

Abstract

β-Adrenoceptor antagonists boast a 50-year use for symptomatic control in numerous cardiovascular diseases. One might expect highly selective antagonists are available for the human β-adrenoceptor subtype involved in these diseases, yet few truly β1-selective molecules exist. To address this clinical need, we re-evaluated LK 204-545 (1),1 a selective β1-adrenoceptor antagonist, and discovered it possessed significant partial agonism. Removal of 1’s aromatic nitrile afforded 19, a ligand with similar β1-adrenoceptor selectivity and partial agonism (log KD of −7.75 and −5.15 as an antagonist of functional β1- and β2-mediated responses, respectively, and 34% of the maximal response of isoprenaline (β1)). In vitro β-adrenoceptor selectivity and partial agonism of 19 were mirrored in vivo. We designed analogues of 19 to improve affinity, selectivity, and partial agonism. Although partial agonism could not be fully attenuated, SAR suggests that an extended alkoxyalkoxy side chain, alongside substituents at the meta- or para-positions of the phenylurea, increases ligand affinity and β1-selectivity.
Introduction
β-Adrenoceptor ligands comprise one of the most commonly used classes of drugs in clinical practice. β-Agonists have been used since the 1940s for their beneficial effects in respiratory disease2,3 and β-antagonists (β-blockers) since the 1960s for the control of cardiovascular disease,4,5 and both classes remain among the most commonly prescribed drugs today.
The major therapeutic aim of β-blockade is antagonism of endogenous catecholamines at the cardiac β1-adrenoceptors to reduce myocardial demand and workload.6 This reduces mortality following myocardial infarction,7 and therefore β-blockers are used for both symptomatic control and life prolongation in ischemic heart disease and acute coronary syndrome and are increasingly being used earlier in the disease process.8 β-Blockers also reduce mortality in patients with heart failure9−14 and are used in the management of arrhythmias, hypertension, portal hypertension, benign essential tremor, thyrotoxicosis, anxiety, migraine, and glaucoma.15
The major therapeutic aim of β-agonist administration is to mimic adrenaline and stimulate the β2-adrenoceptors in bronchial smooth muscle, thus increasing airway caliber and relieving the breathlessness and wheeze of bronchospasm.16 β-Agonists are used in the emergency setting for short-term rescue of bronchospasm in asthma and COPD (e.g., salbutamol inhalers and nebulizers) as well as in the daily prevention of bronchospasm, particularly using longer acting molecules (LABAs, e.g., salmeterol, formoterol, indacaterol, and vilanterol).
As β-ligands have been used in clinical practice for around for 70 years, and a great many different agonist and antagonist molecules have been generated, it would be expected that good tool agonist and antagonist compounds for each of the human β-adrenoceptor subtypes are available. However, it is surprising that there are actually very few truly selective molecules, especially for the β1-adrenoceptor. Drug discovery efforts driven by the clinical need have yielded several highly selective β2-adrenoceptor agonists for the treatment of asthma and COPD (e.g., salmeterol, formoterol, indacaterol, and vilanterol) and one highly selective β2-antagonist (ICI 118551) that has become a very useful tool in pharmacological studies of β-adrenoceptors. For the β1-adrenoceptor, despite the fact that more β1-selective β-blockers would be preferable to minimize the β2-mediated respiratory side effect of bronchospasm in the clinical setting, the selectivity of the clinically available β-blockers for the human β1-adrenoceptor over the human β2-adrenoceptor is poor.17−21 For this reason, β-blockers remain absolutely contraindicated in patients with asthma and relatively contraindicated in people with COPD. Our desire to address this significant unmet clinical need initially led us to assess reported ligands that displayed very high β1-adrenoceptor versus β2-adrenoceptor selectivity. From the literature, one of the most highly β1-selective antagonists described to date was 1 (LK 204-545).1 Although this ligand provided an ideal structural template to investigate further, there was no published synthetic route and, in reality, very little pharmacological data available for this compound. Furthermore, it was not commercially available and therefore not readily accessible for pharmacological and other studies. As we also observed, there were also very few β1-selective β-agonists, with xamoterol and ICI 89406 being the most selective β1-partial agonists reported to date.17−22
Here, we report a synthesis and the in vitro pharmacological characterization of 1 and several novel, highly β1-selective β-adrenoceptor ligands, most of which display significant agonist activity.
