

Abstract
Hypoxia Ischemia (HI) and associated brain injuries are seen in premature as well as term infants with birth complications. Resulting impairments involve deficits in many cognitive domains, including language development. Poor rapid auditory processing is hypothesized to be one possible underlying factor leading to subsequent language delays. Mild hypothermia treatment for HI injuries in term infants is widely used as an intervention, but can be costly and time consuming. Data suggest that the effectiveness of hypothermia treatment following HI injury declines beyond six hours post injury. Thus, the availability of a therapeutic alternative without these limitations could allow doctors to treat HI injured infants more effectively, and thus reduce deleterious cognitive and language outcomes. Evidence from both human studies and animal models of neonatal HI suggests that erythropoietin (Epo), an endogenous cytokine hormone, may be a therapeutic agent that can ameliorate HI brain injury and preserve subsequent cognitive development and function.. The current study sought to investigate the therapeutic effectiveness of Epo when administered immediately after HI injury, or delayed at intervals following post injury, in neonatal rodents. Rat pups received an induced HI injury on postnatal day 7, followed by an intraperitoneal injection of Epo (1,000 U/kg) either immediately, 60 minutes, or 180 minutes following induction of injury. Subjects were tested on rapid auditory processing tasks in juvenile (P38–42) and adult periods (P80–85). Ventricular and cortical size was also measured from post mortem tissue. Results from the current study show a therapeutic benefit of Epo when given immediately following induction of HI injury, with diminished benefit from a 60-minute delayed injection of Epo, and no protection following a 180 minute delayed injection. The current data thus show that the effectiveness of a single dose of Epo in ameliorating auditory processing deficits following HI injury decreases precipitously as treatment is delayed following injury. These data may have important implications for experimental human neonatal intervention with Epo.
Keywords: neuroprotection, Hypoxic Ischemic Encephalopathy, neonatal, language impairment, auditory processing
Introduction
Hypoxia-Ischemia (HI) refers to a disruption in blood and oxygen delivery to the brain. In neonates, incidents such as intraventricular-periventricular hemorrhage (IVH-PVH) - an injury observed in very preterm infants (gestation age <32 weeks) or at very low birth weight (VLBW; < 1,500 grams [1]) – can result in HI injury. Periventricular leukomalacia (PVL) is a related ischemic HI injury also seen in premature/VLBW infants [2]. Hypoxic-ischemic encephalopathy (HIE) is diagnosed in full-term infants suffering from birth complications such as cord prolapse or placental abruption [1]. Despite varying etiologies for HI across pre-term and term populations, these early HI injuries uniformly lead to an increase in behavioral sequelae such as learning and language deficits at later ages [3,4,5,6,7]. VLBW children exhibit deficits on standardized language and cognitive tasks in early childhood [7] and sustained deficits in both language comprehension and language production at four years of age [8]. A meta-analysis revealed that premature/VLBW children also display poorer performance on spelling, reading, and verbal fluency tasks as compared to term control children [3]. Term infants diagnosed with HIE show verbal IQ deficits at four years of age, and lower scores are seen with increasing degree of injury as reflected by MRI [9]. With specific regard to developmental language deficits, it has been suggested that disruptions in underlying rapid auditory processing (RAP) or the ability to discriminate between rapidly presented auditory cues (such as those that occur in speech) may be one factor leading to delayed and/or disordered language learning in HI injured children. Specifically, studies of children diagnosed with an HI injury in infancy show RAP deficits later in childhood [10] (see [7]), and other researchers have shown that deficits in RAP (measured in infancy) correlate with and are predictive of language outcomes in later years [11,12,13]. Children diagnosed with a developmental language impairment concurrent to rapid auditory processing deficits also show abnormal oscillatory EEG activity during a RAP task [14]. Furthermore, children with a family history of language impairment show abnormal maturation of long-latency auditory evoked potentials from six months to four years of age, as compared to children without a family history, and these scores correlate with subsequent language and cognitive abilities for both groups [15].
Studies using the neonatal rodent models of early HI allow for the experimental assessment of outcomes following early HI injury (i.e., short and long-term neuropathology, long-term behavior), as well as variables that may modulate these outcomes. Using the Vannucci method, induction of HI injury on postnatal day 7 (P7) in rodents (roughly correlating to a 34–36 week old human in terms of brain development) produces a pattern of neuropathology that parallels injuries seen in term infants with HIE [16, 17] and provides a model for the study of the developmental trajectory of a term-like HI injury [16,17]. Moreover, male rats that receive an HI injury on P7 are found to show subsequent deficits on RAP tasks at both juvenile and adult ages -- possibly reflecting outcomes related to language deficits in human populations [18, 19, 20]. These P7 HI injured rats also show gray matter damage, particularly in the cortex and hippocampus, along with increased ventricular size as measured by post mortem histological analysis (all similar to the patterns of brain injury seen in term-HI injured children; [1, 2, 18, 19, 21]). Using the neonatal rodent HI model as described here, studies have also examined possible therapeutic variables that could be employed to improve long-term outcomes.
Erythropoietin (Epo) has been proposed as an intervention to reduce deleterious outcomes following an HI injury. Epo is a cytokine hormone expressed endogenously in the brain, kidney and liver [22, 23]. Systemically, Epo regulates the production of new red blood cells, and has been shown to increase the number of erythrocytes in times of chronic hypoxia (thus improving the amount of oxygen delivery to affected tissues [24]). It has also been administered to increase red blood cell production for the treatment of anemia. However, this mechanism seems unlikely to underlie any protective effects of a single dose of Epo such as was used here. Epo has been shown to readily cross the blood brain barrier, and binds to the Epo receptor (EpoR). Epo and EpoR are highly expressed in gray matter, including the cortex and hippocampus, which are particularly vulnerable to hypoxic/ischemic insult [22]. Furthermore, expression of endogenous Epo has been found to increase in the cortex within 24 hours following neonatal HI insult in rodent models [25]. The endogenous expression of Epo in response to HI insult, coupled with the distribution of EpoR in grey matter, makes it a promising candidate for therapeutic intervention following HI injuries.
Historically, Epo has been successfully used to help treat anemia in premature infants. Retrospective studies of outcomes among treated infants reveal that Epo may also have some neuroprotective properties [22, 26, 27, 28, 29]. In a study investigating the therapeutic effects of Epo in extremely low birth weight infants, infants treated with 400 U/kg three times per week and who later showed high Epo plasma concentrations had better Mental Development Index scores (18–22 months) than infants who were treated but showed lower Epo plasma concentrations [26]. In another study (reviewed in McPhearson and Juul, 2010) where preterm infants were treated with multiple doses of Epo at birth, treated subjects showed improvements in gross motor, fine motor and language tasks at 6–12 months when compared to matched controls [27]. Furthermore, high dose of Epo administration in VLBW infants (25,000 U/kg the first three days of life), positively correlated with cognitive and motor scores at 4–36 months [28]. In term infants diagnosed with moderate HIE who were treated with repeated doses of Epo (300 U/kg or 500 U/kg), better Mental Development Index scores and lower incidence of disability were seen as compared to the control group at 18 months [29]. Finally, when term infants diagnosed with HIE were treated with multiple doses of Epo at birth they showed fewer incidence of seizures at five days, improved EEG scores two weeks after birth, and improved neurological scores as measured by the Denver Developmental Screening Test II at six months of age [30].
