

Abstract
Ribbon synapses of tonically releasing sensory neurons must provide a large pool of releasable vesicles for sustained release, while minimizing spontaneous release in the absence of stimulation. Complexins are presynaptic proteins that may accomplish this dual task at conventional synapses by interacting with the molecular machinery of synaptic vesicle fusion at the active zone to retard spontaneous vesicle exocytosis yet facilitate release evoked by depolarization. However, ribbon synapses of photoreceptor cells and bipolar neurons in the retina express distinct complexin subtypes, perhaps reflecting the special requirements of these synapses for tonic release. To investigate the role of ribbon-specific complexins in transmitter release, we combined presynaptic voltage clamp, fluorescence imaging, electron microscopy, and behavioral assays of photoreceptive function in zebrafish. Acute interference with complexin function using a peptide derived from the SNARE-binding domain increased spontaneous synaptic vesicle fusion at ribbon synapses of retinal bipolar neurons without affecting release triggered by depolarization. Knockdown of complexin by injection of an antisense morpholino into zebrafish embryos prevented photoreceptor-driven migration of pigment in skin melanophores and caused the pigment distribution to remain in the dark-adapted state even when embryos were exposed to light. This suggests that loss of complexin function elevated spontaneous release in illuminated photoreceptors sufficiently to mimic the higher release rate normally associated with darkness, thus interfering with visual signaling. We conclude that visual system-specific complexins are required for proper illumination-dependent modulation of the rate of neurotransmitter release at visual system ribbon synapses.
Introduction
Neurotransmitter release via synaptic vesicle exocytosis depends on the formation of the SNARE complex, a tight association of four α helices contributed by three presynaptic proteins: the vesicle protein synaptobrevin/vesicle-associated membrane protein (VAMP) and the plasma-membrane-associated proteins syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) (Südhof and Rothman, 2009). In addition, SNARE function is regulated by complexin (Cplx), which is a small presynaptic protein that binds to the four-helix bundle of an assembled SNARE complex. Although effects of Cplx knock-outs on synaptic transmission vary with species and cell type (Brose, 2008), it seems clear that Cplx can serve dual roles as a clamp for spontaneous neurotransmitter release and an activator of fast calcium-triggered vesicle fusion (Maximov et al., 2009; Xue et al., 2010; Yang et al., 2010; Hobson et al., 2011; Martin et al., 2011). Ribbon synapses of tonically releasing sensory neurons maintain large pools of releasable vesicles to support both fast, phasic and slower, sustained transmitter release (Matthews and Fuchs, 2010), and they may therefore have special demands for facilitating evoked release and preventing spontaneous vesicle fusion, the dual roles postulated for Cplx. Indeed, conventional synapses express principally Cplx subtypes 1 and 2, but two different subtypes—Cplx3 and Cplx4—are expressed selectively at retinal ribbon synapses (Reim et al., 2005), whereas ribbon synapses of cochlear hair cells lack Cplx altogether (Strenzke et al., 2009). Knock-out of both Cplx3 and Cplx4 causes moderate visual deficits in mice (Reim et al., 2009), consistent with a role in regulation of transmitter release. However, the details of the function of ribbon-specific complexins at retinal synapses are largely unknown.
The zebrafish genome contains two orthologs of mammalian Cplx3 and three orthologs of mammalian Cplx4 (Zanazzi and Matthews, 2010). Although all of the Cplx isoforms, except for Cplx3b, are expressed in photoreceptors and bipolar cells during larval zebrafish development, Cplx4a appears to be the predominant isoform (Zanazzi and Matthews, 2010; Zanazzi, 2010). To establish the roles of the ribbon-specific Cplx in regulating calcium-triggered and spontaneous vesicle exocytosis at ribbon synapses, we used two complementary approaches to interfere with Cplx3/4 function in zebrafish. First, we acutely dialyzed isolated retinal bipolar neurons with a peptide derived from the conserved SNARE-binding domain of Cplx3/4, to compete with endogenous Cplx. Second, we knocked down expression of Cplx4a, the principal subtype at retinal and pineal ribbon synapses in zebrafish (Zanazzi and Matthews, 2010), using a translation-blocking antisense morpholino. Our results indicate that one role of ribbon-specific Cplx is to prevent an unacceptably high rate of spontaneous release that would otherwise interfere with visual signaling.
Materials and Methods
Animals and tissue preparation.
The Institutional Animal Care and Use Committee of the Stony Brook University approved all animal procedures. Husbandry of zebrafish and procedures for immunofluorescence staining were as described by Zanazzi and Matthews (2010). The procedure for acutely isolating bipolar cells from adult zebrafish retina was similar to that described for goldfish by Heidelberger and Matthews (1992). Briefly, retinas were dissected from eyecups of either sex in saline [(in mm) 115 NaCl, 2.5 KCl, 0.5 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4], treated for 25 min with hyaluronidase (500 U/ml; Worthington Biochemical), and then transferred to cold saline containing 2.7 mm dl-cysteine HCl (EMD Biosciences), cut into small pieces, and incubated 25–30 min in saline containing dl-cysteine and 15–30 U/ml papain (Sigma-Aldrich) at 25°C. A piece of retina was triturated via a flame-polished Pasteur pipette, and dissociated cells were plated onto glass-bottomed dishes containing saline with CaCl2 increased to 2.5 mm for patch-clamp recording. ON bipolar cells were identified by their characteristic morphology.
Electrophysiology and activity-dependent loading with FM dye.
Whole-cell patch-clamp recordings were made from acutely dissociated bipolar cells, with the patch pipette placed directly on the synaptic terminal, which had a diameter of ∼5 μm for the large-terminal ON cells, which are equivalent to class Mb1 in goldfish (Heidelberger and Matthews, 1992). The pipette solution contained the following (in mm): 120 Cs-gluconate, 10 tetraethylammonium-Cl, 3 MgCl2, 0.2 N-methyl-d-glucamine-EGTA, 2 Na2ATP, 0.5 Na2GTP, 20 HEPES, pH 7.2. Peptides were added at the concentrations indicated in relevant parts of Results. Before recording, bipolar cell terminals were loaded with FM4-64 (Invitrogen) by exchanging the bath solution with a high-K+ solution (80 mm KCl replacing NaCl) plus 5 μm FM4-64 for 60 s, followed by multiple washes with standard solution, 60 s in standard solution plus 1 mm Advasep-7 (Biotium), and again multiple washes with standard solution. Bipolar cells were then selected for recording based on good viability, intact morphology, and FM4-64 uptake restricted to synaptic terminals. Membrane currents were recorded under voltage clamp using a HEKA EPC-9 amplifier controlled by PatchMaster software (HEKA). Holding potential was −60 or −65 mV, which is more negative than the threshold for activation of calcium channels (−55 to −50 mV), and calcium channels were activated by pulses of varying duration to 0 mV. Membrane capacitance, series conductance, and membrane conductance were measured using the sine+DC method of the PatchMaster lock-in extension and a 1600 Hz sinusoidal stimulus with peak-to-peak amplitude of 10 mV centered on the holding potential. Pipettes were coated with dental wax to reduce stray capacitance.
Imaging.
Fluorescence images were acquired using an Olympus FV-1000 laser-scanning confocal system connected to an Olympus IX-81 inverted microscope, controlled by Olympus FV10-ASW software. For live-cell imaging, care was taken to minimize photobleaching and phototoxicity by using fast scan speed (2 μs/pixel), low laser intensity (0.1–0.5% of maximum), and low pixel density (frame size, 64 × 64–256 × 256 pixels). Images were analyzed using ImageJ (imagej.nih.gov).
Electron microscopy.
For electron microscopy of individually identified bipolar cells after whole-cell patch-clamp recording, cells were plated onto chambers constructed using Aclar film (Electron Microscopy Sciences) rather than glass. After recording, the patch pipette was removed and bipolar cells were fixed by local superfusion with fixative containing 2.5% paraformaldehyde and 2.5% glutaraldehyde in 0.13 m phosphate buffer, using an application pipette placed near the cell. After 10 min, the application pipette was removed, the entire bath solution was exchanged for fixative, and a surrounding rectangle was etched in the Aclar around the recorded cell using a broken patch pipette to facilitate subsequent identification of the cell in the electron microscope. Following fixation at 4°C overnight, cells were postfixed in osmium tetroxide, dehydrated, embedded, and sectioned for electron microscopy as described previously (Paillart et al., 2003; Matthews and Sterling, 2008). For electron microscopy of morphant zebrafish embryos, whole embryos injected with control or Cplx4a morpholino were fixed overnight at 4°C in 2.5% paraformaldehyde/2.5% glutaraldehyde in 0.13 m phosphate buffer, and then prepared as described for isolated cells. In experiments to measure the diameter of synaptic vesicles, bipolar cells were fixed conventionally as described above, or were rapidly frozen followed by freeze-substitution fixation. For the latter, isolated bipolar cells on small pieces of Aclar were frozen by freeze-slamming using a Leica EM-CPC device, and frozen samples were transferred to cryotubes containing distilled acetone plus 1% osmium tetroxide frozen in liquid nitrogen. The tubes were then placed in dry ice in a sealed Styrofoam container, which was kept in a −80°C freezer for 24 h and then moved to a 4°C cold room to warm slowly over the course of ∼48 h. Temperature within the box was monitored periodically via a thermistor within a cryotube kept alongside the sample-containing tubes. After reaching 4°C, the box was moved to a laboratory bench and allowed to warm to room temperature, after which the samples were embedded and sectioned for electron microscopy.