Results and Discussion
Synthesis
Whereas the synthesis of 1 is not described in the literature, a synthetic route to the related compound, 1-(2-cyano-4-(2-cyclopropylmethoxyethoxy)phenoxy)-3-(2-(3-phenylureido)ethylamino)-2-propanol, is outlined as an example of this class of compound in a patent.23 The introduction of the cyano group employed an undesirable cyanation step, alongside several protection and deprotection steps, complicating the synthesis and compromising overall yield. Conscious that we would require appreciable quantities of this ligand to benchmark any newer ligands against, we embarked upon a new and more expeditious route to its synthesis (Scheme 1).
Scheme 1. Synthesis of 1-(2-(3-(2-Cyano-4-(2-(cyclopropylmethoxy)ethoxy)phenoxy)-2-hydroxypropylamino)ethyl)-3-(4-hydroxyphenyl)urea (1) and 1-(2-(3-(4-(2-(Cyclopropylmethoxy)ethoxy)phenoxy)-2-hydroxypropylamino)ethyl)-3-(4-hydroxyphenyl)urea Hydroformate (19).

Reagents and conditions: (a) Et3N, pivaloyl chloride, DMF, 0 °C → rt, 72%; (b) (i) NaH, DMF; (ii) p-methoxybenzyl bromide, 0 °C → rt, 33%; (c) 37% NH3 (aq), I2, THF, 98%; (d) sodium tert-butoxide, MeOH, 34%; (e) allyloxyethanol, DIAD, PPh3, DCM, 100%; (f) Et2Zn, CH2I2, toluene 0 °C → rt, 25% 8 and 29% 9; (g) CAN, H2O, MeCN; (h) Et3N, rac-epichlorohydrin, 80 °C, 100%; (i) 4-(benzyloxy)phenylisocyanate, DCM, 94%; (j) (i) concd HCl; (ii) 2 M NaOH (aq), neutralization, 73%; (k) 13, propan-2-ol, reflux, 15%; (l) allyloxyethanol, DIAD, Ph3P, DCM, rt, 71%; (m) Et2Zn, CH2I2, toluene, 0 °C → rt, 97%; (n) H2, 10% Pd/C, EtOH, 100%; (o) (i) 2 M NaOH(aq); (ii) rac-epichlorohydrin, 60 °C, 62%; (p) 13, propan-2-ol, reflux, 21%.
The new synthesis of 1 relied upon an alternative synthesis of epoxide 10, with subsequent aminolysis of 10 using amine 13. The availability of methods to easily convert an aryl aldehyde to the corresponding nitrile24 allowed 2,5-dihydroxybenzaldehyde (2) to be selected as an appropriate starting material in the synthesis of epoxide 10.
A procedure reported by Chen et al.25 facilitated selective monoprotection of 2 using pivaloyl chloride to generate pivaloate ester 3 in good yield. The remaining hydroxy group then required protection orthogonal with respect to the pivaloate ester; hence, the p-methoxybenzyl (PMB) group was selected for this purpose. The ability to cleave a PMB ether under oxidative conditions, using reagents such as ceric(IV) ammonium nitrate (CAN), was anticipated,26 because this would allow selective removal of the PMB group in the presence of the nitrile later in the synthesis.