The use of Epo as a neuroprotectant in neonatal rodent HI models has also revealed promising findings. For example, treatment with Epo immediately following HI injury in P7 rat pups has been shown to reduce infarct size in cortex [31, 32, 33, 34, 35, 36, 37] and hippocampus [21, 36] as well as to reduce the volume of the lateral ventricles [36, 37]. Treatment with Epo immediately following HI injury was also found to reduce white matter damage by protecting developing pre-oligodendrocytes [38, 39] and preserving the volume of the sub-ventricular zone [40]. At the behavioral level, Epo has been shown to ameliorate later sensory/motor deficits [33, 40], as well as spatial memory deficits [35, 36, 37] in P7 HI injured rats. Additionally, Epo has been shown to ameliorate RAP deficits in P7 HI injured rats as measured in both juvenile and adult periods [36, 37]. This latter result suggests that Epo could possibly be used to help ameliorate later language deficits (which are correlated with RAP indices) in HI injured children.
Although other neuroprotective regiments are currently being used in clinical trails to reduce negative outcomes following HI injuries in children -- such as mild head or full-body hypothermia -- the delay time between injury and the initiation of treatment (as defined by target reduction in core body or head temperature) can frequently reduce the efficacy of treatment [41, 42]. Thus, the implementation of a more widely available and speedy form of neuroprotective treatment (such as an injection) could improve intervention options and outcomes for HI injured infants. Given this background, the current study was designed to address two aims: 1) to replicate previously reported protective effects of Epo treatment immediately following P7 HI injury in rats; and 2) to examine whether Epo delivered at a clinically-relevant post-injury delay (60 or 180 minutes) could afford similar (or any) long-term protection. Thus we assessed the effectiveness of immediate and delayed administration of Epo on long-term behavioral and histological outcomes in a P7 rodent model of early HI injury. We hypothesized that HI-saline treated animals would show significantly worse RAP scores and higher pathology indices as compared to sham controls (as previously demonstrated; [18, 19, 20]). Next we hypothesized that immediate post-injury treatment with Epo would significantly improve RAP scores and pathology outcomes, again replication previous data [36, 37] and that HI animals treated with a comparable dose of Epo at 60 and 180 minute delays might demonstrate a partial therapeutic benefit.
Experimental Procedures
Subjects
Subjects were male Wistar rats, born to time-mated dams (Charles River Laboratories, Wilmington, MA) at the University of Connecticut. Males were used based on evidence that behavioral deficits following early HI injury may be less severe in females, making the assessment of neuroprotection in females more difficult [21]. Pups were culled and cross-fostered as necessary on P1 to yield litters comprised of 8 males and 2 females (female pups were retained to maintain normal maternal behavior). Subjects were weaned at P21 and pair-housed with like-treated littermates. At P55, animals were single housed for further testing. All subjects were housed using a 12 hour light/dark cycle with food and water available ad libitum.
Induction of HI
Male pups were randomly assigned within-litter to each treatment group: HI vehicle (sterile saline); HI Epo immediate (1,000 U/kg); HI Epo 60 minute delay (1,000 U/kg); Epo 180 minute delay (1,000 U/kg); or corresponding sham (no HI or ligation) saline-treated groups. Within the sham and HI saline group, subsets of animals received saline injections at time points equivalent volume to each of the Epo groups to control for the possible effects of injection timing (however, no differences were found and these subsets were subsequently pooled). Pups receiving HI surgery on P7 were anesthetized with isoflurane (2.5%) and a midline incision was made longitudinally in the neck. The right common carotid artery was located and separated from surrounding tissue, cauterized, and the incision was sutured. Pups were marked with a footpad injection to identify their treatment condition at weaning. Pups were placed under a warming lamp and allowed to recover from anesthesia, then were returned to their dams for two hours in order to feed. Animals assigned to the sham condition were similarly placed under anesthesia, but only a mid-line incision was made with no manipulation of the carotid artery. After 2 hours with dams, HI pups were then placed in an air-tight container in which they were exposed to 8% humidified oxygen (balanced with nitrogen) for a period of 120 minutes. Sham animals were placed in an open container with normal air exposure for the same period.
Erythropoietin Administration
Following removal from the hypoxic chamber, animals received either Epo (intraperitoneal (i.p.) injection; Protein Sciences Corporation, 1,000 U/kg in sterile saline; 36,37) or vehicle only (sterile saline) according to predetermined assigned conditions (HI Saline, n=15); HI Epo immediate (HI Epo 0, n=15); HI Epo one hour delay (HI Epo 60, n=15); HI Epo three hour delay (HI Epo 180, n=15); or corresponding sham saline treatment (Sham, n=16). Saline treated animals (HI or sham) received a comparable volume i.p. injection of saline at the same designated time points (although immediate, 60 min and 180 delay saline injections were not found to differ and were pooled within both saline treated shams and HI subjects for further analysis). All pups were then returned to their dams, and remained undisturbed until weaning on P21. All behavioral testing was done blind to Treatment condition.
Behavioral Testing: startle reduction
The startle reduction paradigm utilizes the natural acoustic startle reflex (ASR), or motor response, to a startle eliciting stimulus (SES). When the SES is preceded by a pre-stimulus, a reduction is seen in the ASR (startle reduction), also known as pre-pulse inhibition or PPI. In this study, the SES was a 105 dB, 50 ms white noise burst for all auditory tasks. Comparisons between the ASR amplitude on trials with no pre-pulse (uncued trials), versus presence of a pre-pulse cue (cued trials), were made for each subject on each task to calculate attenuation score as an objective measure of sensory detection on that task (as previously described in [43]). The inter-trial interval between each SES ranged from 16 s-24 s, and averaged 20 s.
Behavioral Testing Apparatus
During startle reduction testing, each subject was placed on an individual load-cell platform (Med Associated PHM-252B, Georgia, VT) in a black polypropylene cage. The output from the platforms was amplified (linear amp PHM-250–60 Med Associates) and acquired by a Biopac MP 100A-CE acquisition system. This system was connected to a Power Macintosh G3, which displayed the output in a waveform with the computer program AcqKnowledge. Auditory stimuli were generated on a Pentium III Dell PC with custom programmed software and a Tucker Davis Technologies (RP2) real-time processor, amplified by a Niles SI-1260 System Integration Amplifier. Sound files were delivered via 10 Cambridge Soundworks MC100 loudspeakers placed 53 cm above the platforms. Each animal’s ASR amplitude was recorded (in mV) by extracting the maximum peak value from the 150 ms signal epoch following the SES. Cued and uncued trials were coded and compared to assess the animal’s absolute and attenuated response amplitude across trials. Scores were analyzed by calculating the attenuation (ATT) score as a function of the average cued trials divided by the average uncued trials and multiplied by 100 to get a percentage (cued trials/uncued trials *100).
