reduces the edema seen within the kidney. This effect was partially phenocopied in ift80 morphants by treatment with roscovitine, a cyclin-dependent kinase inhibitor. These data suggest that the cysts forming in the zebrafish models of cystic kidney disease result from a mechanism similar to that which occurs in humans. Thus the identification of new compounds that suppress cyst formation in the zebrafish should translate to an improvement in kidney function in other vertebrates. To this end, a chemical modifier screen was performed, using a custom library of well-defined compounds, to identify those that modify the body axis curvature and/or laterality defects that are present in pkd2hi4166 and ift172hi2211 mutants (Cao et al. 2009)). There were a total of six compounds that modified the axis phenotype and fourteen that affected laterality, with an overlap of only three compounds. One of these compounds, tricostatin A (TSA), was particularly interesting because of its potent ability to inhibit the activity of HDACs. Using pkd2 morphants, it was shown that TSA could effectively reduce kidney cyst formation and this was phenocopied by a second HDAC inhibitor, valproic acid (VPA). Interestingly, a similar effect was observed in Pkd1flox/flox;Pkhd1-Cre mouse kidneys following injection of VPA. The overall size of the kidney as well as the cystic area was reduced with a concomitant improvement in kidney function.
Since innate renal tubular regeneration occurs after tissue injury, there has been considerable interest in developing treatments that enhance the regenerative capacity of the kidney when administered post-injury. This would be a significant advance in the field since most therapies for AKI, which have shown potential when administered prior to the onset of renal injury in experimental models, have failed to show therapeutic benefit in humans. Therefore, the identification of pluripotent renal progenitor cells in zebrafish could be important in identifying the latent regenerative pathways that exist in the human kidney (Romagnani 2009). Likewise, studying self-renewing nephron progenitor cells in zebrafish will allow a better understanding of how to increase the capacity to generate new nephrons. In one zebrafish AKI study, a renal protective effect in zebrafish of the intracellular β-amino acid taurine on gentamicin nephrotoxicity, and of Ucf-101, an inhibitor of the serine protease Omi/HtrA2, on cysplatin nephrotoxicity was shown (Hentschel et al. 2005). In addition, since both zebrafish and mammalian AKI models show reactivation of markers of kidney organogenesis, screens that increase the expression of the embryonic kidney genes could potentially enhance the repair process. Towards this end, a chemical screen in zebrafish identified a new HDAC inhibitor, 4-(phenylthio)butanoic acid (PTBA), that was able to expand the pool of lhx1a-positive embryonic renal progenitor cells (de Groh et al. 2010). Importantly, another HDAC inhibitor, TSA, has been shown to attenuate or partially reverse nephritic serum nephritis (Imai et al. 2007) and decrease the onset of fibrosis in mice (Pang et al. 2009). Testing these compounds in AKI models thus represent a promising strategy to augment kidney regeneration. Taken together, these studies solidify the zebrafish as a successful model system for studying the broad spectrum of ciliopathies and AKI that affect millions of humans worldwide, and point to a very promising future of zebrafish drug discovery.
Acknowledgments
Supported by grants from the NIDDK/NIH (R01DK069403, RC4DK090770)
The authors would like to thank Maria Cecilia Cirio for critical reading of the manuscript. The authors apologize to those whose work we were not able to include in this review due to space restrictions.