Conversion of aldehyde 4 to the desired nitrile was achieved cleanly by oxidative amination24,27 in nearly quantitative yield to give 5. Alkoxide-mediated removal of the pivaloate ester then revealed phenol 6, which was subsequently submitted to Mitsunobu coupling with 2-(allyloxy)ethanol to generate ether 7. Cyclopropanation of 7 using Simmons–Smith methodology gave a mixture of products.28 The desired PMB-protected product 8 was isolated and subjected to CAN-mediated oxidative PMB group cleavage conditions. Unfortunately, this did not yield the desired phenol 8, possibly due to instability of the molecule to CAN, for example, through dealkylation of the aralkyl ether.29 Re-examination of crude reaction products from the cyclopropanation step revealed that apart from the expected PMB-protected product 9, the oxidative conditions of the Simmons–Smith procedure had caused partial removal of the PMB group to afford phenol 8 directly, although in modest yield. Consequently, phenol 8 was recovered, albeit as a side product and in relatively low yield. Alkylation of phenol 8 with rac-epichlorohydrin in the presence of TEA afforded epoxide 10 in quantitative yield, with no further purification required. In this preliminary study, although it was well understood that the S-form was the more pharmacologically active of the two aryloxypropanolamine enantiomers, we elected to synthesize racemic derivatives throughout, as a more expeditious and cost-effective route to explore early SAR, with the intention that favorable pharmacology would prompt synthesis of individual stereoisomers if warranted.
Construction of aminophenol 13 required initial addition of 1,2-ethanediamine (11) to 4-(benzyloxy)phenyl isocyanate. Dropwise introduction of a solution of the isocyanate into a solution containing an excess of 11 allowed selective mono-addition to occur, as the product urea 12 was found to precipitate on formation, allowing facile isolation and purification by filtration. Conversely, the poor solubility of 12 in a variety of appropriate solvents meant that attempted hydrogenolysis progressed slowly. Acidolytic O-benzyl deprotection was therefore chosen as an alternative to give aminophenol 13 in good yield. Finally, aminolysis of epoxide 10 with 13 under neutral conditions, by reflux in propan-2-ol, afforded 1 after purification by flash column chromatography.
Pharmacological analysis of 1 in our hands (cf. Table 1 and pharmacology discussion) indicated a slightly lower β1/β2-selectivity than previously reported.1 In addition to this, we noted with interest that previous studies indicated that the presence of substituents of the phenyl ring ortho to the oxypropanolamine moiety did not improve β1/β2-selectivity but led to either increased or decreased affinity at both receptors simultaneously.30−35 Because our aim was to devise β1-selective ligands, we were therefore encouraged to explore whether removal of the cyano group was a worthwhile modification because, if successful, it would allow a more expeditious route into a range of new analogues. We therefore embarked upon the more efficient synthesis of the decyanated analogue of 1 (19), which had not previously been described, to assess the impact of the nitrile group on β1/β2-receptor pharmacology (Scheme 1).
Table 1. Human β1- and β2-Adrenoceptor Binding Affinities for Established β-Adrenoceptor Ligands, 1 and 19a.
| compd | log KD β1 | n | log KD β2 | n | selectivity ratio (β1/β2) |
|---|---|---|---|---|---|
| CGP 20712A | –8.79 ± 0.07 | 9 | –5.82 ± 0.04 | 8 | 933 |
| ICI 118551 | –6.62 ± 0.01 | 9 | –9.17 ± 0.03 | 8 | 354 β2 selective |
| xamoterol | –7.09 ± 0.04 | 8 | –5.76 ± 0.04 | 8 | 21 |
| ICI 89406 | –8.75 ± 0.03 | 9 | –6.84 ± 0.03 | 9 | 81 |
| 1 | –8.04 ± 0.03 | 9 | –5.29 ± 0.04 | 11 | 562 |
| 19 | –7.49 ± 0.03 | 11 | b | 11 |
Values represent the mean ± SEM of n separate experiments. Log KD values were obtained from 3H-CGP 12177 whole cell binding studies in CHO cells stably expressing the human β1 or β2-adrenoceptors.