Single Tone procedure (P35)
The single tone task consisted of 103 cued or uncued trials. Trials were presented in random order, and animals were tested for one session. The single tone procedure was used to assess for underlying processing deficits or hearing deficits that might confound further testing results. Uncued trials consisted of a silent background followed by the SES. On cued trials, a 75 dB, 7 ms, 2300 Hz tone was presented. On cued trials, the cue to burst interval (the time between when the cue was presented and when the SES was presented) varied (25 ms, 50 ms, 75 ms, and 100 ms). This was to ensure all animals could perform the pre-pulse inhibition task at differing difficulty levels.
Silent Gap (SG) procedure (P38–P42; Juvenile)
The silent gap procedure consisted of 299 trials per session, and animals were tested on one session/day for four consecutive days. Testing occurred in the juvenile period (age P38 – P42). For all trials, a continuous background broad-band white noise (75 dB) was played. Uncued trials consisted of the background white noise followed by the SES (a burst of white noise at 105 dB). On cued trials, a gap in the white noise varying in duration (between 2–100 ms) was provided as a cue 50 ms before the presentation of the SES. In prior research we have found the silent gap task to provide a good evaluation of acoustic processing thresholds in young animals as compared to more difficult tasks that are effective in eliciting deficits from adults (41).
FM Sweep procedure (P80–P85; Adult)
The FM sweep procedure consisted of 102 trials per session. Animals were tested on one session/day for five consecutive days. The background comprised a repeated presentation consisted of a 75 dB downward frequency sweep (2300–1900 Hz), which varied in duration (275ms, 225ms, 175ms, 125ms, 75ms) for each session on different days (one duration per session). Sweeps were separated by a between stimulus ISI, always 200 ms greater than the sweep duration. The uncued trials consisted of the same downward sweep presented 50 ms prior to the SES. Cued trials consisted of a reversal of the previous sweep (or “oddball,” i.e., low to high instead of high to low) presented 50 ms before the SES. The FM Sweep task provides a more challenging task as compared to the SG task. In fact, by adulthood most animals (including HI subjects) can successfully perform SG 0–100, thus revealing no Treatment differences on this task. Introducing a more difficult task in adulthood, such as the FM Sweep, thus allows us to investigate persistent HI effects.
Histology
At P100 animals were weighed, anesthetized with Ketamine (100 mg/kg) and Xylazine (15 mg/kg), and transcardially perfused with 0.09% saline and 10% buffered formalin phosphate. Brains were removed and placed in formalin. Before slicing, brains were placed in a 30% sucrose solution for cryoprotection and then were sliced on a coronal plane at 60μm using a cryostat. Every third section was mounted on a slide and stained using cresyl violet. Volumes of the lateral ventricles and right cortical volume were examined using a Zeiss Imager A.2 microscope and Micro Bright Field (MBF) Stereo Investigator computer software (Williston, VT). Cavalieri’s estimator of volume was calculated using the MBF computer software. All measurements were performed blind to Treatment group.
Statistical Analyses
All statistical analyses were performed using SPSS 15.0 software, and an alpha criterion of .05. Two-tailed analyses were used unless otherwise stated. For initial analyses, for each task we compared behavioral scores of subsets of the sham saline and HI saline treated animals that received injections at the 3 different time points. No differences were found, and therefore these subsets were grouped into HI saline (n=15) and Sham saline (n=16) for further analyses. ANOVAs were performed for each task using multiple levels of Treatment. Based on specific a priori hypotheses as described above, planned comparisons were performed (as a function of Treatment) between paired groups. Specific comparisons were made between HI saline vs Sham; HI Epo 0 delay vs Sham; HI Epo 60 minute delay vs Sham; and HI Epo 180 delay vs Sham. Additionally, a separate comparison of each of the 3 Epo groups to HI saline treated was performed.
Repeated measures analysis of variance (ANOVA) was used to analyze ATT scores for each of the RAP tasks. Variables included Treatment (5 levels; Sham, HI Saline, HI Epo 0, HI Epo 60, and HI Epo 180), Day (4 levels for Silent Gap and 5 levels for FM sweep), and Gap (9 levels for SG 0–100 task only). Univariate ANOVA was also used to analyze histology, specifically for right and left ventricular size and right cortical size, which we have previously shown to provide a reliable index of pathology [18, 19, 21]. We note here that tissue from several brains could not be accurately measured as a result of histologic factors (loss of tissue integrity in severely damaged brains leading to inability to confidently identify tissue borders), and the final n used for anatomic analysis in each group was: HI Saline, n=12; HI Epo 0, n=13; HI Epo 60, n=13; HI Epo 180, n=12; Sham, n=14. Specific follow up independent samples t-tests were used to compare ventricular scores between each HI group relative to shams, as described above. Graphs presented in the results section show mean ATT scores for auditory tasks; note that a higher score indicates poorer performance (100% equals chance performance levels, with no difference between cued and uncued trials).
Results
Normal Single Tone
A 4 (Cue) × 5 (Treatment) repeated measure ANOVA revealed no main effect of Treatment, [F (4,71) = .945, p>.05], indicating no differences in subjects simple pre-pulse inhibition with a single tone (data not shown). Additional paired analyses further failed to reveal significant differences for any comparison. This confirms an absence of differences in basic startle reduction behavior or auditory processing abilities between groups, which is important in allowing attribution of Treatment effects on more complex tasks to a failure to process the stimulus, and not hearing or basic PPI deficits.
Silent Gap 0–100 (P38–P42)
A 4 (Day) × 9 (Gap) × 5 (Treatment) repeated measures ANOVA was performed, and this revealed a trend toward an overall main effect of Treatment [F (4,71) = 2.281, p=.06] though it did not reach significance, as well as a significant Gap x Day x Treatment interaction [F (4,71) = 1.495, p<.05]. The triple interaction likely reflects the fact that all scores improved over Days, and particularly so at the longer Gaps, but this improvement was more marked in the high-performing Treatment groups. A follow up 9 (Gap) × 2 (Treatment) × 4 (Day) one-tailed repeated measures ANOVA was performed specifically between Sham and HI saline groups, and this also revealed a trend toward a Treatment effect [F (1,29) = 1.980, p=.085; one-tailed], though it did not each significance (see Figure 1), with HI saline subjects performing worse. Further analysis did reveal significant HI effects at specific gaps including 40 ms, t(29) = 1.182, p<.05, and 75 ms, t(29) = 1.982, p<.05, on the third day of testing (data not shown). A 9 (Gap) × 2 (Treatment) × 4 (Day) repeated measures ANOVA was performed between the Sham and Epo 0 groups, and this revealed no significant difference between groups [F (1,29) = .226, p>.05]. However, a significant Treatment effect was seen between Sham and HI Epo 60 [F (1,29) = 6.575, p<.05], as well as a significant Treatment effect between Sham and HI Epo 180 [F (1,29) = 6.974, p<.01] (see Figure 2). Thus scores were significantly worse in HI saline treated, HI Epo 60 delay, and HI Epo 180 delay groups as compared to the Sham and Epo 0 groups.