Antisense morpholino knockdown of Cplx expression.
To knock down complexin 4a protein expression, a morpholino antisense oligonucleotide (Gene Tools) (5′-AAACGCCATTATTTACCACGCCGGA-3′) complementary to the translation start site of the complexin 4a gene was used. A morpholino antisense oligonucleotide (5′-CCTCTTACCTCAGTTACAATTTATA-3′) directed against a human β-globin intron mutation was used as a negative control. Morpholino solutions were diluted to 20 ng/nl with 0.2 m KCl and 2 mg/ml phenol red. Then, 0.5 nl of a morpholino solution was pressure-injected into one-cell-stage, dechorionated zebrafish embryos. Zebrafish embryos were then raised at 28.5°C in embryo medium (0.346 mg/ml sodium bicarbonate supplemented with HBSS #1, HBSS #2, HBSS #4, HBSS #5) with penicillin-streptomycin. The day after injection, embryos were switched to embryo water [10 ml of reverse osmosis water plus 3 mg of Instant Ocean plus 10 μl of methylene blue (1 mg/ml)], which was replaced daily thereafter. Embryos and larvae were maintained on a controlled cycle of 13 h of light and 11 h of darkness, and were killed with 0.168 mg/ml tricaine (Sigma-Aldrich).
Melanophore responses to light and dark adaptation.
Zebrafish embryos [2 d postfertilization (dpf)] or larvae (5 dpf) were initially dark-adapted or light-adapted for a minimum of 1 h at 28.5°C. Representative zebrafish were individually immobilized by placing them in small wells cut out of a thin, 2% agar layer in a 100-mm-diameter Petri dish (the “imaging chamber”). The zebrafish were examined on a Zeiss SteREO Discovery.V20 stereomicroscope equipped with an Axiocam camera and AxioVision 4.8 software. Dorsal views were photographed. The embryos and larvae were then light-adapted or dark-adapted for a minimum of 1 h at 28.5°C. At the end of this period, embryos and larvae were again photographed in the imaging chambers. Since tricaine leads to the dispersion of melanosomes (Sheets et al., 2007), all of the imaging experiments were performed on nonanesthetized zebrafish. Photographs were then analyzed using ImageJ to trace the outlines of pigmented regions and measure the percentage of the dorsal surface of the head covered by pigmented cells.
Results
An inhibitory peptide derived from the SNARE-binding domain of Cplx3 increases spontaneous synaptic vesicle fusion
To acutely interfere with Cplx function at ribbon synapses, we designed an inhibitory peptide containing the SNARE-binding domain (SBD) of mouse Cplx3 but lacking other domains required for function. In mammals, there are four Cplx proteins, Cplx1–Cplx4, whose amino-acid sequences are aligned in Figure 1A. A variety of lines of evidence point to the importance of the region highlighted in gray in Figure 1A for binding of Cplx to the assembled SNARE complex. First, a peptide consisting of the homologous region of squid Cplx (shown beneath the corresponding sequences of mouse Cplx in Fig. 1A) competes with full-length Cplx for binding to syntaxin and inhibits neurotransmitter release when injected into squid giant synapse (Tokumaru et al., 2001), indicating that the peptide acts as a dominant negative. Second, a synthetic peptide corresponding to this region has been shown directly to bind to assembled SNARE complexes incorporated into lipid bilayers, based on single-molecule FRET measurements (Li et al., 2007). Third, biochemical analysis of SNARE-binding by Cplx1 demonstrated that the residues shown in red in Figure 1A are critical for binding (Xue et al., 2007). In keeping with their importance for binding, these amino acids are invariant across Cplx subtypes and across vertebrate and invertebrate species (Tokumaru et al., 2001; Xue et al., 2007; Martin et al., 2011; Fig. 1C, comparison with zebrafish Cplx-family members). Fourth, crystal structure of Cplx1 bound to the four-helix SNARE bundle (Kümmel et al., 2011) shows that binding of the central helix of Cplx1 is mediated by the portion shaded yellow in Figure 1B, which fits within a groove formed by α helices of synaptobrevin/VAMP and syntaxin. This part of Cplx1 (Fig. 1A, yellow box) corresponds to the gray-highlighted sequence of Cplx3 in Figure 1A. We therefore used this portion of mouse Cplx3 to construct a 15-residue synthetic peptide (ATLRSHFRDKYRLPK), designated Cplx-SBD, which we introduced into zebrafish bipolar neurons via whole-cell dialysis to inhibit functions mediated by the central helix of Cplx. The sequence of Cplx-SBD is highly conserved among the five Cplx3/4 isoforms found in zebrafish (Fig. 1C), including Cplx4a, which is the principal subtype found in visual-system ribbon synapses (Zanazzi and Matthews, 2010).
Figure 1.
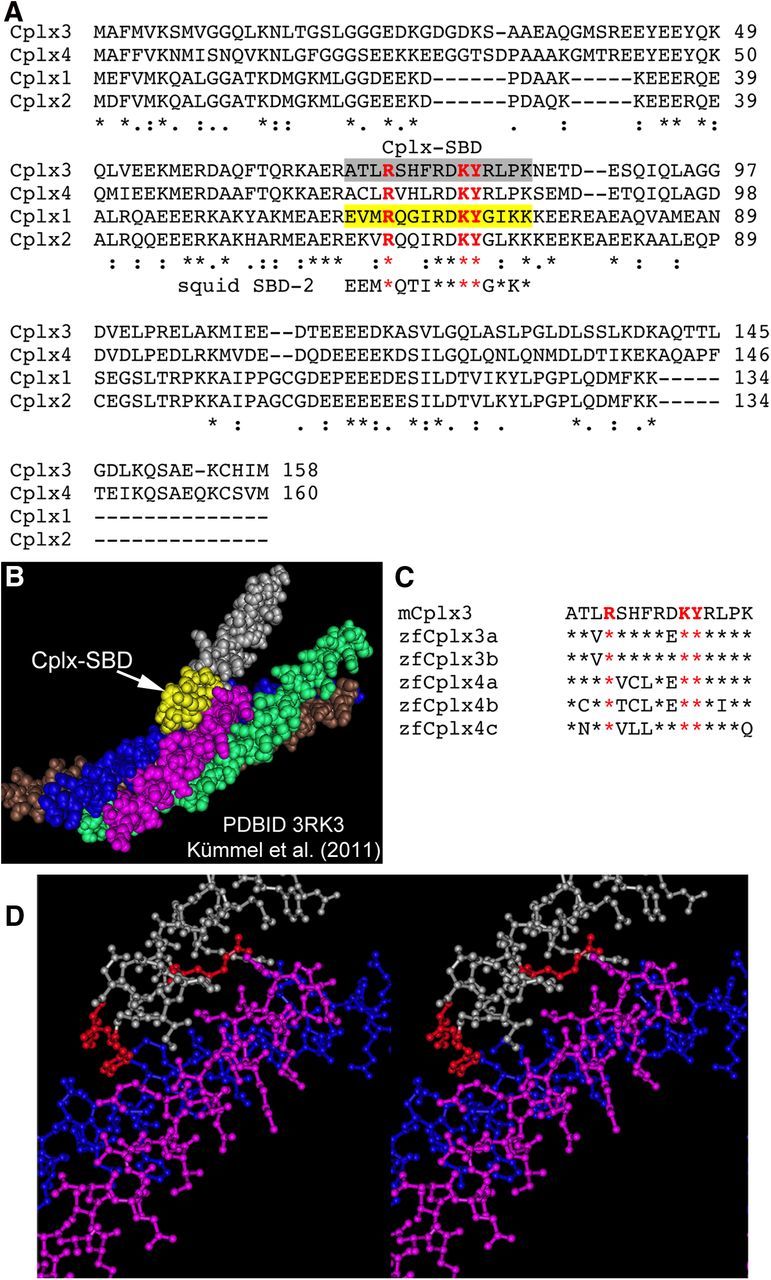
Design of an inhibitory peptide based on the SNARE-binding domain of Cplx3. A, Clustal alignment of mouse Cplx1–Cplx4. Gray highlight indicates the 15-residue SNARE-binding domain (Cplx-SBD) used as an inhibitory peptide in whole-cell recordings from bipolar neurons. Conserved residues required for binding (Xue et al., 2007) are shown in red. These were substituted by alanine to generate a non-SNARE-binding control peptide (Cplx-SBD/3A). The region of Cplx1 shown in yellow, which corresponds to Cplx-SBD, contacts the assembled SNARE complex as revealed by crystal structure (Kümmel et al., 2011). The sequence of the SBD-2 peptide shown by Tokumaru et al. (2001) to interfere with binding of squid Cplx to syntaxin is also included for comparison with Cplx-SBD. B, Space-filling model of the four-helix SNARE bundle bound with Cplx1, generated using Cn3D (Wang et al., 2000) from crystallographic data in Protein Data Bank ID 3RK3 (Kümmel et al., 2011). The region of Cplx1 corresponding to Cplx-SBD is shown in yellow. Silver and yellow, Cplx1; magenta, synaptobrevin/VAMP2; blue, syntaxin1a; green and brown, two helices of SNAP-25. C, Alignment of the Cplx3-SBD from mouse with the five Cplx subtypes of zebrafish (Zanazzi and Matthews, 2010). Residues shown in red are critical for SNARE binding and indicate the positions substituted by alanine in the inactive control peptide (Cplx-SBD/3A). D, Stereogram of ball-and-stick model of Cplx1 (silver) interacting with syntaxin1a (blue) and VAMP2 (magenta). The amino acids of Cplx1 shown in red correspond to the three critical residues (A and C) whose side-chains interact with the binding groove formed by α helices of syntaxin1a and synaptobrevin/VAMP2. The helices of SNAP-25 are omitted for clarity.