Incomplete inhibition of specific 3H-CGP12177 binding meant that generation of an affinity value (log KD value) by this method was not possible.
Mitsunobu coupling of 2-(allyloxy)ethanol with commercially available 4-(benzyloxy)phenol (14) furnished the desired phenylether 15, whose terminal allyl ether function was efficiently converted to the corresponding cyclopropylmethyl ether 16 using Simmons–Smith cyclopropanation.28 Subsequent benzyl ether hydrogenolysis of 16 over Pd(0) on charcoal, using standard conditions, afforded phenol 17 in quantitative yield. Alkylation of 17 was achieved in moderate yield by initial deprotonation in aqueous 2 M NaOH solution, followed by heating in excess rac-epichlorohydrin to afford oxirane 18. Finally, oxirane 18 was subjected to aminolysis with 13 under basic conditions by refluxing in propan-2-ol. The desired aryloxypropanolamine 19 was obtained as the hydroformate salt, after PLC and subsequent preparative RP-HPLC purification.
Pharmacological and in vivo analysis of our early lead ligand 19 gave mixed results. Whereas its selectivity was somewhat diminished compared to that of 1 (cf. pharmacology results), both compounds possessed significant partial agonism, rendering them unsuitable for future therapeutic development as a selective β1-adrenoceptor antagonist. Therefore, to explore the SARs of this class of molecule further and to ascertain if selectivity could be enhanced while decreasing partial agonism, the more facile synthesis of 19 now allowed access to new analogues in a more expeditious manner. Initially we tried to ascertain the effect of the alkoxyalkylether and the phenylurea termini on affinity, selectivity, and partial agonism. To explore the influence of the alkoxyalkoxy terminus on pharmacology, and on the basis of established literature precedent, we synthesized oxiranes 28a–c in moderate yield (Scheme 2),36 essentially in the same manner as for 10. Although 2-ethoxyethanol (25) was commercially available, the analogous 2-(cyclopentyloxy)ethanol (21) and 2-(4-fluorophenethyloxy)ethanol (24) starting materials required independent synthesis (Scheme 2).
Scheme 2. Synthesis of Hydroxyphenylureas, Varied at the Alkoxyethyl Terminus.

Reagents and conditions: (a) NaBH4, ZrCl4, THF 0–5 °C, 81%; (b) (i) NaH, DMF 60 °C; (ii) chloroacetic acid, 60 °C, 50%; (c) LiAlH4, THF, 0 °C, 67%; (d) Ph3P, 14, di-tert-butyl azodicarboxylate or diethyl azodicarboxylate, DCM, 35–85%; (e) H2, 10% Pd/C, EtOH, 68–100%; (f) (i) NaH, DMF, 0 °C → rt; (ii) rac-epichlorohydrin, 71–84%; (g) (i) 2 M NaOH(aq); (ii) rac-epichlorohydrin, 60 °C, 84%; (h) 13, propan-2-ol, reflux, 21–29%.
2-(Cyclopentyloxy)ethanol (21) was obtained in good yield from ketal 20, according to the NaBH4/ZrCl4 reductive cleavage conditions reported by Chary et al.37 In the case of 2-(4-fluorophenethyloxy)ethanol (24), the route described by Machin et al.38 was employed. This entailed initial alkylation of 4-fluorophenethyl alcohol (22) with chloroacetic acid to give alkoxyacetic acid 23. Subsequent reduction was achieved with LiAlH4, to give alcohol 24 in acceptable yield.
The alcohols 21, 24, and 25 were subjected to Mitsunobu coupling with 4-(benzyloxyphenol) (14) in a similar manner as previously described, to give O-benzyl ethers 26a–c. Hydrogenolysis under standard conditions afforded phenols 27a–c. Subsequent alkylation of phenol 27c was carried out using the previously described method to give oxirane 28c. In the case of phenols 27a,b, NaH was used as a base in anhydrous DMF, with subsequent alkylation proceeding at room temperature to furnish oxiranes 28a,b. The target aryloxypropanolamines 29a–c were obtained in the free base form, from oxiranes 28a–c through aminolysis with 13 as previously detailed.