Figure 1.
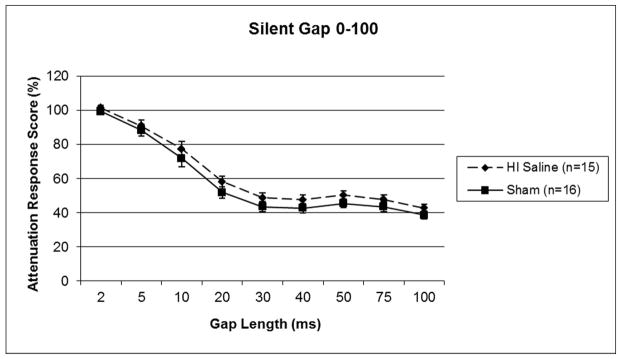
A repeated measures ANOVA revealed a trend towards a Treatment effect (p=.085, one-tail) on silent gap detection, with HI saline animals performing worse than shams. Scores represent mean attenuated response (cued/uncued*100), and lower scores indicate better performance.
Figure 2.
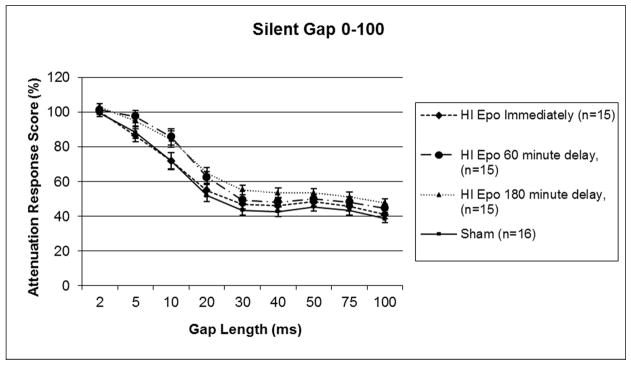
A repeated measures ANOVA revealed no differences in Treatment between HI Epo immediate groups and Sham groups, but did show a significant Treatment effect between HI Epo 60 minute delay and shams (p<.05), as well as significant Treatment effect between HI Epo 180 minute delay and shams (p<.01) on silent gap detection. Scores represent mean attenuated response (cued/uncued*100), and lower scores indicate better performance.
FM Sweep (P80–P85)
Based on a priori planned comparisons, a 5 (Day) × 2 (Treatment) one-tailed repeated measures ANOVA was performed with Sham and HI saline treated groups. This analysis revealed a significant main effect of Treatment [F (1,29) = 3.361, p<.05] (see Figure 3), with HI saline subjects performing worse than shams. A similar 5 (Day) × 2 (Treatment) repeated measures ANOVA comparing Sham treatment to Epo 0 was performed, and this revealed no significant difference between groups [F (1,29) = 2.879, p>.05]. However, a non-significant effect of Treatment was seen between Sham and HI Epo 60 groups [F (1,29) = 3.773, p=.06], as well as a significant effect of Treatment between Sham and HI Epo 180 groups [F (1,29) = 4.387, p<.05] (see Figure 4). Thus ATT response scores for HI saline treated and HI 180 minute delay groups were again significantly worse than the Sham and Epo 0 treated groups, with an intermediate difference for Epo 60.
Figure 3.
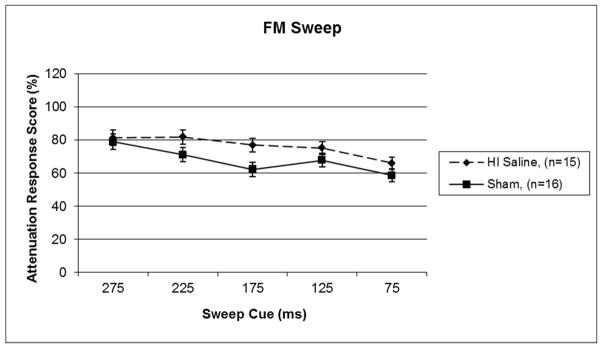
A repeated measures ANOVA revealed a significant overall Treatment effect between HI saline and sham groups (p<.05, one-tail) on FM sweep detection. Scores represent mean attenuated response (cued/uncued*100), and lower scores indicate better performance.
Figure 4.
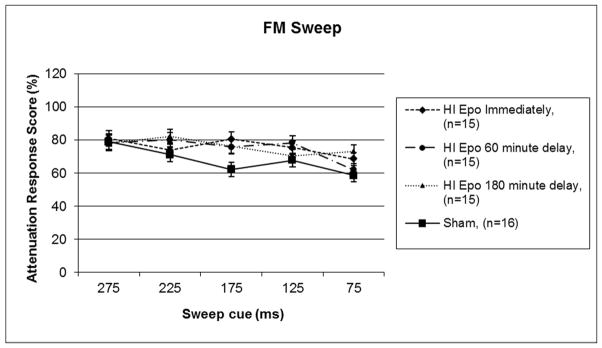
A repeated measures ANOVA revealed no significant differences in Treatment between HI Epo immediate and sham groups, but did reveal a trend towards a Treatment effect (p =.062) between HI Epo 60 minute delay and sham groups, and a significant Treatment effect (p <.05) between HI Epo 180 minute delay and sham groups on FM sweep detection. Scores represent mean attenuated response (cued/uncued*100), and lower scores indicate better performance.
Histological Results
Ventricular volumes (measured in mm3) were assessed for both left and right hemispheres in all subjects (data not shown). Paired Samples T-Tests were performed for each Treatment group individually and these revealed no difference between the left and right ventricle in HI saline animals, (t(11)=−1.582, p=.07; one-tail), with the right ventricular volume larger than the left. There was no difference in the Sham group (t(13)=−0.297, p>.05), the HI Epo 0 group, (t(12)=−1.65, p>.05) or the HI Epo 60 group, (t(12)=−1.06, p>.05) when comparing left and right ventricular volume (all comparisons were two-tail comparisons). There was, however, a significant difference in the HI Epo 180 group, (t(11)=−2.16, p<.05), with right ventricular volume larger than left ventricular volume. These indices indicate ventriculomegaly of the right side in injured subjects (HI saline and HI Epo 180) with amelioration of this effect in the Epo 0 group. The HI Epo 60 group showed a similar trend as the HI Epo 0 and HI Epo 180 group, with the right ventricular volume larger than the left, but this did not reach significance. We found right cortical volume to be significantly smaller in HI saline animals as compared to shams (t(14.7)=−1.827, p<.05 (one-tailed)). Right cortical volumes in HI Epo 0 animals did not differ from shams. Comparisons for HI Epo 60 and 180 versus shams were in the expected direction (smaller than shams), these comparisons did not reach significance.