References
- Akimenko MA, Johnson SL, Westerfield M, Ekker M. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development. 1995;121(2):347–357. doi: 10.1242/dev.121.2.347. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, Townsend K, Farrington S, Haldi M, Hopkins N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13(20):2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S, Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439(7074):326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Bai Q, Garver JA, Hukriede NA, Burton EA. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007;35(19):6501–6516. doi: 10.1093/nar/gkm608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes JD, Crosby JL, Jones CM, Wright CV, Hogan BL. Embryonic expression of Lim-1, the mouse homolog of Xenopus Xlim-1, suggests a role in lateral mesoderm differentiation and neurogenesis. Dev Biol. 1994;161(1):168–178. doi: 10.1006/dbio.1994.1018. [DOI] [PubMed] [Google Scholar]
- Bayliss PE, Bellavance KL, Whitehead GG, Abrams JM, Aegerter S, Robbins HS, Cowan DB, Keating MT, O’Reilly T, Wood JM, Roberts TM, Chan J. Chemical modulation of receptor signaling inhibits regenerative angiogenesis in adult zebrafish. Nat Chem Biol. 2006;2(5):265–273. doi: 10.1038/nchembio778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belleri M, Ribatti D, Nicoli S, Cotelli F, Forti L, Vannini V, Stivala LA, Presta M. Antiangiogenic and vascular-targeting activity of the microtubule-destabilizing trans-resveratrol derivative 3,5,4′-trimethoxystilbene. Mol Pharmacol. 2005;67(5):1451–1459. doi: 10.1124/mol.104.009043. [DOI] [PubMed] [Google Scholar]
- Benigni A, Morigi M, Remuzzi G. Kidney regeneration. Lancet. 2010;375(9722):1310–1317. doi: 10.1016/S0140-6736(10)60237-1. [DOI] [PubMed] [Google Scholar]
- Bisgrove BW, Snarr BS, Emrazian A, Yost HJ. Polaris and Polycystin-2 in dorsal forerunner cells and Kupffer’s vesicle are required for specification of the zebrafish left-right axis. Dev Biol. 2005;287(2):274–288. doi: 10.1016/j.ydbio.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol. 2003;14(Suppl 1):S55–61. doi: 10.1097/01.asn.0000067652.51441.21. [DOI] [PubMed] [Google Scholar]
- Bouchard M, Souabni A, Mandler M, Neubuser A, Busslinger M. Nephric lineage specification by Pax2 and Pax8. Genes Dev. 2002;16(22):2958–2970. doi: 10.1101/gad.240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CG, Milan DJ, Grande EJ, Rottbauer W, MacRae CA, Fishman MC. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1(5):263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- Cameron DA, Carney LH. Cell mosaic patterns in the native and regenerated inner retina of zebrafish: implications for retinal assembly. J Comp Neurol. 2000;416(3):356–367. [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83(2):170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LG. Adult stem cells in the repair of the injured renal tubule. Nat Clin Pract Nephrol. 2005;1(1):22–32. doi: 10.1038/ncpneph0021. [DOI] [PubMed] [Google Scholar]
- Cao Y, Park A, Sun Z. Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J Am Soc Nephrol. 2010;21(8):1326–1333. doi: 10.1681/ASN.2009091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, Sun Z. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci U S A. 2009;106(51):21819–21824. doi: 10.1073/pnas.0911987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C(4):263–280. doi: 10.1002/ajmg.c.30227. [DOI] [PubMed] [Google Scholar]
- Carney TJ, Feitosa NM, Sonntag C, Slanchev K, Kluger J, Kiyozumi D, Gebauer JM, Coffin Talbot J, Kimmel CB, Sekiguchi K, Wagener R, Schwarz H, Ingham PW, Hammerschmidt M. Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes. PLoS Genet. 2010;6(4):e1000907. doi: 10.1371/journal.pgen.1000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Streisinger G, Singer F, Walker C. Frequency of gamma-Ray Induced Specific Locus and Recessive Lethal Mutations in Mature Germ Cells of the Zebrafish, BRACHYDANIO RERIO. Genetics. 1983;103(1):109–123. doi: 10.1093/genetics/103.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Bayliss PE, Wood JM, Roberts TM. Dissection of angiogenic signaling in zebrafish using a chemical genetic approach. Cancer Cell. 2002;1(3):257–267. doi: 10.1016/s1535-6108(02)00042-9. [DOI] [PubMed] [Google Scholar]
- Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol. 2010;191(4):701–710. doi: 10.1083/jcb.201006173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim KY, Huang J, Elbedour K, Carmi R, Slusarski DC, Casavant TL, Stone EM, Sheffield VC. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proceedings of the National Academy of Sciences of the United States of America. 2006;103(16):6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianciolo Cosentino C, Roman BL, Drummond IA, Hukriede NA. Intravenous microinjections of zebrafish larvae to study acute kidney injury. J Vis Exp. 2010;(42) doi: 10.3791/2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ. Uncharted waters: nephrogenesis and renal regeneration in fish and mammals. Pediatr Nephrol. 2011 doi: 10.1007/s00467-011-1795-z. [DOI] [PubMed] [Google Scholar]
- de Groh ED, Swanhart LM, Cosentino CC, Jackson RL, Dai W, Kitchens CA, Day BW, Smithgall TE, Hukriede NA. Inhibition of histone deacetylase expands the renal progenitor cell population. J Am Soc Nephrol. 2010;21(5):794–802. doi: 10.1681/ASN.2009080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep CQ, Ma D, Deo RC, Holm TM, Naylor RW, Arora N, Wingert RA, Bollig F, Djordjevic G, Lichman B, Zhu H, Ikenaga T, Ono F, Englert C, Cowan CA, Hukriede NA, Handin RI, Davidson AJ. Identification of adult nephron progenitors capable of kidney regeneration in zebrafish. Nature. 2011;470(7332):95–100. doi: 10.1038/nature09669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Schultz PG. A role for chemistry in stem cell biology. Nat Biotechnol. 2004;22(7):833–840. doi: 10.1038/nbt987. [DOI] [PubMed] [Google Scholar]
- Drews C, Senkel S, Ryffel GU. The nephrogenic potential of the transcription factors osr1, osr2, hnf1b, lhx1 and pax8 assessed in Xenopus animal caps. BMC Dev Biol. 2011;11:5. doi: 10.1186/1471-213X-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Drummond I. The pronephros. Results Probl Cell Differ. 2002;40:322–345. doi: 10.1007/978-3-540-46041-1_16. [DOI] [PubMed] [Google Scholar]
- Drummond I. Making a zebrafish kidney: a tale of two tubes. Trends in cell biology. 2003;13(7):357–365. doi: 10.1016/s0962-8924(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16(2):299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- Drummond IA, Majumdar A, Hentschel H, Elger M, Solnica-Krezel L, Schier AF, Neuhauss SC, Stemple DL, Zwartkruis F, Rangini Z, Driever W, Fishman MC. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125(23):4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, Bonventre JV. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest. 2005;115(7):1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duldulao NA, Lee S, Sun Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development. 2009;136(23):4033–4042. doi: 10.1242/dev.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton K, Abbas L, Spencer J, Brannon C, Mowbray C, Nikaido M, Kelsh RN, Whitfield TT. A zebrafish model for Waardenburg syndrome type IV reveals diverse roles for Sox10 in the otic vesicle. Dis Model Mech. 2009;2(1–2):68–83. doi: 10.1242/dmm.001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger M, Hentschel H, Litteral J, Wellner M, Kirsch T, Luft FC, Haller H. Nephrogenesis is induced by partial nephrectomy in the elasmobranch Leucoraja erinacea. J Am Soc Nephrol. 2003;14(6):1506–1518. doi: 10.1097/01.asn.0000067645.49562.09. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, Wilson SW. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Human molecular genetics. 2009;18(2):289–303. doi: 10.1093/hmg/ddn356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio) Brain. 2009;132(Pt 6):1613–1623. doi: 10.1093/brain/awp108. [DOI] [PubMed] [Google Scholar]