In designing a control peptide, we exploited previous results on the interaction between Cplx and the SNARE complex. Biochemical studies (Xue et al., 2007) have shown that substitution of alanine for arginine 63 or double substitution of alanine for lysine 69 and tyrosine 70 eliminated binding of Cplx1 to the assembled SNARE complex. Based on crystallographic data (Kümmel et al., 2011), the side chains of these three critical amino acids of Cplx project into the binding groove formed by α helices of syntaxin and synaptobrevin/VAMP, as shown in the stereogram in Figure 1D (critical residues of Cplx shown in red), which is consistent with the importance of these residues demonstrated in biochemical binding assays (Xue et al., 2007). Therefore, we synthesized a control peptide, designated Cplx-SBD/3A, containing alanine at all three critical sites but otherwise identical to Cplx-SBD (ATLASHFRDAARLPK).
Isolated bipolar neurons were dialyzed in whole-cell patch-clamp recordings with an intracellular solution containing 10 μm Cplx-SBD or Cplx-SBD/3A, labeled at the N terminus with 5-carboxyfluorescein (5-FAM) to track dialysis. After break-in with a patch pipette placed directly on the synaptic terminal, peptide fluorescence rose rapidly to asymptote with a time constant of ∼5 s (Fig. 2, inset), which is similar to the τ of ∼4 s calculated from equations given in Pusch and Neher (1988) using the conditions of our recordings (molecular weight, ≈2000; series resistance, ≈20 MΩ; terminal radius, ≈2.3 μm). Before whole-cell recording, synaptic vesicles were preloaded with the activity-dependent dye, FM4-64, and spontaneous release was monitored using the rate of FM-dye destaining in the absence of stimulation (voltage-clamped at −60 mV). Figure 2 shows that in the presence of Cplx-SBD, FM4-64 fluorescence declined approximately linearly with a slope of −0.18 ± 0.03%/s, indicating ongoing synaptic vesicle exocytosis. However, FM4-64 fluorescence was relatively stable in the presence of the control peptide, Cplx-SBD/3A, with a slope of −0.05 ± 0.02%/s (p = 0.009, two-tailed t test), which was not significantly different from the decline due to photobleaching alone (−0.03 ± 0.01%/s; p = 0.42). We interpret these results to indicate that Cplx-SBD destabilizes docked and primed synaptic vesicles, so that they undergo exocytosis in the absence of calcium influx.
Figure 2.

Inhibitory peptide Cplx-SBD caused spontaneous destaining of FM4-64 in bipolar cell synaptic terminals. Synaptic vesicles were preloaded by activity-dependent uptake of FM4-64, and then voltage-clamped at −60 mV in whole-cell patch clamp. Data points show FM4-64 fluorescence of four terminals dialyzed with Cplx-SBD (open circles) and four terminals dialyzed with the control peptide, Cplx-SBD/3A (filled circles). Error bars indicate ±1 SEM. Both peptides were labeled with green-fluorescent 5-FAM to monitor effective dialysis of the synaptic terminal (inset).
To determine whether Cplx-SBD altered spontaneous release by increasing basal intracellular calcium concentration, synaptic terminals were dialyzed with 100 μm calcium indicator fluo-2 (KD, 390 nm) with or without unlabeled Cplx-SBD. The fluorescence reported by fluo-2 in single confocal optical sections through the terminal was not significantly different (p = 0.33; 2-tailed t test with unequal variance) in the presence [836 ± 129 arbitrary units (A.U.); N = 7] and absence (1015 ± 121 A.U.; N = 11) of Cplx-SBD, indicating that Cplx-SBD does not affect intracellular calcium concentration at rest. Therefore, a likely mechanism for the increased spontaneous FM-dye destaining is that Cplx-SBD, but not Cplx-SBD/3A, competes with the central helix native Cplx for binding to assembled SNARE complexes but lacks the clamping effect of full-length Cplx on SNARE-mediated fusion.
As an independent measure of vesicle exocytosis, we monitored the membrane capacitance of the synaptic terminal, which reflects the increase in surface area as synaptic vesicles fuse with the plasma membrane. In terminals dialyzed with 10 μm Cplx-SBD, capacitance rose steadily in the absence of stimulation, reaching a plateau ∼40 s after break-in, without correlated changes in series conductance. By contrast, terminals dialyzed with the inactive control peptide Cplx-SBD/3A showed little consistent change in either capacitance or series conductance after break-in. Examples of individual experiments for each peptide are shown in Figure 3A,B and averaged capacitance responses for seven cells each with Cplx-SBD and Cplx-SBD/3A are shown in Figure 3C. At asymptote (i.e., 40 s after onset of capacitance measurements), capacitance increased by 222 ± 51 fF with Cplx-SBD versus −62 ± 46 fF with Cplx-SBD/3A, which is a statistically significant difference (p = 0.001). Therefore, capacitance measurements are also consistent with an increase in spontaneous release in the presence of Cplx-SBD peptide but not Cplx-SBD/3A. Although membrane capacitance reached a plateau within ∼40 s after onset of dialysis with Cplx-SBD, FM4-64 destaining continued unabated throughout the recording (Fig. 2). We take this to mean that the plateau of capacitance represents a steady-state balance between exocytosis and endocytosis, rather than cessation of exocytosis.
Figure 3.
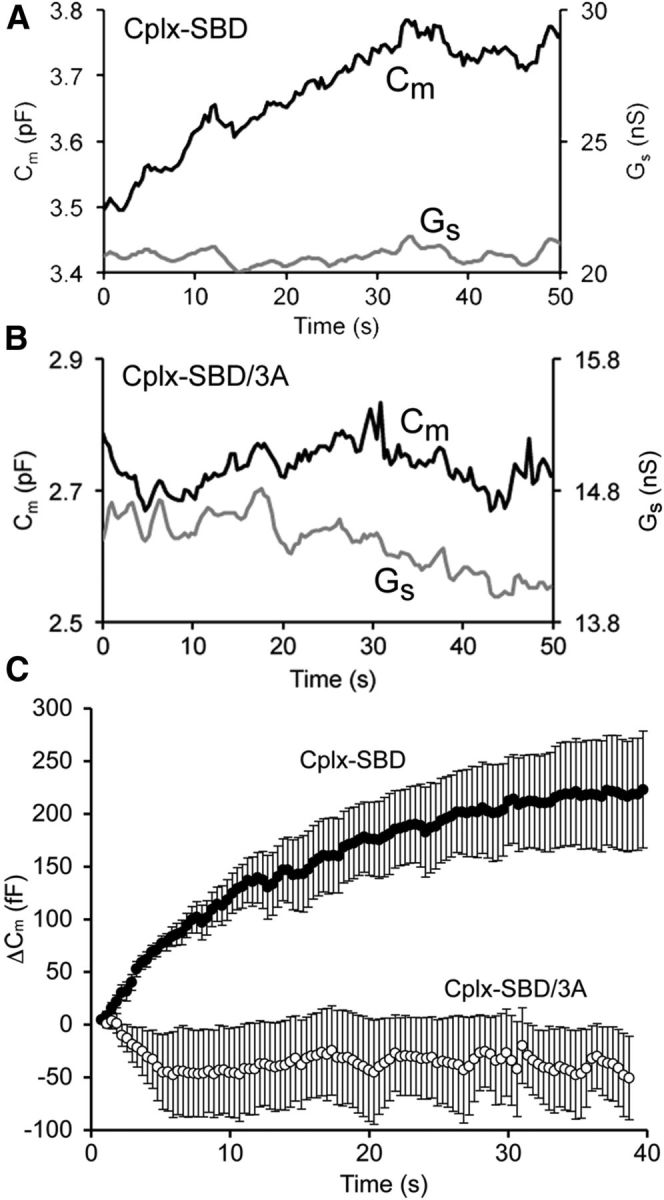
Inhibitory peptide Cplx-SBD caused a spontaneous rise in membrane capacitance of bipolar cell synaptic terminals. A, Example of the change in membrane capacitance (Cm) and series conductance (Gs) for a bipolar cell synaptic terminal dialyzed with Cplx-SBD. B, Example of a bipolar cell terminal dialyzed with the control peptide, Cplx-SBD/3A. C, Average change in membrane capacitance for seven terminals dialyzed with Cplx-SBD (filled circles) and seven terminals dialyzed with Cplx-SBD/3A (open circles). Error bars indicate ±1 SEM. Holding potential under voltage clamp, −60 mV.