Encouraged by the pharmacological data of the novel compounds possessing the 2-(cyclopentyloxy)ethoxyphenyl terminus (cf. Table 4 and pharmacology discussion), we kept this group constant to investigate the pharmacological effects of modifying the phenylurea substituent at the opposite pole of the molecule. This required synthesis of a small set of phenyl-substituted 1-(2-aminoethyl)-3-phenylureas (Scheme 3).
Table 4. Human β1- and β2-Adrenoceptor Binding Affinity and Receptor Selectivity of 1-(2-(3-(4-(2-(Alkyloxy)ethoxy)phenoxy)-2-hydroxypropylamino)ethyl)-3-((4-hydroxy)phenyl)ureas 28a–ca.
| compd | R1 | log KD β1 | n | log KD β2 | n | selectivity ratio (β1/β2) |
|---|---|---|---|---|---|---|
| 19 | –7.49 ± 0.03 | 11 | b | 11 | ||
| 29a | cyclopentyl | –8.13 ± 0.05 | 12 | –5.45 ± 0.07 | 7 | 478 |
| 29b | p-FC6H4CH2CH2 | –8.50 ± 0.05 | 9 | –5.91 ± 0.05 | 9 | 389 |
| 29c | CH3CH2 | –7.04 ± 0.04 | 16 | b | 16 |
Values represent the mean ± SEM of n separate experiments. Log KD values were obtained from 3H-CGP 12177 whole cell binding studies in CHO cells stably expressing the human β1- or β2-adrenoceptors.
Incomplete inhibition of specific 3H-CGP12177 binding meant that generation of an affinity value (log KD value) by this method was not possible.
Scheme 3. Synthesis of Aryl Monosubstituted 3-(2-((3-(4-(2-(Cyclopentyloxy)ethoxy]phenoxy)-2-hydroxypropyl)amino]ethyl)-1-phenylureas.

Reagents and conditions: (a) o-tolylisocyanate (30), DCM, 0 °C → rt, 64%; (b) di-tert-butyl dicarboxylate, DCM, 87%; (c) substituted phenylisocyanate, DCM, 0 °C → rt, 46 – 95%; (d) MeOH/concd HCl, 80–100%; (e) phthalic anhydride, 150 °C, 94%; (f) (i) diphenylphosphoryl azide, TEA, toluene; (ii) reflux; (iii) 2-aminophenol or 3-aminophenol, 75–76%; (g) hydrazine monohydrate, EtOH, reflux; (ii) acidic workup, 51–60%; (h) 33a–r, 10 M NaOH(aq), propan-2-ol, reflux, 7–31%; (i) 33t–u, Et3N, propan-2-ol, reflux, 21–25%.
In accordance with the synthesis of 12, attempted condensation of o-tolylisocyanate (30) with an excess of ethylenediamine (11) afforded urea 33b in only modest yield, alongside the undesirable bis-urea side product. To avoid this complication with the remaining analogues, tert-butyl-2-aminoethylcarbamate (31) was employed instead of 11. Selective mono-Boc protection of ethylenediamine (11) was accomplished in excellent yield using a literature method39 to give amine 31 (Scheme 3). The stoichiometric condensation of 31 with corresponding substituted phenylisocyanates permitted preparation of the Boc-protected intermediates 32a and 32c–r in moderate to good yield, with the pure products being easily isolated by filtration following precipitation with hexanes. Boc-deprotection of these intermediates with concentrated HCl in methanol proceeded smoothly and in excellent yields, affording the desired amines 33a and 33c–r as their respective hydrochloride salts. The remaining 1-(2-aminoethyl)-3-(hydroxyphenyl)urea analogues (33t–u) required an alternative synthetic strategy, due to the lack of commercially available phenylisocyanate starting materials. However, the availability of 2-amino and 3-aminophenol allowed urea synthesis via condensation with the appropriate alkylisocyanates (Scheme 3).