- Fu X, Wang Y, Schetle N, Gao H, Putz M, von Gersdorff G, Walz G, Kramer-Zucker AG. The subcellular localization of TRPP2 modulates its function. J Am Soc Nephrol. 2008;19(7):1342–1351. doi: 10.1681/ASN.2007070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatric nephrology. 1989;3(1):50–55. doi: 10.1007/BF00859626. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39(11):1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg R, Dennen P. Long-term outcomes of acute kidney injury. Adv Chronic Kidney Dis. 2008;15(3):297–307. doi: 10.1053/j.ackd.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topcu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83(5):559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hader C, Marlier A, Cantley L. Mesenchymal-epithelial transition in epithelial response to injury: the role of Foxc2. Oncogene. 2010;29(7):1031–1040. doi: 10.1038/onc.2009.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Nusslein-Volhard C. Large scale genetics in a small vertebrate, the zebrafish. Int J Dev Biol. 1996;40(1):221–227. [PubMed] [Google Scholar]
- Harris JA, Cheng AG, Cunningham LL, MacDonald G, Raible DW, Rubel EW. Neomycin-induced hair cell death and rapid regeneration in the lateral line of zebrafish (Danio rerio) J Assoc Res Otolaryngol. 2003;4(2):219–234. doi: 10.1007/s10162-002-3022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PC. Genetic complexity in Joubert syndrome and related disorders. Kidney Int. 2007;72(12):1421–1423. doi: 10.1038/sj.ki.5002577. [DOI] [PubMed] [Google Scholar]
- Hentschel DM, Park KM, Cilenti L, Zervos AS, Drummond I, Bonventre JV. Acute renal failure in zebrafish: a novel system to study a complex disease. Am J Physiol Renal Physiol. 2005;288(5):F923–929. doi: 10.1152/ajprenal.00386.2004. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(1):23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18(6):1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2(3):284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Imai N, Hishikawa K, Marumo T, Hirahashi J, Inowa T, Matsuzaki Y, Okano H, Kitamura T, Salant D, Fujita T. Inhibition of histone deacetylase activates side population cells in kidney and partially reverses chronic renal injury. Stem Cells. 2007;25(10):2469–2475. doi: 10.1634/stemcells.2007-0049. [DOI] [PubMed] [Google Scholar]
- James RG, Schultheiss TM. Patterning of the avian intermediate mesoderm by lateral plate and axial tissues. Dev Biol. 2003;253(1):109–124. doi: 10.1006/dbio.2002.0863. [DOI] [PubMed] [Google Scholar]
- Jopling C, Sleep E, Raya M, Marti M, Raya A, Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464(7288):606–609. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. Shaping the vertebrate body plan by polarized embryonic cell movements. Science. 2002;298(5600):1950–1954. doi: 10.1126/science.1079478. [DOI] [PubMed] [Google Scholar]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, MacDonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attie-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41(6):739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464(7288):601–605. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Terada Y, Kuwana H, Tanaka H, Okado T, Kuwahara M, Tohda S, Sakano S, Sasaki S. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 2008;73(11):1240–1250. doi: 10.1038/ki.2008.74. [DOI] [PubMed] [Google Scholar]
- Kopan R, Cheng HT, Surendran K. Molecular insights into segmentation along the proximal-distal axis of the nephron. J Am Soc Nephrol. 2007;18(7):2014–2020. doi: 10.1681/ASN.2007040453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Zucker AG, Olale F, Haycraft CJ, Yoder BK, Schier AF, Drummond IA. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development. 2005;132(8):1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- Kuure S, Vuolteenaho R, Vainio S. Kidney morphogenesis: cellular and molecular regulation. Mech Dev. 2000;92(1):31–45. doi: 10.1016/s0925-4773(99)00323-8. [DOI] [PubMed] [Google Scholar]
- Lameire N, Van Biesen W, Vanholder R. The changing epidemiology of acute renal failure. Nat Clin Pract Nephrol. 2006;2(7):364–377. doi: 10.1038/ncpneph0218. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Curr Opin Genet Dev. 2009;19(3):220–229. doi: 10.1016/j.gde.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, Lin S, Prochownik E, Trede NS, Zon LI, Look AT. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299(5608):887–890. doi: 10.1126/science.1080280. [DOI] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40(4):443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest. 2005;115(7):1756–1764. doi: 10.1172/JCI23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH. Regrow or repair: potential regenerative therapies for the kidney. J Am Soc Nephrol. 2006;17(9):2390–2401. doi: 10.1681/ASN.2006030218. [DOI] [PubMed] [Google Scholar]
- Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, Drummond IA, Beier DR. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development. 2002;129(24):5839–5846. doi: 10.1242/dev.00173. [DOI] [PubMed] [Google Scholar]
- Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134(6):1111–1122. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- Lopez-Schier H, Hudspeth AJ. A two-step mechanism underlies the planar polarization of regenerating sensory hair cells. Proc Natl Acad Sci U S A. 2006;103(49):18615–18620. doi: 10.1073/pnas.0608536103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Jiang YJ. Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genet. 2007;3(1):e18. doi: 10.1371/journal.pgen.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, Liu A, Drummond IA. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Disease models & mechanisms. 2010;3(5–6):354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumo T, Hishikawa K, Yoshikawa M, Fujita T. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J Am Soc Nephrol. 2008;19(7):1311–1320. doi: 10.1681/ASN.2007091040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch TJ, Yang G, Wright M, Smith D, Schoenwolf GC. Signals from trunk paraxial mesoderm induce pronephros formation in chick intermediate mesoderm. Dev Biol. 2000;220(1):62–75. doi: 10.1006/dbio.2000.9623. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Saijoh Y, Tsuchiya K, Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H, Nakatsuji N, Overbeek PA, Hamada H, Yokoyama T. Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature. 1998;395(6698):177–181. doi: 10.1038/26006. [DOI] [PubMed] [Google Scholar]
- Molina G, Vogt A, Bakan A, Dai W, Queiroz de Oliveira P, Znosko W, Smithgall TE, Bahar I, Lazo JS, Day BW, Tsang M. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol. 2009;5(9):680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina GA, Watkins SC, Tsang M. Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev Biol. 2007;7:62. doi: 10.1186/1471-213X-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Turnpenny L, Goodship J, Dai W, Majumder K, Matthews L, Gardner A, Schuster G, Vien L, Harrison W, Elder FF, Penman-Splitt M, Overbeek P, Strachan T. Inversin, a novel gene in the vertebrate left-right axis pathway, is partially deleted in the inv mouse. Nat Genet. 1998;20(2):149–156. doi: 10.1038/2450. [DOI] [PubMed] [Google Scholar]
- Murphey RD, Stern HM, Straub CT, Zon LI. A chemical genetic screen for cell cycle inhibitors in zebrafish embryos. Chem Biol Drug Des. 2006;68(4):213–219. doi: 10.1111/j.1747-0285.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- Obara T, Mangos S, Liu Y, Zhao J, Wiessner S, Kramer-Zucker AG, Olale F, Schier AF, Drummond IA. Polycystin-2 immunolocalization and function in zebrafish. J Am Soc Nephrol. 2006;17(10):2706–2718. doi: 10.1681/ASN.2006040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34(4):413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297(4):F996–F1005. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Schmid B, Haass C. Transgenic zebrafish as a novel animal model to study tauopathies and other neurodegenerative disorders in vivo. Neurodegener Dis. 2010;7(1–3):99–102. doi: 10.1159/000285515. [DOI] [PubMed] [Google Scholar]
- Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CD, Aster JC, Granter SR, Look AT, Lee C, Fisher DE, Zon LI. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15(3):249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, I, Veland R, Schroder JM, Christensen ST. Assembly of primary cilia. Dev Dyn. 2008;237(8):1993–2006. doi: 10.1002/dvdy.21521. [DOI] [PubMed] [Google Scholar]
- Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12(11):938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Link BA, Dowling JE, Schreiber SL. Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc Natl Acad Sci USA. 2000;97(24):12965–12969. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RT, Shaw SY, Peterson TA, Milan DJ, Zhong TP, Schreiber SL, MacRae CA, Fishman MC. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22(5):595–599. doi: 10.1038/nbt963. [DOI] [PubMed] [Google Scholar]