Discussion
The current study evaluated the therapeutic effects of immediate and delayed Epo administration following a P7 HI injury in male rats, using performance outcomes on several RAP tasks (administered in juvenile and adult periods) with clinical relevance to language outcomes. We hypothesized that HI saline animals would show worse performance on RAP tasks, as well as higher pathology (ventricular) indices, as compared to Sham treated animals (as previously reported; [18, 19, 20]). Additionally, we hypothesized that HI Epo immediate subjects would show comparable RAP scores and pathology measures relative to sham groups, again based on prior findings [36, 37]. The current study did in fact confirm deficits on RAP attenuation scores (using silent gap detection task in the juvenile period, as well as a more difficult FM Sweep task in adulthood) when HI saline treated subjects were compared to sham treated groups, thus replicating previous research [18, 19, 20]. We note that although the overall effects on the SG 0–100 task were marginal between HI saline and sham animals, we did find a significant HI effect specifically at the 40 and 75 msec gaps. Additionally, the current study showed a therapeutic benefit of immediate Epo administration following HI injury in P7 rat pups on these same tasks, again replicating prior work [36, 37].
However, the current study demonstrated little to no therapeutic benefit following a 60 or 180 minute delayed injection of Epo following P7 HI injury when treated subjects were compared to sham treated animals on rapid auditory processing tasks in juvenile and adulthood periods. Additional studies are needed to evaluate the potential therapeutic effects of a single delayed injection of Epo on other behavioral tasks, including other cognitive domains or sensorimotor tasks. Additionally, ventricular pathology scores revealed a trend in the difference between left and right ventricular volume in the HI saline group and a significant difference in the HI Epo 180 group. For both groups the right ventricle was larger than the left, as expected (given unilateral HI injury), noting that in several cases, tissue with a high degree of ventriculomegaly and cortical atrophy was compromised and therefore dropped. We suggest that if these more severely damaged HI brains had been included, the ventricular differences between HI and sham subjects likely would have reached significance. In the sham and HI Epo 0, there was no significant difference found between left and right ventricular volume. For the HI Epo 60 group, although the right ventricular volume was larger than the left, this comparison did not reach significance.
Mechanisms of Epo Action
Previous research has shown therapeutic effects of Epo administration following neonatal brain injury specifically in the central nervous system for both human infants and animal models. In the central nervous system, Epo appears to mediate protective effects via the endogenous Epo-receptor [23, 44]. Moreover, Epo receptors are expressed in both neonatal and adult brains [44], and are up-regulated in ischemic areas in a neonatal focal ischemic injury model in rodents. Specifically, EpoR expression increases on neurons, microglia, and endothelium of blood vessels in the ischemic areas within 24 hours post insult following focal cerebral ischemia in 7 day old rat pups [45]. Increased Epo expression is also found in the cerebral spinal fluid of infants diagnosed with IVH as compared to controls [46].
Studies have further shown that cell death following neonatal HI injury in a rodent model seems to occur in two phases: necrosis (an early or immediate response to HI) and apoptosis (secondary or delayed response to HI; [47]). Once in the central nervous system, Epo seems to exert beneficial effects primarily by preventing the apoptotic cascade, although some early anti-inflammatory effects are also seen. Specifically, when Epo binds to its receptor, Janus kinase 2 (Jak 2) is activated, leading to the phosphorylation of signal transducer and activator of signal transduction and transcription 5 (Stat 5; [23, 48]). These events lead to the up-regulation of anti-apoptotic genes such as Bcl-xL, a part of the Bcl family [23]. Additionally, Epo treatment following a P7 HI injury in rodents has been found to down-regulate pro-apoptotic genes Bax and DP5 [34] and to prevent DNA fragmentation associated with apoptosis [25]. Lastly, Epo, when bound to its receptor, modulates calcium influx into the cell, further preserving the cell’s integrity [23].
Timing of cell death cascades following HI injury
Further research has shown that apoptotic cell death markers are heightened at different time points regionally within the rat brain following injury. Areas such as cortex, striatum, globus pallidus, and CA1 show a high density of apoptotic cells approximately 6–12 hours following a P7 HI injury in the rat, and these remain elevated for up to 7 days post injury [49]. Other areas, however, such as CA3, dentate gyrus, subiculum, and thalamus show more of a bell-shaped expression of apoptotic cells over time, where peak density is seen between 24 and 72 hours, followed by a decline in expression [48]. Following a 5,000 U/kg i.p. injection, rodent brains have shown a peak expression of Epo at ten hours, with concentrations elevated for about 24 hours post injection [50]. Thus an immediate post-injury i.p. injection of Epo should be expressed in the brain 10 hours after injury -- around the same time apoptotic cells are first seen to be highly expressed.
Based on the above, a 60 minute delayed injection of Epo would be expressed in the brain just 11 hours after HI injury (and 180 minute delayed injection expressed in the brain 13 hours post injection), which certainly falls in the spectrum of apoptotic cell expression following HI injury. However, it may be that once critical aspects of the apoptotic cell death cascade are initiated, the process becomes increasingly difficult to reverse. Thus an immediate injection of Epo may reach the brain in time to interfere with the very early events of the cell death cascade following HI - specifically, the inflammatory response (a part of necrosis) as well as activation of N-Methyl-D-aspartic acid (NMDA) glutamate receptors [51], and early phases of apoptosis. In fact, in vitro models reveal that neurons pretreated with Epo showed increased survival in hypoxic neuronal cultures as compared to cells with no Epo treatment, an effect that appears to be due to Epo inhibiting glutamate release stimulated by hypoxia [52]. In vivo, treatment with Epo prior to induction of P7 HI injury also was shown to attenuate brain injury and decrease the amount of inflammatory mediators (such as interleukin 1β and tumor necrosis factor –α; [53]). Additionally, pretreatment with Epo (300 U/kg) prior to induction of hypoxia has been shown to inhibit apoptosis (with the use of DNA ladder and TUNNEL staining; [54]); to protect both astrocytes and glia (Epo at 100 U/kg, [55]); decrease the expression of caspase-3 (Epo at 30,000 U/kg; [56]); and decrease the expression of pro-apoptotic genes Bax and DP5 (Epo at 1,000 U/kg; [57]). Thus, although Epo has been shown to work on multiple facets of the apoptotic cascade, interference with the very early factors might also be necessary to a fully therapeutic benefit of immediate Epo treatment following HI injury.