Using β-alanine (34) as a starting material, the conversion of the primary amine to the phthalimide eliminated possible side reactions during isocyanate formation; monoacylated amine protecting groups still have the propensity to attack the isocyanate functionality.40 Phthalimide protection of 34 was effected through a solvent-free method, by heating directly with phthalic anhydride to give acid 35,41 which was converted to the corresponding acyl azide with the aid of diphenylphosphoryl azide in the presence of TEA; careful reflux of this intermediate promoted Curtius rearrangement to the isocyanate. The isocyanate solution was divided into two equal portions, prior to addition of either 2- or 3-aminophenol, producing protected intermediates 36a and 36b. The desired amines 33t and 33u were once again isolated as the hydrochloride salts after standard hydrazinolytic cleavage of the phthalimide group and subsequent acidic workup. Finally, aminolysis of oxirane 28a with amines 33a–u and subsequent PLC purification afforded the target aryloxypropanolamine compounds 37a–u (Scheme 3).
Pharmacology
Ligand Affinity Measurements: 3H-CGP 12177 Whole Cell Binding
We initially evaluated and compared the affinity of 1 with that of the decyanated analogue 19 at the human β1-, β2-, and β3-adrenoceptors, each stably expressed in Chinese hamster ovary cells. The KD values for β-adrenoceptor binding of 3H-CGP 12177 in these cell lines have previously been established and are 0.42, 0.17, and 109.2 nM for the β1-, β2-, and β3-adrenoceptors, respectively.201 completely inhibited specific binding to the human β1-adrenoceptor to yield a log KD of −8.04 ± 0.03, n = 9, and −5.29 ± 0.04, n = 11, for the human β2-adrenoceptor, thus giving a selectivity for the β1-adrenoceptor of 562-fold (Figure 1; Table 1). Likewise, 19 was also shown to inhibit specific binding to the human β1-adrenoceptor to yield a log KD of −7.49 ± 0.03. However, its affinity for the human β2-adrenoceptor was so low that measurement of complete inhibition of specific 3H-CGP 12177 binding was not possible (Figure 1d), so that affinity (KD values) could not be calculated by this method. The affinity of both ligands for the human β3-adrenoceptor was also too low to establish reliable KD values from this method.
Figure 1.

Inhibition of 3H-CGP 12177 binding to whole cells by (a, b) 1 and (c, d) 19 in (a, c) CHO β1 cells and (b, d) CHO β2 cells. Bars represent total 3H-CGP 12177 binding, and nonspecific binding was determined in the presence of 10 μM propranolol. The concentration of 3H-CGP 12177 present in each case was 0.73 nM. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (a) 9 and (b–d) 11 separate experiments.
Antagonist Affinity Measurements: 3H-cAMP Accumulation
Because quantifiable antagonist affinity was difficult to determine for 19 at the human β2- and β3-adrenoceptors, an alternative assay was employed to establish accurate values via measurement from the shift of an agonist response (Figure 2; Table 2).
Figure 2.

3H-cAMP accumulation in response to cimaterol in (a) CHO β1 cells, (b) CHO β2 cells, and (c) CHO β3 cells in the absence and presence of 19. Bars show basal 3H-cAMP accumulation, that in response to 10 μM isoprenaline, and that in response to 30 nM, 100 nM, 300 nM, 1 μM, 3 μM, and 10 μM 19 alone. Data points are the mean ± SEM of triplicate values from single experiments that are representative of seven separate experiments in each case.