- Pfeffer PL, Gerster T, Lun K, Brand M, Busslinger M. Characterization of three novel members of the zebrafish Pax2/5/8 family: dependency of Pax5 and Pax8 expression on the Pax2.1 (noi) function. Development. 1998;125(16):3063–3074. doi: 10.1242/dev.125.16.3063. [DOI] [PubMed] [Google Scholar]
- Pichler FB, Laurenson S, Williams LC, Dodd A, Copp BR, Love DR. Chemical discovery and global gene expression analysis in zebrafish. Nat Biotechnol. 2003;21(8):879–883. doi: 10.1038/nbt852. [DOI] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- Reimschuessel R. A fish model of renal regeneration and development. ILAR J. 2001;42(4):285–291. doi: 10.1093/ilar.42.4.285. [DOI] [PubMed] [Google Scholar]
- Reimschuessel R, Bennett RO, May EB, Lipsky MM. Development of newly formed nephrons in the goldfish kidney following hexachlorobutadiene-induced nephrotoxicity. Toxicol Pathol. 1990;18(1 Pt 1):32–38. doi: 10.1177/019262339001800105. [DOI] [PubMed] [Google Scholar]
- Reimschuessel R, Williams D. Development of new nephrons in adult kidneys following gentamicin-induced nephrotoxicity. Ren Fail. 1995;17(2):101–106. doi: 10.3109/08860229509026246. [DOI] [PubMed] [Google Scholar]
- Romagnani P. Toward the identification of arenopoietic system? Stem Cells. 2009;27(9):2247–2253. doi: 10.1002/stem.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnani P, Kalluri R. Possible mechanisms of kidney repair. Fibrogenesis Tissue Repair. 2009;2(1):3. doi: 10.1186/1755-1536-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, Tan PL, Phillips HM, Leroux MR, Henderson DJ, Murdoch JN, Copp AJ, Eliot MM, Lupski JR, Kemp DT, Dollfus H, Tada M, Katsanis N, Forge A, Beales PL. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37(10):1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- Sachidanandan C, Yeh JR, Peterson QP, Peterson RT. Identification of a novel retinoid by small molecule screening with zebrafish embryos. PLoS ONE. 2008;3(4):e1947. doi: 10.1371/journal.pone.0001947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos N, Reiter JF. Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn. 2008;237(8):1972–1981. doi: 10.1002/dvdy.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38(6):674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Schafer T, Putz M, Lienkamp S, Ganner A, Bergbreiter A, Ramachandran H, Gieloff V, Gerner M, Mattonet C, Czarnecki PG, Sayer JA, Otto EA, Hildebrandt F, Kramer-Zucker A, Walz G. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum Mol Genet. 2008;17(23):3655–3662. doi: 10.1093/hmg/ddn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottenfeld J, Sullivan-Brown J, Burdine RD. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development. 2007;134(8):1605–1615. doi: 10.1242/dev.02827. [DOI] [PubMed] [Google Scholar]
- Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- Shawlot W, Behringer RR. Requirement for Lim1 in head-organizer function. Nature. 1995;374(6521):425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
- Shepard JL, Amatruda JF, Finkelstein D, Ziai J, Finley KR, Stern HM, Chiang K, Hersey C, Barut B, Freeman JL, Lee C, Glickman JN, Kutok JL, Aster JC, Zon LI. A mutation in separase causes genome instability and increased susceptibility to epithelial cancer. Genes Dev. 2007;21(1):55–59. doi: 10.1101/gad.1470407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37(5):537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RL. High-throughput screening in academia: the Harvard experience. J Biomol Screen. 2003;8(6):615–619. doi: 10.1177/1087057103260741. [DOI] [PubMed] [Google Scholar]
- Stern HM, Zon LI. Cancer genetics and drug discovery in the zebrafish. Nat Rev Cancer. 2003;3(7):533–539. doi: 10.1038/nrc1126. [DOI] [PubMed] [Google Scholar]
- Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda P, Hamel C, de Ravel TJ, Lewis RA, Friederich E, Thibault C, Danse JM, Verloes A, Bonneau D, Katsanis N, Poch O, Mandel JL, Dollfus H. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet. 2007;80(1):1–11. doi: 10.1086/510256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streets AJ, Moon DJ, Kane ME, Obara T, Ong AC. Identification of an N-terminal glycogen synthase kinase 3 phosphorylation site which regulates the functional localization of polycystin-2 in vivo and in vitro. Hum Mol Genet. 2006;15(9):1465–1473. doi: 10.1093/hmg/ddl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio) Nature. 1981;291(5813):293–296. doi: 10.1038/291293a0. [DOI] [PubMed] [Google Scholar]
- Sullivan-Brown J, Schottenfeld J, Okabe N, Hostetter CL, Serluca FC, Thiberge SY, Burdine RD. Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Dev Biol. 2008;314(2):261–275. doi: 10.1016/j.ydbio.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131(16):4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- Tayeh MK, Yen HJ, Beck JS, Searby CC, Westfall TA, Griesbach H, Sheffield VC, Slusarski DC. Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet. 2008;17(13):1956–1967. doi: 10.1093/hmg/ddn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada Y, Tanaka H, Okado T, Shimamura H, Inoshita S, Kuwahara M, Sasaki S. Expression and function of the developmental gene Wnt-4 during experimental acute renal failure in rats. J Am Soc Nephrol. 2003;14(5):1223–1233. doi: 10.1097/01.asn.0000060577.94532.06. [DOI] [PubMed] [Google Scholar]
- Thornhill P, Bassett D, Lochmuller H, Bushby K, Straub V. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP) Brain. 2008;131(Pt 6):1551–1561. doi: 10.1093/brain/awn078. [DOI] [PubMed] [Google Scholar]
- Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol. 2008;23(11):2095–2099. doi: 10.1007/s00467-008-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JL, Beales PL. The nonmotile ciliopathies. Genet Med. 2009;11(6):386–402. doi: 10.1097/GIM.0b013e3181a02882. [DOI] [PubMed] [Google Scholar]
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama R, I, Dawid B. lim6, a novel LIM homeobox gene in the zebrafish: comparison of its expression pattern with lim1. Dev Dyn. 1997;209(4):406–417. doi: 10.1002/(SICI)1097-0177(199708)209:4<406::AID-AJA8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Tran TC, Sneed B, Haider J, Blavo D, White A, Aiyejorun T, Baranowski TC, Rubinstein AL, Doan TN, Dingledine R, Sandberg EM. Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Cancer Res. 2007;67(23):11386–11392. doi: 10.1158/0008-5472.CAN-07-3126. [DOI] [PubMed] [Google Scholar]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, Adams M, Szymanska K, Mazzotta A, Lee JE, Tolentino JC, Swistun D, Salpietro CD, Fede C, Gabriel S, Russ C, Cibulskis K, Sougnez C, Hildebrandt F, Otto EA, Held S, Diplas BH, Davis EE, Mikula M, Strom CM, Ben-Zeev B, Lev D, Sagie TL, Michelson M, Yaron Y, Krause A, Boltshauser E, Elkhartoufi N, Roume J, Shalev S, Munnich A, Saunier S, Inglehearn C, Saad A, Alkindy A, Thomas S, Vekemans M, Dallapiccola B, Katsanis N, Johnson CA, Attie-Bitach T, Gleeson JG. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42(7):619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen E, Giles RH, Voest EE, van Rooijen C, Schulte-Merker S, van Eeden FJ. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J Am Soc Nephrol. 2008;19(6):1128–1138. doi: 10.1681/ASN.2007080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111(3):p39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese E, Weidenfeld R, Bertram JF, Ricardo SD, Deane JA. Renal cilia display length alterations following tubular injury and are present early in epithelial repair. Nephrol Dial Transplant. 2008;23(3):834–841. doi: 10.1093/ndt/gfm743. [DOI] [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000;44(3):289–307. doi: 10.1002/1097-4695(20000905)44:3<289::aid-neu1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Villanueva S, Cespedes C, Vio CP. Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am J Physiol Regul Integr Comp Physiol. 2006;290(4):R861–870. doi: 10.1152/ajpregu.00384.2005. [DOI] [PubMed] [Google Scholar]
- Vincensini L, Blisnick T, Bastin P. 1001 model organisms to study cilia and flagella. Biol Cell. 2011;103(3):109–130. doi: 10.1042/BC20100104. [DOI] [PubMed] [Google Scholar]