Cplx-SBD peptide does not affect evoked synaptic vesicle fusion
At a variety of synapses, loss of Cplx increases spontaneous release and also inhibits fast neurotransmitter release evoked by stimulation (Brose, 2008; Maximov et al., 2009; Yang et al., 2010; Hobson et al., 2011; Martin et al., 2011). To determine whether the inhibitory peptide similarly inhibits evoked release as well as enhancing spontaneous release at bipolar-cell ribbon synapses, we examined the effect of Cplx-SBD on exocytosis elicited by activation of presynaptic calcium channels under voltage clamp, using capacitance measurements to monitor vesicle fusion. Figure 4A shows the average capacitance response to a 500 ms depolarization from −60 to 0 mV in terminals dialyzed with 10 μm Cplx-SBD, 10 μm Cplx-SBD/3A, or no peptide. In all three conditions, the response averaged ∼150 fF, corresponding to the release of ∼4500 synaptic vesicles during depolarization. As in goldfish bipolar cell terminals (von Gersdorff and Matthews, 1994; von Gersdorff et al., 1996), this population of vesicles represents the entire readily releasable pool in zebrafish bipolar neurons, which can be exhausted within ∼200 ms during strong depolarization (Vaithianathan and Matthews, unpublished observations). Although the initial response was unaffected by the inhibitory peptide, vesicles that refill the releasable pool may become sensitive to inhibition by Cplx-SBD while being prepared for release. However, subsequent responses to 500 ms depolarizations given after allowing >30 s for refilling of the readily releasable pool were also unaffected by Cplx-SBD, as shown in Figure 4B. Therefore, it appears that Cplx-SBD did not alter release triggered by calcium influx, even though it was able to destabilize spontaneous vesicle fusion.
Figure 4.
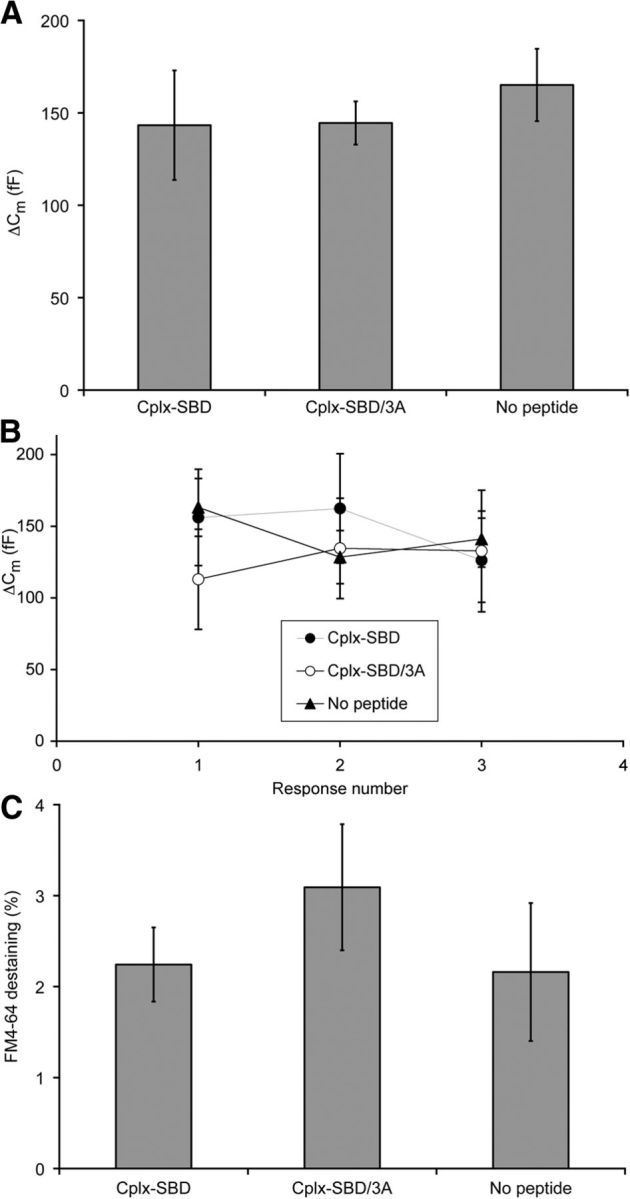
Inhibitory peptide Cplx-SBD had no effect on exocytosis evoked by depolarization in bipolar cell synaptic terminals. A, Change in membrane capacitance (ΔCm) elicited by 500 ms depolarization in terminals dialyzed with Cplx-SBD (N = 10), Cplx-SBD/3A (N = 10), or no peptide (N = 14). The differences are not statistically significant (p = 0.38 to 0.97; 2-tailed t test with unequal variance). B, Average capacitance changes in response to a series of three 500 ms depolarizations were similar in terminals dialyzed with Cplx-SBD (N = 11), Cplx-SBD/3A (N = 5), or no peptide (N = 15). None of the differences are statistically significant. C, Loss of FM4-64 fluorescence triggered by 500 ms depolarization in terminals dialyzed with Cplx-SBD (N = 10), Cplx-SBD/3A (N = 7), or no peptide (N = 5). The differences are not statistically significant (p = 0.32 to 0.93; 2-tailed t test with unequal variance). Error bars indicate ±1 SEM.
To provide an independent index of vesicle fusion elicited by stimulation, we measured the decrease in FM4-64 fluorescence evoked by 500 ms depolarizations, which stimulated exocytosis of synaptic vesicles and loss of trapped dye. As shown in Figure 4C, there was no significant difference in the amount of FM4-64 destaining triggered by depolarization in terminals dialyzed with 10 μm Cplx-SBD, 10 μm Cplx-SBD/3A, or no peptide, which is consistent with the results from capacitance measurements. Therefore, Cplx-SBD did not affect release of the readily releasable pool of vesicles during depolarization, monitored using either capacitance responses or FM4-64 destaining.
Cplx-SBD peptide depletes reserve synaptic vesicles but not vesicles associated with synaptic ribbons
Steady-state capacitance increased by ∼220 fF in the presence of Cplx-SBD peptide (Fig. 3), which we take to represent an increased rate of spontaneous vesicle fusion induced by the active peptide but not the control peptide. To determine how many synaptic vesicles must remain incorporated into the plasma membrane to reach this steady-state capacitance increase, we measured the diameter of synaptic vesicles using electron microscopy of zebrafish bipolar cells fixed by local application of conventional aldehyde fixative after whole-cell recording, or by rapid freezing followed by freeze-substitution fixation. Only vesicles associated with synaptic ribbons were measured, to avoid possible confusion of synaptic vesicles with other membrane-bound structures present in the cytoplasm of the terminal. The observed diameter of synaptic vesicles was the same after both mechanisms of fixation, averaging 32.2 ± 0.4 nm (N = 142) with conventional fixation and 32.5 ± 0.4 nm (N = 230) in frozen cells. Synaptic vesicles in zebrafish bipolar cell terminals are therefore similar in diameter to vesicles of goldfish bipolar cells (∼29 nm; von Gersdorff et al., 1996) and mouse bipolar cells (∼33 nm; LoGiudice et al., 2008). Assuming 10 fF/μm2, a single vesicle would have a capacitance of ∼33 aF, and ∼6700 vesicles would be required to reach the plateau of elevated capacitance during spontaneous release evoked by Cplx-SBD peptide.
Steady-state transfer of this number of vesicles from cytoplasmic pools to the plasma membrane might cause detectable reduction in cytoplasmic pools of synaptic vesicles. To examine this, the density of synaptic vesicles was determined at nonribbon locations within the synaptic terminal using electron microscopy of terminals fixed after whole-cell dialysis with either Cplx-SBD (Fig. 5A) or Cplx-SBD/3A (Fig. 5B). Vesicle-free zones, such as areas occupied by glycogen or mitochondria, were excluded from analysis. Figure 5C shows that cytoplasmic vesicle density was in fact significantly reduced in the presence of the active peptide, which stimulated spontaneous release, compared with the inactive control peptide. Nevertheless, Cplx-SBD had no effect on the number of vesicles associated with synaptic ribbons (Fig. 5C–E), despite the partial depletion of cytoplasmic reserve pools. This lack of depletion of the ribbon-associated vesicles may indicate that the refilling rate of the ribbon from the remaining reserve pool is sufficient to compensate for spontaneous release of vesicles on ribbons induced by Cplx-SBD. Alternatively, the spontaneous release generated by Cplx-SBD may occur at nonribbon locations and therefore fails to affect the ribbon-associated population of vesicles.
Figure 5.