Table 2. Antagonist Affinities of Established Ligands and 19 at the Human β1-, β2-, and β3-Adrenoceptorsa.
| ligand | log KD β1 | n | log KDβ2 | n | log KDβ3 | n | selectivity β1 vs β2 |
|---|---|---|---|---|---|---|---|
| CGP 20712A | –9.48 ± 0.06 | 12 | –5.98 ± 0.03 | 4 | –5.26 ± 0.04 | 4 | 3162 |
| xamoterol | –7.75 ± 0.05 | 11 | –6.18 ± 0.05 | 4 | –4.70 ± 0.08 | 4 | 37 |
| ICI 89406 | –9.48 ± 0.04 | 12 | –7.25 ± 0.05 | 4 | –5.72 ± 0.05 | 4 | 170 |
| 1 | –8.77 ± 0.05 | 11 | –5.54 ± 0.05 | 4 | >10 μM | 4 | 1698 |
| 19 | –7.75 ± 0.06 | 21 | –5.15 ± 0.06 | 7 | >10 μM | 7 | 398 |
Values represent the mean ± SEM of n separate experiments. Log KD values were obtained from 3H-cAMP accumulation assays following inhibition of the cimaterol agonist response in CHO cells stably expressing the human β1-, β2-, or β3-adrenoceptors.
Cimaterol was chosen as the agonist as it is a chemically stable nonselective agonist and thus could be used across all three human β-adrenoceptors and because cimaterol agonist responses are also readily inhibited by classical β-blockers, suggesting that its primary action is agonism of the catecholamine conformation of the human β1-adrenoceptor.42 Cimaterol stimulated responses were 96.9 ± 1.1% (log EC50 = −8.13 ± 0.03, n = 9), 98.9 ± 1.7% (log EC50 = −8.78 ± 0.06, n = 9), and 89.6 ± 1.9% (log EC50 = −6.62 ± 0.06, n = 9) of the isoprenaline maximum response at the human β1-, β2-, and β3-adrenoceptors, respectively (Figure 2). These agonist responses were antagonized by both 1 and 19 (Table 2). As both ligands examined demonstrated clear partial agonist stimulatory effects (e.g., Figure 2), the data were analyzed by the partial agonist method of Stephenson.43
Agonist Responses: 3H-cAMP Accumulation
Given the significant degree of agonist activity seen above, full concentration response relationships were investigated for the ligands. 1 caused an agonist response at the human β1-adrenoceptor that was 37.1 ± 2.2% of the maximum response to isoprenaline (n = 5). It is thus a partial agonist at the human β1-adrenoceptor. Agonist responses were also seen at the human β2- and β3-adrenoceptors; however, the top of the concentration response curve was not reached in each case. At the maximum concentration of 10 μM, 1 stimulated responses that were 15.2 ± 0.7% (β2) and 8.6 ± 0.4% (β3) of the isoprenaline maximum at each receptor. Similar responses were seen for 19 (Table 3).
Table 3. Agonist Responses of Established Ligands and 19 at the Human β1-, β2-, and β3-Adrenoceptorsa.
| β1 |
β2 |
β3 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | log EC50 β1 | % isop max | n | log EC50 β2 | % isop max | n | log EC50 β3 | % isop max | n |
| CGP 20712A | no response | 0 | 4 | 10 μMb | 1.7 ± 0.0 | 4 | no response | 0 | 4 |
| xamoterol | –7.96 ± 0.03 | 47.6 ± 1.7 | 5 | –6.15 ± 0.06 | 5.1 ± 2.5 | 4 | 100 μMb | 38.3 ± 1.4 | 4 |
| ICI 89406 | –9.09 ± 0.03 | 27.3 ± 0.05 | 5 | –7.22 ± 0.08 | 4.9 ± 0.2 | 4 | 10 μMb | 11.7 ± 0.5 | 4 |
| 1 | –8.64 ± 0.04 | 37.1 ± 2.2 | 5 | 10 μMb | 15.2 ± 0.7 | 4 | 10 μMb | 8.6 ± 0.4 | 4 |
| 19 | –8.01 ± 0.04 | 33.6 ± 2.4 | 6 | 10 μMb | 8.0 ± 1.1 | 7 | 10 μMb | 12.5 ± 2.3 | 7 |
Values represent the mean ± SEM of n separate experiments. Log EC50 values and % isoprenaline maximal responses were obtained from 3H-cAMP accumulation for cells expressing the human β1-adrenoceptor.