- Vize PD, Woolf AS, Bard JBL. The Kidney from Normal Development to Congenital Diseases. Academic Press; 2003. [Google Scholar]
- Vogetseder A, Palan T, Bacic D, Kaissling B, Le Hir M. Proximal tubular epithelial cells are generated by division of differentiated cells in the healthy kidney. Am J Physiol Cell Physiol. 2007;292(2):C807–813. doi: 10.1152/ajpcell.00301.2006. [DOI] [PubMed] [Google Scholar]
- Vogetseder A, Picard N, Gaspert A, Walch M, Kaissling B, Le Hir M. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am J Physiol Cell Physiol. 2008;294(1):C22–28. doi: 10.1152/ajpcell.00227.2007. [DOI] [PubMed] [Google Scholar]
- Vogt A, Cholewinski A, Shen X, Nelson SG, Lazo JS, Tsang M, Hukriede NA. Automated image-based phenotypic analysis in zebrafish embryos. Dev Dyn. 2009;238(3):656–663. doi: 10.1002/dvdy.21892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C, Streisinger G. Induction of Mutations by gamma-Rays in Pregonial Germ Cells of Zebrafish Embryos. Genetics. 1983;103(1):125–136. doi: 10.1093/genetics/103.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Kato M, Suzuki N, Inoue C, Fedorova S, Hashimoto H, Maruyama S, Matsuo S, Wakamatsu Y. Kidney regeneration through nephron neogenesis in medaka. Dev Growth Differ. 2009;51(2):135–143. doi: 10.1111/j.1440-169X.2009.01090.x. [DOI] [PubMed] [Google Scholar]
- White JA, Boffa MB, Jones B, Petkovich M. A zebrafish retinoic acid receptor expressed in the regenerating caudal fin. Development. 1994;120(7):1861–1872. doi: 10.1242/dev.120.7.1861. [DOI] [PubMed] [Google Scholar]
- White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, Chen F, Long HK, Kramer M, Datta S, Neuberg D, Granter S, Young RA, Morrison S, Wheeler GN, Zon LI. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O’Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert RA, Davidson AJ. The zebrafish pronephros: a model to study nephron segmentation. Kidney Int. 2008;73(10):1120–1127. doi: 10.1038/ki.2008.37. [DOI] [PubMed] [Google Scholar]
- Wingert RA, Selleck R, Yu J, Song HD, Chen Z, Song A, Zhou Y, Thisse B, Thisse C, McMahon AP, Davidson AJ. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007;3(10):1922–1938. doi: 10.1371/journal.pgen.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest. 1994;93(5):2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H, Jr, Kucherlapati R, Edelmann W, Somlo S. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- Wu G, Markowitz GS, Li L, D’Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H, Jr, Kucherlapati R, Edelmann W, Somlo S. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24(1):75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- Yalavarthy R, Edelstein CL. Therapeutic and predictive targets of AKI. Clin Nephrol. 2008;70(6):453–463. doi: 10.5414/cnp70453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Kutok JL, Lee NH, Piao HY, Fletcher CD, Kanki JP, Look AT. Targeted expression of human MYCN selectively causes pancreatic neuroendocrine tumors in transgenic zebrafish. Cancer Res. 2004;64(20):7256–7262. doi: 10.1158/0008-5472.CAN-04-0931. [DOI] [PubMed] [Google Scholar]
- Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum Mol Genet. 2006;15(5):667–677. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]
- Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18(5):1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4(1):33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119(3):428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul NA, Liu Y, Gerdes JM, Gascue C, Oh EC, Leitch CC, Bromberg Y, Binkley J, Leibel RL, Sidow A, Badano JL, Katsanis N. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2010;107(23):10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Boucher RC, Bollig F, Englert C, Hildebrandt F. Characterization of mesonephric development and regeneration using transgenic zebrafish. Am J Physiol Renal Physiol. 2010;299(5):F1040–1047. doi: 10.1152/ajprenal.00394.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Dai J, Attanasio M, Hildebrandt F. Nephrocystin-3 is required for ciliary function in zebrafish embryos. Am J Physiol Renal Physiol. 2010;299(1):F55–62. doi: 10.1152/ajprenal.00043.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]