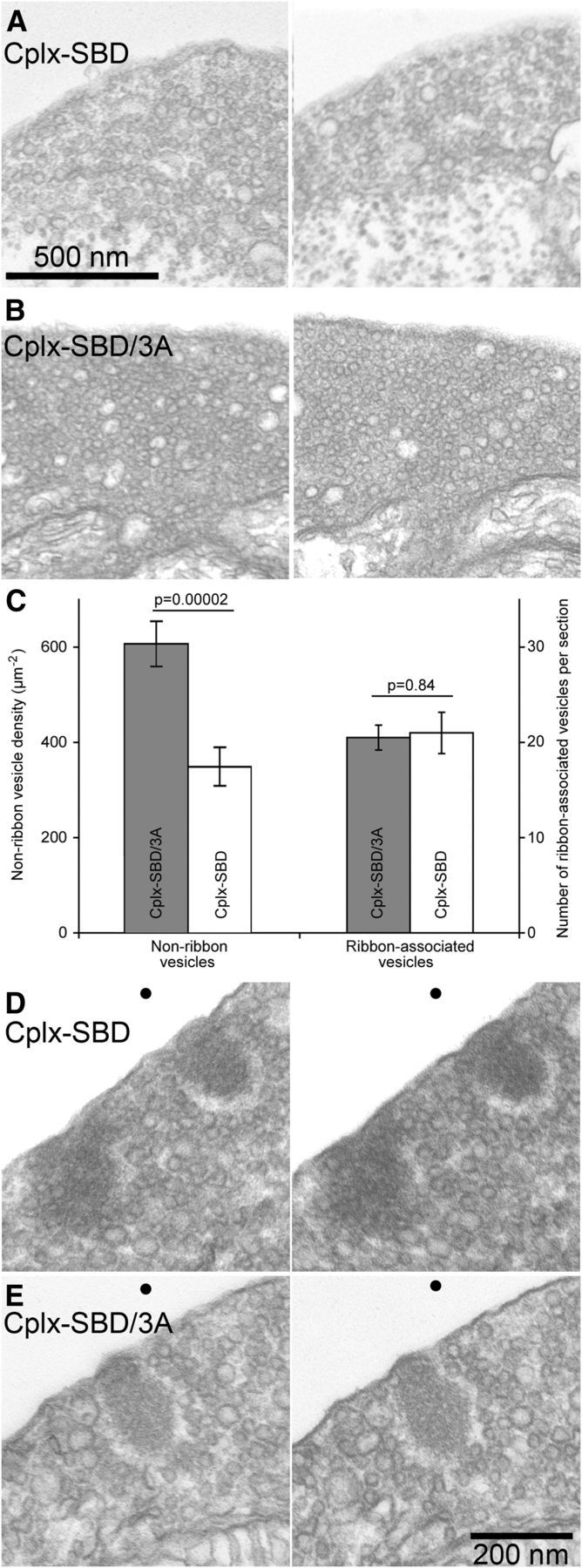
Inhibitory peptide Cplx-SBD depleted nonribbon synaptic vesicles but not ribbon-associated synaptic vesicles in bipolar cell terminals. A, Two examples of electron micrographs from terminals dialyzed with 10 μm Cplx-SBD. The regions near the bottom of each image containing glycogen particles and lacking vesicles were not included in determining vesicle density. B, Two examples of electron micrographs from terminals dialyzed with 10 μm Cplx-SBD/3A. The regions containing mitochondria were not included in analysis of vesicle density. C, Summary of vesicle counts in electron micrographs of thin sections taken from two terminals fixed after dialysis with Cplx-SBD and two terminals fixed after dialysis with Cplx-SBD/3A. The density of nonribbon vesicles (number per square micrometer) was measured in 24 cytoplasmic regions for each peptide, taken >400 nm from the nearest ribbon. Ribbon-associated vesicles were counted in the first row surrounding ribbons in stereoscopic images (±15° tilt) of single thin sections (7 ribbons, Cplx-SBD; 15 ribbons, Cplx-SBD/3A). Error bars show ±1 SEM. D, E, Stereo pairs (±15° tilt) showing examples of ribbon-associated vesicles in a synaptic terminal dialyzed with Cplx-SBD (D) or Cplx-SBD/3A (E). Dots at the top are to facilitate vergence eye movements to bring the two images into register. To obtain the stereoscopic depth effect, view the figure from a distance of ∼40 cm and diverge the eyes until the two dots superimpose.
Morpholino knockdown of Cplx expression affects melanophore pigment migration in response to illumination
We next asked whether interfering with Cplx function would affect visual system function in zebrafish. Cplx4a is the dominant subtype expressed at ribbon synapses in retinal and pineal photoreceptive systems of zebrafish (Zanazzi and Matthews, 2010), and expression of this subtype can be dramatically reduced by injection of an antisense morpholino into zebrafish embryos at the one-cell stage (Zanazzi and Matthews, 2010). Therefore, we used photoreceptor-driven changes in pigment distribution in skin melanophores (Neuhauss et al., 1999; Logan et al., 2006) to assess visual function after knockdown of Cplx4a in morpholino-injected embryos. Ambient light levels regulate the trafficking and overall quantity of pigment-containing melanosomes within melanophores (for review, see Yamaguchi and Hearing, 2009), and at 2 dpf, this melanophore response is controlled by pineal photoreceptors (Shiraki et al., 2010), which differentiate substantially earlier than retinal photoreceptors. Teleost embryos whiten during the night and darken during the day due in large part to melanosome aggregation and dispersal, respectively (for review, see Fujii, 2000). In the light-adapted control morphants, melanosomes were dispersed within melanophores, which therefore covered much of the integument with pigment. After dark adaptation, melanosomes of control morphants aggregated within melanophores, causing a statistically significant decrease in the percentage of the dorsal surface covered by black pigment (Fig. 6A, left bars). Examples of this light-dependent pigment migration response of a control embryo are shown in Figure 6B,C, in which the arrows point to the same cluster of melanophores in light-adapted and dark-adapted conditions.
Figure 6.
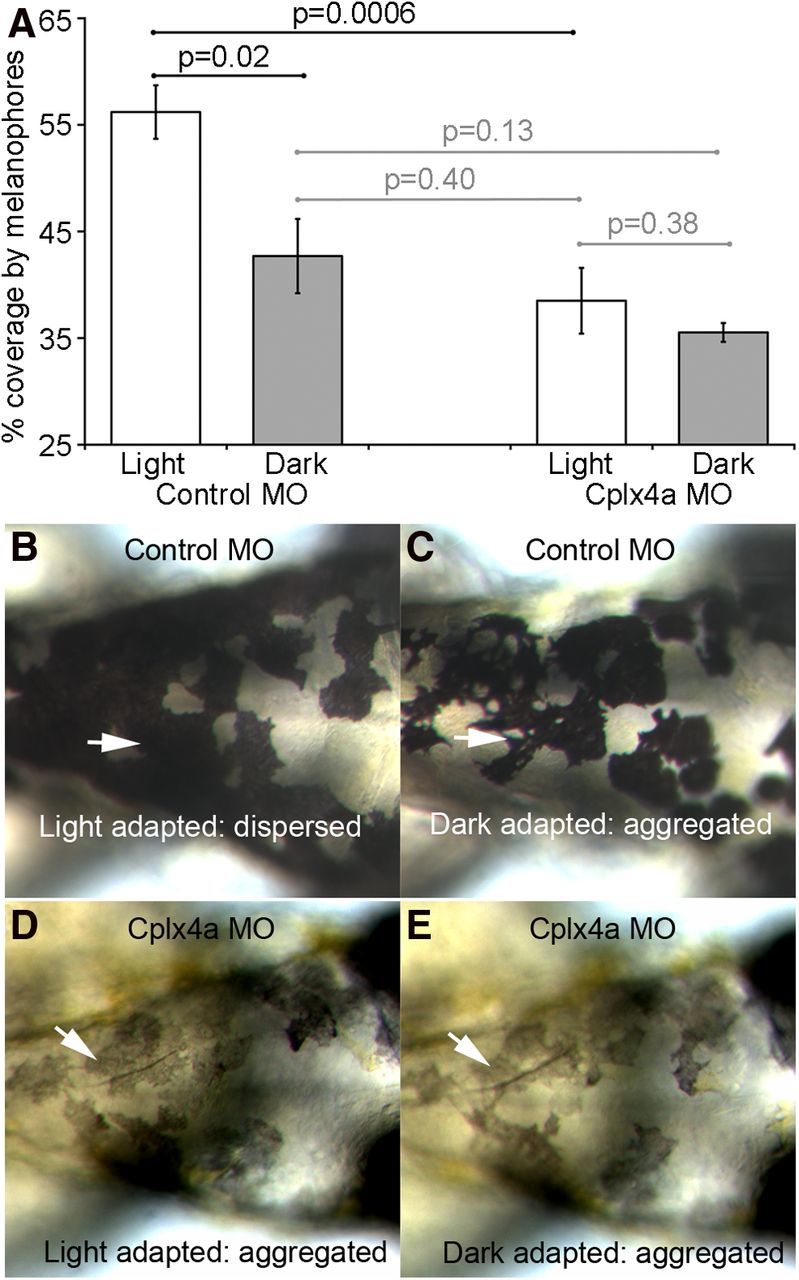
Knockdown of Cplx4a prevented pineal-dependent pigment migration in skin melanophores during light adaptation of 2-dpf zebrafish embryos. A, Percentage of the dorsal surface of the head covered by black-pigmented melanophores in light-adapted and dark-adapted embryos injected with a control morpholino (left) or an antisense morpholino against Cplx4a (right). The pigment migration response was normal in control morphants but failed to occur in Cplx4a morphants. Results are shown from four control and four Cplx4a morphant embryos. Error bars indicate ±1 SEM. Horizontal lines indicate paired comparisons, with the indicated t test probabilities. Black lines show significant differences, and gray lines show statistically insignificant differences. Light-adapted and dark-adapted Cplx4a morphants were similar to dark-adapted control morphants. B, C, Dorsal views of an example embryo injected with the control morpholino, showing normal pigment migration in response to light adaptation (A; dispersed pigment granules) and dark adaptation (B; aggregated pigment granules). Arrows point to the same cluster of melanophores in both conditions. D, E, Example of a Cplx4a morphant embryo in which pigment migration failed to occur. Pigment granules remained aggregated in dorsal melanophores in both light-adapted (D) and dark-adapted (E) conditions. Arrows point to the same cluster of melanophores in the two conditions.