Given these findings, it remains difficult to ascertain a basis for a specific critical temporal deadline, after which therapeutic benefits can no longer be obtained. Our behavioral findings, however, suggest that if Epo levels do not “reach the brain” within 10 hours following injury – the time frame seen with immediate injection, and corresponding to peak apoptosis in cortical tissues – then the activation of cellular apoptotic cascades may reach levels at which subsequent Epo intervention can no longer reverse the process. Notably, some data suggests that when subsequent injections are combined WITH an immediate injection, additional benefits from later administration can be obtained. Although speculative, this may reflect the fact that once Epo effectively “blunts” critical neural apoptotic cascades, then additional Epo may provide a further ameliorative benefit. Alternately, multiple or higher doses of Epo (up to 30,000 U/Kg) at a delay following injury could provide therapeutic benefits where the single does of Epo provided here at 1,000 U/Kg was not effective. For example, clinical studies have used multiple injections of Epo over days in both preterm and HIE infants [26, 27, 28, 29, 30], and have shown improvements in a variety of cognitive and motor tasks. Animal studies have also shown an improvement with multiple Epo injections in neonatal brain injured rodent models. One study investigating the therapeutic effect of multiple doses of Epo (1 U/g at 0h, 24h, and 7 days post injury) following a P10 medial cerebral artery occlusion revealed that animals treated with multiple doses scored better on a spatial learning and memory tasks, and also showed increased regional and hemispheric volumes compared to non-treated animals and animals treated with a single dose of Epo [58]. Furthermore, when Epo (1,000 U/kg) was administered at multiple time points starting 48 hours post P7 HI injury, treated animals showed increased oligodendrogenesis and maturation of oligodendrocytes, enhanced neurogenesis, attenuated white matter injury and improvements on sensorimotor tasks as compared to non-treated animals [59]. Clearly, future studies with repeated injections are needed to determine the full therapeutic window for Epo at multiple or higher doses.
Nonetheless, it is important to note that our results are consistent with a study by Keller et al., (2006). Here, researchers used an intracranial injection of ibotenic acid in P5 mice to mimic the effects of PVL in preterm neonates. Animals who received a single injection of Epo (at 5,000 U/kg) showed attenuated lesion size when the drug was administered less than an hour after injury. Delayed administration (4, 8, or 12 hours post injury), however, was found to have no therapeutic effect [60]. Our current study showed similar evidence of a therapeutic time window for efficacy of Epo following neonatal brain injury in rodent models. Specifically, we showed beneficial effects of Epo when administered immediately following the induction of injury, with little to no benefit when administration was delayed for an hour or more post injury. Additional studies are needed to fully understand the effects of delayed administration of Epo following HI injury along with the possible importance of these finding to clinical populations.
Finally, future studies should include assessment of RAP in female HI injured animals treated with Epo, noting that prior studies have failed to show a robust RAP deficit as typically seen in males [21], thus providing little platform to identify any “improvements” following treatment.
Conclusions
The current study extends previous findings showing that immediate treatment with Epo following P7 HI injury in a rodent models yields positive therapeutic effects on rapid auditory processing tasks, as well as reducing gross brain injury as measured by ventricular indices. We report here that delaying treatment with Epo for one to three hours hour results in no measurable benefits on RAP and histological measures. Future studies will need to more carefully examine the effects of Epo administration during the time window under 60 minutes following injury. Future studies should also investigate the use of both multiple doses after a delay, higher doses (up to 30,000 U/kg) after a delay, and potential protective effects of Epo in females following neonatal HI. In closing, these findings may have critical implications in suggesting that time is truly “of the essence” in providing clinical therapeutic interventions for neonates with HI, and this issue should be further studied in human clinical trials. Further, while the results of the study presented here are specifically relevant to the clinical efficacy of delayed Epo treatment in term HIE populations, future work should explore the relevance of the findings for preterm and other neonatal HI populations as well.
Acknowledgments
We would like to thank the undergraduate students in our lab with their help in behavioral testing for this study. We would also like the thank Dr. Glenn D. Rosen and the laboratory of Dr. John Salamone for advice on histological preparation and analysis. This research was funded by NIH Grant HD049792.
References
- 1.Volpe JJ. Neurology of the newborn. Vol. 5. W.B. Saunders Company; 2008. pp. 347–400. [Google Scholar]
- 2.Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Aarnoudse-Moens CS, Weisglas-Kuperus N, van Goudoever JB, Oosterlaan J. Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatr. 2009;124:717–728. doi: 10.1542/peds.2008-2816. [DOI] [PubMed] [Google Scholar]
- 4.Anderson CA, Arciniegas DB. Cognitive sequelae of hypoxic-ischemic brain injury: A review. NeuroRehabilitation. 2010;26(1):47–63. doi: 10.3233/NRE-2010-0535. [DOI] [PubMed] [Google Scholar]
- 5.Badawi N, Keogh JM, Dixon G, Kurinczuk JJ. Developmental outcomes of newborn encephalopathy in the term infant. Indian Journal of Pediatrs. 2001;68(6):527–530. doi: 10.1007/BF02723247. [DOI] [PubMed] [Google Scholar]
- 6.de Vries LS, Jongmans MJ. Long-term outcome after neonatal hypoxic-ischaemic encephalopathy. Archives of Disease in Childhood Fetal and Neonatal Edition. 2010;95(3):220–224. doi: 10.1136/adc.2008.148205. [DOI] [PubMed] [Google Scholar]
- 7.Ortiz-Mantilla S, Choudhury N, Leevers H, Benasich AA. Understanding language and cognitive deficits in very low birth weight children. Dev Psychobiol. 2008;50(2):107–126. doi: 10.1002/dev.20278. [DOI] [PubMed] [Google Scholar]