By contrast, Cplx4a morphants were hypopigmented (Zanazzi, 2010), and the distribution of the pigment in melanophores was relatively insensitive to dark or light adaptation, as illustrated in Figure 6D,E. Arrows indicate the same cluster of melanophores under the two conditions. To determine whether the constant pigment distribution in Cplx4a morphants represents aggregation (i.e., the dark-adapted state) or dispersion (i.e., the light-adapted state), we measured the percentage of skin on the dorsal surface of the head covered by melanophores in dark-adapted and light-adapted states (Fig. 6A, right bars). Light-adapted and dark-adapted Cplx4a morphant embryos were not significantly different from dark-adapted control morphants, whereas light-adapted control morphants had significantly greater coverage than either light-adapted or dark-adapted Cplx4a morphant embryos (Fig. 6A). Therefore, melanophores of Cplx4a morphants remain in the dark-adapted, aggregated state regardless of ambient light levels. We suggest that when Cplx4a expression is reduced, pineal photoreceptors provide a signal consistent with darkness, even when exposed to light.
The absence of the pineal-driven melanophore response upon illumination in Cplx4a morphants could be due to failure of pineal photoreceptors to differentiate properly without normal expression of Cplx4a. To determine whether the morphology of pineal photoreceptors was affected by knocking down expression of Cplx4a, we examined pineal organs of control and Cplx4a morphants using immunofluorescence and electron microscopy. First, we confirmed that the Cplx4a morpholino, but not the control morpholino, substantially reduced anti-Cplx4a immunostaining in the pineal organ of 2-dpf embryos (Fig. 7A,B), as reported previously (Zanazzi and Matthews, 2010). This indicates that the morpholino was effective in knocking down expression of Cplx4a protein in the pineal. Immunostaining with antibody zpr1, which labels pineal photoreceptors (Zanazzi and Matthews, 2010), demonstrated that photoreceptors were nevertheless present in the pineal organ of Cplx4a morphants. Electron microscopy of control and Cplx4a morphant embryos showed that cone-like outer segments appeared to be normal in Cplx4a morphants (Fig. 7C,D). Furthermore, active zones of photoreceptor synapses were comparable in both morphants, including size, shape, and proper targeting of synaptic ribbons at the active zone (Fig. 7E,F). Based on these morphological features, then, we conclude that the deficit in pigment migration in Cplx4a morphants is not likely attributable to aberrant phototransduction or failure of synapse formation. Furthermore, since melanophores of Cplx4a morphants remain in the dark state after light adaptation, we suggest that pineal photoreceptors continue to release transmitter, mimicking darkness, even when hyperpolarized by illumination.
Figure 7.
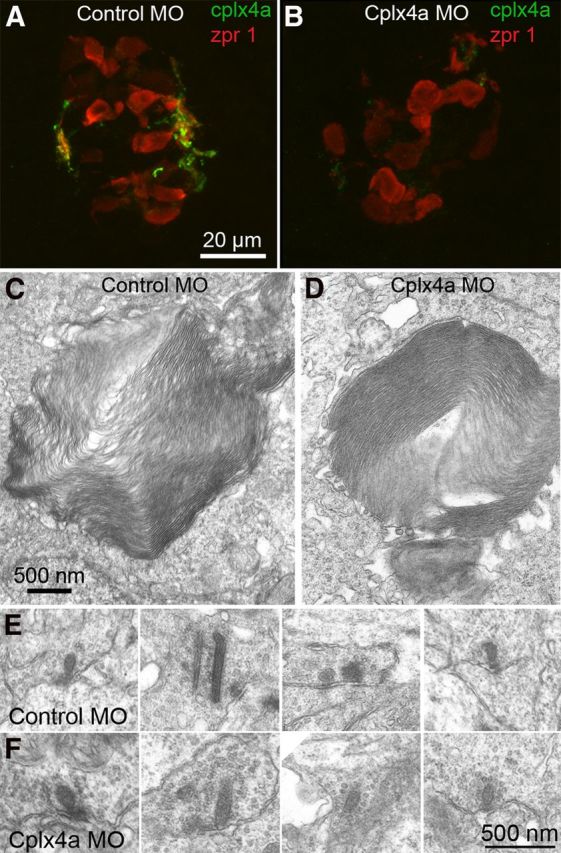
Knockdown of Cplx4a in pineal photoreceptors did not affect either outer segments or photoreceptor ribbon synapses. A, Cplx4a immunostaining (green) in the synaptic plexus of 2-dpf zebrafish pineal body after injection of control morpholino (MO) at the one-cell stage. The cone-like photoreceptors of the pineal organ are labeled with antibody zpr 1 (red). B, Cplx4a immunostaining was largely absent in the pineal body of 2-dpf zebrafish injected at the one-cell stage with an antisense morpholino directed against Cplx4a mRNA. C, D, Cone-like outer segments of pineal photoreceptors were unaffected by Cplx4a knockdown. E, F, Photoreceptor synapses marked by ribbons in the synaptic plexus of the pineal organ of 2 dpf embryos injected with control morpholino or Cplx4a morpholino. Synaptic ultrastructure was similar in both morphants.
Although retinal photoreceptors develop later than pineal photoreceptors, the Cplx4a morpholino also effectively eliminated Cplx4a expression in the retina at 4.3 dpf (Zanazzi and Matthews, 2010; see their Fig. 11), a stage at which functional cone-mediated vision can be assessed using the robust optokinetic response (OKR) to a rotating striped drum (Brockerhoff, 2006). We found that Cplx4a morphant larvae failed to show OKR at 4–5 dpf, whereas control morphants were normal. Although this suggests that Cplx4a morphants are blind, these larvae also had no spontaneous eye movements, unlike control morphants. Therefore, we could not rule out an oculomotor defect rather than blindness as an explanation for the absence of OKR, and so we turned to the melanophore pigment response to assess retina-dependent visual function. After ∼4 dpf, control of pigment migration in melanophores switches from pineal photoreceptors to retinal photoreceptors, via a retina–hypothalamus–pituitary pathway, and the direction of the response reverses so that pigment aggregates in the light and disperses in the dark (Shiraki et al., 2010). As at 2 dpf, light-adapted melanophores of 5-dpf Cplx4a morphant larvae remained in the dark-adapted state, but pigment remained dispersed rather than aggregated, consistent with the reversal of the migration response when under control of the retina. However, unlike the pineal photoreceptors earlier in development, retinal cones failed to develop properly in Cplx4a morphants. Cones labeled with antibody zpr1 were present in both control and Cplx4a morphants at 5 dpf (Fig. 8A,B), and their synaptic terminals contained ribbons labeled with an antibody against RIBEYE-b (Fig. 8A,B), suggesting that synaptic connections were not grossly defective when Cplx4a was knocked down. However, although cones were present in Cplx4a morphants, they appeared to be shorter, and no outer segments were detectable with antibody zpr1 (Fig. 8A,B, arrows).
Figure 8.

Absence of photoreceptor outer segments in the retina after knockdown of Cplx4a in larval zebrafish. A, B, Cone photoreceptors were immunostained with antibody zpr1 (red) in sections of 5-dpf retina from control morphants (A) and Cplx4a morphants (B). Synaptic ribbons were stained with an antibody against the ribbon protein, RIBEYE-b (green). Outer segments of cone photoreceptors were visible with zpr1 staining in control morphants (A, arrow) but not in Cplx4a morphants (B, arrow). C, D, Low-magnification electron micrographs of the outer retina in eyes from control morphants (C) and Cplx4a morphants (D) at 5 dpf. OPL, outer plexiform layer; ONL, outer nuclear layer; OS, cone photoreceptor outer segments; RPE, retinal pigmented epithelium. Outer segments were not present in photoreceptors of Cplx4a morphants.
To examine this morphological difference in more detail, we used electron microscopy of control and Cplx4a morphant retinas at 5 dpf. As shown in Figure 8C, numerous cone outer segments were present in control morphants, similar to wild-type retina at this developmental stage. In zebrafish, rod development approaches maturity between 15 and 24 dpf, and cone signaling predominates before this time (Branchek and BreMiller, 1984; Bilotta et al., 2001). Consistent with this delayed maturation of rods, we did not observe any rod outer segments in electron micrographs of control morphants at 5 dpf. By contrast, cone outer segments were almost entirely absent in Cplx4a morphants at 5 dpf (Fig. 8D). As with control morphants, no rods were observed in Cplx4a morphant retinas. Inner segments of cones in Cplx4a morphants had grossly normal ultrastructure and showed no evidence of degenerative changes (Fig. 8D), suggesting that the absence of outer segments represents a failure of development rather than cell death. Lack of photoreceptor outer segments would result in retinal blindness, which explains the failure of melanophore pigment migration driven by retinal outputs, as well as the absence of OKR in Cplx4a morphants. This defect in photoreceptor cells therefore precludes any interpretation of Cplx4a knockdown in terms of a possible effect of Cplx4a on neurotransmitter release.
Discussion
Effects of Cplx-SBD peptide on spontaneous and evoked release
Acutely interfering with binding of the Cplx3/4 central helix using an inhibitory peptide increased spontaneous release in zebrafish bipolar cells, without affecting evoked release. This result differs from the recently emerging view that Cplx has two competing roles at synapses, acting as both a clamp to prevent spontaneous vesicle fusion and a facilitator for fast fusion in response to calcium influx (Brose, 2008; Maximov et al., 2009; Yang et al., 2010; Hobson et al., 2011; Martin et al., 2011). In this view, the inhibitory peptide might be expected to both increase spontaneous release and inhibit evoked release. What accounts for the difference between this expectation and our results? A possible explanation is that our experiments were conducted using retinal ribbon synapses, which express distinct isoforms of Cplx (Reim et al., 2005) and syntaxin (Curtis et al., 2008, 2010). As pointed out by Neher (2010), the effect of interfering with Cplx function at a given synapse likely depends on which subtypes of Cplx and its partner proteins are expressed. Because ribbon synapses maintain a large pool of readily releasable vesicles to support sustained release (Matthews and Fuchs, 2010), preventing excessive spontaneous release from this large releasable pool may require special mechanisms. Therefore, Cplx3/4 may be specialized for efficacious clamping of spontaneous release, possibly because these isoforms are efficiently recruited to membranes via C-terminal prenylation (Reim et al., 2005), which is necessary for their inhibitory effect on spontaneous release (Xue et al., 2009; Cho et al., 2010). Recent studies in Drosophila (Buhl et al., 2013) and in C. elegans (Wragg et al., 2013) also indicate that membrane interactions of Cplx, including recruitment to synaptic vesicles themselves (Wragg et al., 2013), are important in the suppression of spontaneous vesicle fusion.
By contrast, evoked release at retinal ribbon synapses may be subject to other regulatory controls involving other domains of complexin or additional molecules. Precedent for such Cplx-independent regulatory mechanisms exists at ribbon synapses of auditory hair cells, which lack expression of Cplx (Strenzke et al., 2009; Zanazzi and Matthews, 2010) yet nevertheless support precisely timed release from a large pool of vesicles. Strenzke et al. (2009) suggested that hair cells might express an as yet unidentified molecule that serves the same function as Cplx, and if this mechanism can be identified, it would be interesting to know whether it is also present at ribbon synapses in the retina.
A possible molecular mechanism for our result is suggested by recent evidence from rescue experiments in C. elegans Cplx mutants. These experiments indicate that tight binding of the central helix to the SNARE complex is not required for the stimulatory effect of Cplx on evoked release, but is important for Cplx's action in inhibiting spontaneous release (Martin et al., 2011). Because our inhibitory peptide targets the binding site for the central helix, it may have no effect on the facilitatory effect of Cplx on evoked release, which apparently depends on interactions of other functional domains of Cplx with the SNARE complex (Martin et al., 2011).
Since native Cplx was present in normal amounts in the peptide experiments, the lack of effect of Cplx-SBD on evoked release might also arise from its inability to compete successfully with native Cplx for binding to SNARE complexes in the vesicle pool mediating release triggered by depolarization. This pool is thought to consist of the vesicles tethered to ribbons (von Gersdorff et al., 1996; Heidelberger, 1998; LoGiudice et al., 2008). Recent structural work (Kümmel et al., 2011) suggests that Cplx forms a bridge between neighboring SNARE complexes and stabilizes them in a nonfusogenic state, with the central helix (corresponding to Cplx-SBD) occupying one complex and the accessory helix, nearer the N terminus, occupying the neighboring complex. If the ribbon acts as a priming station for vesicles (Heidelberger et al., 2002; Snellman et al., 2011), then fusion-ready vesicles tethered to the ribbon might already possess partially assembled SNAREs bridged by Cplx3/4 held in place by binding of both the central helix and the accessory helix. Native Cplx might also be recruited to synaptic terminals via membrane interactions mediated by its C terminus, which would enhance its ability to interact with SNARE complexes (Wragg et al., 2013). In these scenarios, the Cplx-SBD peptide may have little opportunity to occupy the binding groove for the central helix and displace native Cplx.
Experiments in cultured mouse cortical neurons using knock-down of Cplx1 followed by rescue (Kaeser-Woo et al., 2012) showed that the C terminus of Cplx1 is responsible for clamping spontaneous fusion. Furthermore, although Cplx3 was able to substitute for Cplx1 in enhancing evoked release, Cplx3 was ineffective in reducing the rate of spontaneous fusion, which seems to conflict with our conclusion that Cplx3 and/or Cplx4 efficiently stabilize spontaneous release at ribbon synapses. However, because we used retinal ribbon synapses that natively express Cplx3/4, differences in the molecular context could influence the effect of Cplx3 on spontaneous release and make it difficult to compare the two experimental systems. In addition, Cplx4a, not Cplx3, is thought to be the principal subtype expressed at ribbon synapses in zebrafish (Zanazzi and Matthews, 2010). Therefore, the effect of Cplx-SBD on spontaneous release in our experiments is likely to be due to interference with Cplx4, which was not tested in the experiments of Kaeser-Woo et al. (2012). In this regard, it would be useful to examine the effect of Cplx-SBD peptide on spontaneous release in mouse bipolar neurons, where Cplx3 is highly expressed (Reim et al., 2005; Zanazzi and Matthews, 2010).
Effects of Cplx knockdown on photoreceptor morphology
Injection of antisense Cplx4a morpholino into zebrafish embryos has been shown to strongly reduce expression of Cplx4a protein at pineal and retinal photoreceptor synapses (Zanazzi and Matthews, 2010). Here, we showed that this knockdown of Cplx4a did not affect photoreceptor morphology and ultrastructure in the pineal organ (Fig. 7), whereas retinal cones lacked outer segments in the absence of Cplx4a (Fig. 8). Other than the absence of outer segments, Cplx4a-morphant cones appeared ultrastructurally normal (Fig. 8D), and their ribbon synapses were normal as revealed by immunostaining for RIBEYE-b (Fig. 8B). This suggests that outer segments failed to differentiate properly in Cplx4a morphants. However, it is unclear whether the effect of Cplx4a knockdown on outer segment development reflects a direct role for Cplx4a in cone differentiation or is an indirect consequence of interfering with synaptic transmission. In previous work, Wan et al. (2005) used an antisense morpholino against the synaptic protein RIBEYE-a and found no deficits in photoreceptor differentiation when ribbon synapse function was inhibited, which suggests that failure of outer segment formation is not a general feature of altered photoreceptor synaptic function in zebrafish cones. Also, Cplx immunoreactivity in zebrafish hair cells is associated with the base of the ciliary bundle at the apical surface, not with synaptic outputs at the basal surface (Zanazzi and Matthews, 2010), and this expression pattern is set up early in the differentiation of hair cells (Zanazzi and Matthews, 2010). Therefore, by analogy with hair cells, it is feasible that Cplx4a could serve a nonsynaptic function in the development of cone outer segments, possibly related to cilia formation.
Effects of Cplx knockdown on visual function
Morpholino knockdown of Cplx4a expression prevented illumination-dependent changes in melanophore pigment distribution driven by pineal photoreceptors at 2 dpf, and the pigment distribution in Cplx4a morphant fish was consistent with the maintained generation of a darkness signal even in the presence of illumination. This behavior would be expected if knockdown of Cplx4a increases spontaneous release from pineal photoreceptor terminals in the absence of depolarization, in the same manner as the Cplx-SBD peptide in retinal cones and bipolar cells. Photoreceptors hyperpolarize in response to illumination, which causes transmitter release to decrease or cease altogether, depending on illumination intensity. But elevated spontaneous release in the absence of Cplx would mimic darkness in terms of downstream signaling to second-order ganglion cells in the pineal organ. Pineal photoreceptor cells make excitatory connections with ganglion cells (Uchida et al., 1992), which are therefore excited in darkness and cease firing when photoreceptors hyperpolarize in response to illumination. We hypothesize that in the absence of Cplx4a, release continues during illumination, ganglion cells fire, and pigment aggregates in melanophores, as it would in darkness.