- 8.Jansson-Verkasola E, Valkama M, Vainionpaa L, Paakko E, Iikko, E’Lehtihalmes M. Language development in very low birth weight preterm children: a follow up study. Folia Phoniatr Logop. 2004;56:108–119. doi: 10.1159/000076062. [DOI] [PubMed] [Google Scholar]
- 9.Steinman KJ, Gorno-Tempini ML, Glidden DV, Kramer JH, Miller SP, Barkovich AJ, Ferriero DM. Neonatal watershed brain injury on MRI correlates with verbal IQ at four years. Pediatrs. 2009;123(3):1025–1030. doi: 10.1542/peds.2008-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Downie AL, Jakobson LS, Frisk V, Ushycky I. Auditory temporal processing deficits in children with periventricular brain injury. Brain Lang. 2002;80(2):208–225. doi: 10.1006/brln.2001.2594. [DOI] [PubMed] [Google Scholar]
- 11.Benasich AA, Tallal P. Infant discrimination of rapid auditory cues predicts later language impairment. Behav Brain Res. 2002;136(1):31–49. doi: 10.1016/s0166-4328(02)00098-0. [DOI] [PubMed] [Google Scholar]
- 12.Choudhury N, Leppanen PHT, Leevers HJ, Benasich AA. Infant information processing and family history of specific language impairment: converging evidence for RAP deficits from two paradigms. Dev Sci. 2007;10(2):213–236. doi: 10.1111/j.1467-7687.2007.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tallal P. Rapid auditory processing in normal and disordered language development. J Speech Hear Res. 1976;19(3):561–571. doi: 10.1044/jshr.1903.561. [DOI] [PubMed] [Google Scholar]
- 14.Heim S, Friedman JT, Keil A, Benasich AA. Reduced sensory oscillatory activity during rapid auditory processing as a correlate of language-learning impairment. J Neurolinguistics. 2011;24:538–555. doi: 10.1016/j.jneuroling.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choudhury N, Benasich AA. Maturation of auditory evoked potentials from 6 to 48 months: Prediction to 3 and 4 year language and cognitive abilities. Clin Neurophysiol. 2011;122:320–338. doi: 10.1016/j.clinph.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 16.Vannucci RC, Vannucci SJ. A model of perinatal hypoxic-ischemic brain damage. Annals of the New York Academy of Sciences. 1997;835:234–249. doi: 10.1111/j.1749-6632.1997.tb48634.x. [DOI] [PubMed] [Google Scholar]
- 17.Vannucci RC. Hypoxic-ischemic encephalopathy. Am J Perinatol. 2000;17(3):113–120. doi: 10.1055/s-2000-9293. [DOI] [PubMed] [Google Scholar]
- 18.McClure MM, Peiffer AM, Rosen GD, Fitch RH. Auditory processing deficits in rats with neonatal hypoxic-ischemic injury. Int J Dev Neurosci. 2005;23(4):351–362. doi: 10.1016/j.ijdevneu.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 19.McClure MM, Threlkeld SW, Rosen GD, Fitch R. Auditory processing deficits in unilaterally and bilaterally injured hypoxic-ischemic rats. Neuroreport. 2005;16(12):1309–1312. doi: 10.1097/01.wnr.0000175613.16183.6c. [DOI] [PubMed] [Google Scholar]
- 20.McClure MM, Threlkeld SW, Rosen GD, Fitch R. Rapid auditory processing and learning deficits in rats with P1 versus P7 neonatal hypoxic-ischemic injury. Behav Brain Res. 2006;172(1):114–121. doi: 10.1016/j.bbr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill CA, Threlkeld SW, Fitch RH. Early testosterone modulated sex differences in behavioral outcome following neonatal hypoxia ischemia in rats. Int J Dev Neurosci. 2011;29(4):381–388. doi: 10.1016/j.ijdevneu.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buemi M, Cavallaro E, Floccari F, Sturiale A, Aloisi C, Trimarchi M, Crasso G, Corica F, Frisina N. Erythropoietin and the brain: From neurodevelopment to neuroprotection. Clin Trials. 2002;103(3):275–282. doi: 10.1042/cs1030275. [DOI] [PubMed] [Google Scholar]
- 23.Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ. 2004;11:37–44. doi: 10.1038/sj.cdd.4401450. [DOI] [PubMed] [Google Scholar]
- 24.Soliz J. Erythropoietin and respiratory control at adulthood and during early postnatal life. Respir Physiol Neurobiol. 2012 doi: 10.1016/j.resp.2012.07.018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 25.Spandou E, Soubasi V, Papoutsopoulou S, Karkavelas G, Simeonidou C, Kaiki-Astara A, Guiba-Tziampiri O. Erythropoietin prevents hypoxia/ischemia-induced DNA fragmentation in an experimental model of perinatal asphyxia. Neurosci Lett. 2004;366(1):24–28. doi: 10.1016/j.neulet.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 26.Bierer R, Peceny MC, Hartenberger CH, Ohls RK. Erythropoietin concentrations and neurodevelopmental outcome in preterm infants. Pediatrs. 2006;118(3):e635–640. doi: 10.1542/peds.2005-3186. [DOI] [PubMed] [Google Scholar]
- 27.McPhearson RJ, Juul SE. Erythropoietin for infants with hypoxic-ischemic encephalopathy. Curr Opin Pediatr. 2010;22(2):138–145. doi: 10.1097/MOP.0b013e328336eb57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAdams RM, McPhearson RJ, Mayock DE, Juul SE. Outcomes of extremely low birth weight infants given early high-dose erythropoietin. J Perinatol. 2012 Jun; doi: 10.1038/jp.2012.78. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, Ji L, Guo X, Xiong H, Simbruner G, Blomgren K, Wang X. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrs. 2009;124(2):e218–226. doi: 10.1542/peds.2008-3553. [DOI] [PubMed] [Google Scholar]
- 30.Elmahdy H, El-Mashad AR, El-Bahrawy H, El-Gohary T, El-Barbary A, Aly H. Human recombinant erythropoietin in asphyxia neonatorum: Pilot trial. Pediatrs. 2010;125(5):e1135–1142. doi: 10.1542/peds.2009-2268. [DOI] [PubMed] [Google Scholar]
- 31.Aydin A, Genc K, Akhisaroglu M, Yorukoglu K, Gokmen N, Gonullu E. Erythropoietin exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. Brain Dev. 2003;25(7):494–498. doi: 10.1016/s0387-7604(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 32.Chang YS, Mu D, Wendland M, Sheldon RA, Vexler ZS, McQuillen PS, Ferriero DM. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatr Res. 2005;58(1):106–111. doi: 10.1203/01.PDR.0000163616.89767.69. [DOI] [PubMed] [Google Scholar]
- 33.Fan X, Heijnen CJ, van der Koolj MA, Groenendaal F, van Bel F. Beneficial effect of erythropoietin on sensorimotor function and white matter after hypoxia-ischemia in neonatal mice. Pediatr Res. 2011;69(1):56–61. doi: 10.1203/PDR.0b013e3181fcbef3. [DOI] [PubMed] [Google Scholar]
- 34.Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, Ulukus C, Genc S, Ozkan H. Neuroprotective effect of erythropoietin on hypoxic-ischemic brain injury in neonatal rats. Biology of the Neonate. 2003;83(3):224–228. doi: 10.1159/000068926. [DOI] [PubMed] [Google Scholar]
- 35.Kumral A, Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, Kiray M, Genc S, Duman N, Koroglu TF, Ozkan H, Genc K. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res. 2004;153(1):77–86. doi: 10.1016/j.bbr.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 36.McClure MM, Threlkeld SW, Fitch RH. Auditory processing and learning/memory following erythropoietin administration in neonatally hypoxic-ischemic injured rats. Brain Res. 2007;1132(1):203–209. doi: 10.1016/j.brainres.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 37.McClure MM, Threlkeld SW, Fitch RH. The effects of erythropoietin on auditory processing following neonatal hypoxic-ischemic injury. Brain Res. 2006;1087(1):190–195. doi: 10.1016/j.brainres.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Iwai M, Stetler RA, Xing J, Hu X, Gao Y, Zhang W, Chen J, Cao G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke. 2010;41(5):1032–1037. doi: 10.1161/STROKEAHA.109.570325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizuno K, Hida H, Masuda T, Nishino H, Togari H. Pretreatment with low doses of erythropoietin ameliorates brain damage in periventricular leukomalacia by targeting late oligodendrocyte progenitors: A rat model. Neonatology. 2008;94(4):255–266. doi: 10.1159/000151644. [DOI] [PubMed] [Google Scholar]
- 40.Kim SS, Lee KH, Sung DK, Shim JW, Kim MJ, Jeon GW, Chang YS, Park WS. Erythropoietin attenuates brain injury, subventricular zone expansion, and sensorimotor deficits in hypoxic-ischemic neonatal rats. J Korean Med Sci. 2008;23(3):484–491. doi: 10.3346/jkms.2008.23.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunn AJ, Bennet L, Gunning MI, Gluckman PD, Gunn TR. Cerebral hypothermia is not neuroprotective when started after postischemic seizures in fetal sheep. Pediatr Res. 1999;40(3):274–280. doi: 10.1203/00006450-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 42.Karlsson M, Tooley JR, Satas S, Hobbs CE, Chakkarapani E, Stone J, Poter H, Thoresen M. Delayed hypothermia as selective head cooling or whole body cooling does not protect brain or body in newborn pig subjected to hypoxia-ischemia. Pediatr Res. 2008;64(1):74–78. doi: 10.1203/PDR.0b013e318174efdd. [DOI] [PubMed] [Google Scholar]
- 43.Fitch RH, Threlkeld SW, McClure MM, Peiffer AM. Use of a modified prepulse inhibition paradigm to assess complex auditory discrimination in rodents. Brain Res Bull. 2008;70:1–7. doi: 10.1016/j.brainresbull.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dame C, Juul SE, Christensen RD. The biology of erythropoietin in the central nervous system and its neurotrophic and neuroprotective potential. Biology of the Neonate. 2001;79(3–4):228–235. doi: 10.1159/000047097. [DOI] [PubMed] [Google Scholar]
- 45.Wen TC, Rogido M, Genetta T, Sola A. Permanent focal cerebral ischemia activates erythropoietin receptor in the neonatal rat brain. Neuroci Lett. 2004;355(3):165–168. doi: 10.1016/j.neulet.2003.10.078. [DOI] [PubMed] [Google Scholar]
- 46.Juul SE, Stallings SA, Christensen RD. Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatr Res. 1999;46(5):543–547. doi: 10.1203/00006450-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 47.Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8(2):207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- 48.Sola A, Rogido M, Lee BH, Genetta T, Wen TC. Erythropoietin after focal cerebral ischemia activates the janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57(4):481–487. doi: 10.1203/01.PDR.0000155760.88664.06. [DOI] [PubMed] [Google Scholar]
- 49.Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20(21):7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Statler PA, McPhearson RJ, Bauer LA, Kellert BA, Juul SE. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr Res. 2007;61(6):671–675. doi: 10.1203/pdr.0b013e31805341dc. [DOI] [PubMed] [Google Scholar]
- 51.Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induce neuronal apoptosis. Prog Neurobiol. 2000;62:215–249. doi: 10.1016/s0301-0082(00)00011-3. [DOI] [PubMed] [Google Scholar]
- 52.Xu X, Dia H, Shi Y. Erythropoietin protects primary cultures of rat cortical neurons from hypoxia-induced toxicity through attenuating both glutamate release and NMDA receptor evoked neurotoxicity pathway. Pharmazie. 2009;64(3):210–213. [PubMed] [Google Scholar]
- 53.Sun Y, Calvert JW, Zhang JH. Neonatal Hypoxia/Ischemia is associated with decreased inflammatory mediators after Erythropoietin administration. Stroke. 2005;36:1672–1678. doi: 10.1161/01.STR.0000173406.04891.8c. [DOI] [PubMed] [Google Scholar]
- 54.Sun Y, Zhou C, Polk P, Nanda A, Zhang JH. Mechanisms of erythropoietin-induced brain protection in neonatal hypoxia-ischemia rat model. J Cereb Blood Flow Metab. 2004;24:259–270. doi: 10.1097/01.WCB.0000110049.43905.AC. [DOI] [PubMed] [Google Scholar]
- 55.Yamada M, Burke C, Colditz P, Johnson DW, Gobe G. Erythropoietin protects against apoptosis and increases expression of non-neuronal cell markers in the hypoxia-injured developing brain. J Pathol. 2011;224:101–109. doi: 10.1002/path.2862. [DOI] [PubMed] [Google Scholar]
- 56.Brissaud O, Villega F, Pieter Konsman J, Sanchez S, Franconi JM, Chateil JF, Bouzier-Sore AK. Short-term effect of erythropoietin on brain lesions and aquaporin-4 expression in a hypoxic-ischemic neonatal rat model assessed by magnetic resonance diffusion weighted imaging and immunohistochemistry. Pediatr Res. 2010;68(2):123–127. doi: 10.1203/PDR.0b013e3181e67d02. [DOI] [PubMed] [Google Scholar]
- 57.Kumral A, Genc S, Ozer E, Yilmaz O, Gokmen N, Koroglu T, Duman N, Genc K. Erythropoietin downregulates Bax and DP5 proapoptoic gene expression in neonatal hypoxic-ischemic brain injury. Bio Neonate. 2006;89:205–210. doi: 10.1159/000089951. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez FF, Abel R, Almli CR, Mu D, Wendland M, Ferriero DM. Erythropoietin sustains cognitive function and brain volume after neonatal stoke. Dev Neurosci. 2008;31:403–411. doi: 10.1159/000232558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwai M, Stetler RA, Xing J, Hu X, Gao Y, Zhang W, Chen J, Cao G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic ischemic brain injury. Stroke. 2010;41:1032–1037. doi: 10.1161/STROKEAHA.109.570325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keller M, Yang J, Griesmaier E, Gorna A, Sarkozy G, Urbanek M, Gressens P, Simbruner G. Erythropoietin is neuroprotective against NMDA-receptor-mediated excitotoxic brain injury in newborn mice. Neurobiol Dis. 2006;24(2):357–366. doi: 10.1016/j.nbd.2006.07.007. [DOI] [PubMed] [Google Scholar]