Together, our results suggest that Cplx isoforms specific to ribbon synapses of the retina and pineal organ reduce spontaneous release, preventing it from masking the changes in sustained transmission that underlie the coding of visual information by tonically releasing ribbon synapses. This mechanism thus contributes to the precise regulation of synaptic vesicle fusion necessary to encode various aspects of the visual field. Future studies focusing on Cplx3/4 interactions with other components of the exocytotic machinery at ribbons should further clarify the mechanisms that underlie signal transmission at these sensory synapses.
Footnotes
This work was supported by National Institutes of Health Grants R01EY003821 (G.M.), R21EY018095 (G.M.), and F30NS061494 (G.Z.). We thank Dr. Teresa Nicolson (Oregon Health and Science University) for the kind gift of the RIBEYE-b polyclonal antibody and Dr. Howard Sirotkin (Stony Brook University) for use of his zebrafish facility and for much advice about zebrafish experimental methods and husbandry.
References
- Bilotta J, Saszik S, Sutherland SE. Rod contributions to the electroretinogram of the dark-adapted developing zebrafish. Dev Dyn. 2001;222:564–570. doi: 10.1002/dvdy.1188. [DOI] [PubMed] [Google Scholar]
- Branchek T, BreMiller R. The development of photoreceptors in the zebrafish, Brachydanio rerio. I. Structure. J Comp Neurol. 1984;224:107–115. doi: 10.1002/cne.902240109. [DOI] [PubMed] [Google Scholar]
- Brockerhoff SE. Measuring the optokinetic response of zebrafish larvae. Nat Protoc. 2006;1:2448–2451. doi: 10.1038/nprot.2006.255. [DOI] [PubMed] [Google Scholar]
- Brose N. For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–1413. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- Buhl LK, Jorquera RA, Akbergenova Y, Huntwork-Rodriguez S, Volfson D, Littleton JT. Differential regulation of evoked and spontaneous neurotransmitter release by C-terminal modifications of complexin. Mol Cell Neurosci. 2013;52:161–172. doi: 10.1016/j.mcn.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Song Y, Littleton JT. Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Mol Cell Neurosci. 2010;45:389–397. doi: 10.1016/j.mcn.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis LB, Doneske B, Liu X, Thaller C, McNew JA, Janz R. Syntaxin 3b is a t-SNARE specific for ribbon synapses of the retina. J Comp Neurol. 2008;510:550–559. doi: 10.1002/cne.21806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis L, Datta P, Liu X, Bogdanova N, Heidelberger R, Janz R. Syntaxin 3B is essential for the exocytosis of synaptic vesicles in ribbon synapses of the retina. Neuroscience. 2010;166:832–841. doi: 10.1016/j.neuroscience.2009.12.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R. The regulation of motile activity in fish chromatophores. Pigment Cell Res. 2000;13:300–319. doi: 10.1034/j.1600-0749.2000.130502.x. [DOI] [PubMed] [Google Scholar]
- Heidelberger R. Adenosine triphosphate and the late steps in calcium-dependent exocytosis at a ribbon synapse. J Gen Physiol. 1998;111:225–241. doi: 10.1085/jgp.111.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol. 1992;447:235–256. doi: 10.1113/jphysiol.1992.sp019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Sterling P, Matthews G. Roles of ATP in depletion and replenishment of the releasable pool of synaptic vesicles. J Neurophysiol. 2002;88:98–106. doi: 10.1152/jn.2002.88.1.98. [DOI] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. Complexin maintains vesicles in the primed state in C. elegans. Curr Biol. 2011;21:106–113. doi: 10.1016/j.cub.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Südhof TC. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci. 2012;32:2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, Reinisch KM. Complexin cross-links prefusion SNAREs into a zigzag array. Nat Struct Mol Biol. 2011;18:927–933. doi: 10.1038/nsmb.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Augustine GJ, Weninger K. Kinetics of complexin binding to the SNARE complex: correcting single molecule FRET measurements for hidden events. Biophys J. 2007;93:2178–2187. doi: 10.1529/biophysj.106.101220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan DW, Burn SF, Jackson IJ. Regulation of pigmentation in zebrafish melanophores. Pigment Cell Res. 2006;19:206–213. doi: 10.1111/j.1600-0749.2006.00307.x. [DOI] [PubMed] [Google Scholar]
- LoGiudice L, Sterling P, Matthews G. Mobility and turnover of vesicles at the synaptic ribbon. J Neurosci. 2008;28:3150–3158. doi: 10.1523/JNEUROSCI.5753-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol. 2011;21:97–105. doi: 10.1016/j.cub.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G, Fuchs P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci. 2010;11:812–822. doi: 10.1038/nrn2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G, Sterling P. Evidence that vesicles undergo compound fusion on the synaptic ribbon. J Neurosci. 2008;28:5403–5411. doi: 10.1523/JNEUROSCI.0935-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Complexin: does it deserve its name? Neuron. 2010;68:803–806. doi: 10.1016/j.neuron.2010.11.038. [DOI] [PubMed] [Google Scholar]
- Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier H. Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J Neurosci. 1999;19:8603–8615. doi: 10.1523/JNEUROSCI.19-19-08603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart C, Li J, Matthews G, Sterling P. Endocytosis and vesicle recycling at a ribbon synapse. J Neurosci. 2003;23:4092–4099. doi: 10.1523/JNEUROSCI.23-10-04092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Reim K, Wegmeyer H, Brandstätter JH, Xue M, Rosenmund C, Dresbach T, Hofmann K, Brose N. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol. 2005;169:669–680. doi: 10.1083/jcb.200502115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Regus-Leidig H, Ammermüller J, El-Kordi A, Radyushkin K, Ehrenreich H, Brandstätter JH, Brose N. Aberrant function and structure of retinal ribbon synapses in the absence of complexin 3 and complexin 4. J Cell Sci. 2009;122:1352–1361. doi: 10.1242/jcs.045401. [DOI] [PubMed] [Google Scholar]
- Sheets L, Ransom DG, Mellgren EM, Johnson SL, Schnapp BJ. Zebrafish melanophilin facilitates melanosome dispersion by regulating dynein. Curr Biol. 2007;17:1721–1734. doi: 10.1016/j.cub.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T, Kojima D, Fukada Y. Light-induced body color change in developing zebrafish. Photochem Photobiol Sci. 2010;9:1498–1504. doi: 10.1039/c0pp00199f. [DOI] [PubMed] [Google Scholar]
- Snellman J, Mehta B, Babai N, Bartoletti TM, Akmentin W, Francis A, Matthews G, Thoreson W, Zenisek D. Acute destruction of the synaptic ribbon reveals a role for the ribbon in vesicle priming. Nat Neurosci. 2011;14:1135–1141. doi: 10.1038/nn.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T. Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumaru H, Umayahara K, Pellegrini LL, Ishizuka T, Saisu H, Betz H, Augustine GJ, Abe T. SNARE complex oligomerization by synaphin/complexin is essential for synaptic vesicle exocytosis. Cell. 2001;104:421–432. doi: 10.1016/S0092-8674(01)00229-X. [DOI] [PubMed] [Google Scholar]
- Uchida K, Nakamura T, Morita Y. Signal transmission from pineal photoreceptors to luminosity-type ganglion cells in the lamprey, Lampetra japonica. Neuroscience. 1992;47:241–247. doi: 10.1016/0306-4522(92)90136-P. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature. 1994;367:735–739. doi: 10.1038/367735a0. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Vardi E, Matthews G, Sterling P. Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron. 1996;16:1221–1227. doi: 10.1016/S0896-6273(00)80148-8. [DOI] [PubMed] [Google Scholar]
- Wan L, Almers W, Chen W. Two ribeye genes in teleosts: the role of Ribeye in ribbon formation and bipolar cell development. J Neurosci. 2005;25:941–949. doi: 10.1523/JNEUROSCI.4657-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Geer LY, Chappey C, Kans JA, Bryant SH. Cn3D: sequence and structure views for Entrez. Trends Biochem Sci. 2000;25:300–302. doi: 10.1016/S0968-0004(00)01561-9. [DOI] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS. Synaptic vesicles position complexin to block spontaneous fusion. Neuron. 2013;77:323–334. doi: 10.1016/j.neuron.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Reim K, Chen X, Chao HT, Deng H, Rizo J, Brose N, Rosenmund C. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–958. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat Struct Mol Biol. 2010;17:568–575. doi: 10.1038/nsmb.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Hearing VJ. Physiological factors that regulate skin pigmentation. Biofactors. 2009;35:193–199. doi: 10.1002/biof.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC. Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory α helix. Neuron. 2010;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanazzi G. State University of New York at Stony Brook; 2010. Targeting and roles of complexins in zebrafish ribbon-containing neurons. PhD thesis. [Google Scholar]
- Zanazzi G, Matthews G. Enrichment and differential targeting of complexins 3 and 4 in ribbon-containing sensory neurons during zebrafish development. Neural Dev. 2010;5:24. doi: 10.1186/1749-8104-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]