For the human β2- and β3-adrenoceptors, the top of the agonist concentration response curve was not obtained even with the maximum concentration of ligand. In these instances, the percentage of isoprenaline response is given for the response at 10 μM of ligand or, in the case of xamoterol, 100 μM.
In Vivo Selectivity and Agonist Actions of 19
To assess the potential clinical effect of this degree of selectivity and partial agonism, these parameters were investigated in a freely moving conscious rat model. Previous studies with this model have shown that the heart rate response to isoprenaline is solely a β1-mediated response and that the hindquarters vascular conductance is solely a β2-mediated response.44 We employed the hydrochloride salt of 19 (2 mg/kg iv bolus, 1 mg/kg/h iv infusion) and observed that it significantly inhibited the β1-mediated heart rate responses to isoprenaline (at 40 and 120 ng/kg/min, 30–90 min following administration) while having no effect on the β2-mediated hindquarters response (n = 4 rats), consistent with this compound’s β1-adrenoceptor selectivity (Figure 3). In addition, the ligand caused an increase in basal heart rate (from 424 ± 9 to 465 ± 3 beats/min, an increase that corresponds to 44% of the response to 120 ng/kg/min isoprenaline, n = 4 animals), in keeping with the partial agonist actions observed in the in vitro cell studies with 19.
Figure 3.
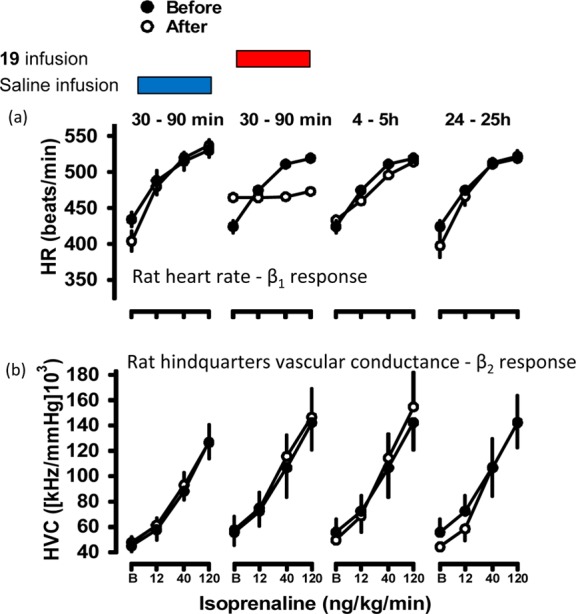
Absolute values for heart rate (a) and hindquarters conductance (b) in conscious freely moving rats (n = 4 per group) before and at the end of 3 min infusions of isoprenaline (12, 40, and 120 ng/kg/min). Isoprenaline responses were measured before and at intervals after saline or 19 (HCl salt) administration. 19 (HCl salt) was given as a 2 mg/kg bolus followed by 1 mg/kg/h infusion for 90 min, and isoprenaline responses were measured during the infusion (30–90 min) and after the 19 (HCl salt) infusion was turned off (4–5 and 24–25 h). Isoprenaline responses measured in saline-treated animals at 30–90 min, 4–5 h, and 24–25 h were highly reproducible, but for the sake of clarity only the 30–90 min time point is shown here.
β-Adrenoceptor Profiling of Second- and Third-Generation Ligands
To investigate whether the structural extremities of our lead compounds could be modified to improve affinity and selectivity while attenuating partial agonism, we employed an iterative screening cascade approach. Initially we used a 3H-CGP 12177 whole cell binding assay to ascertain antagonist affinity measurements (Table 4).
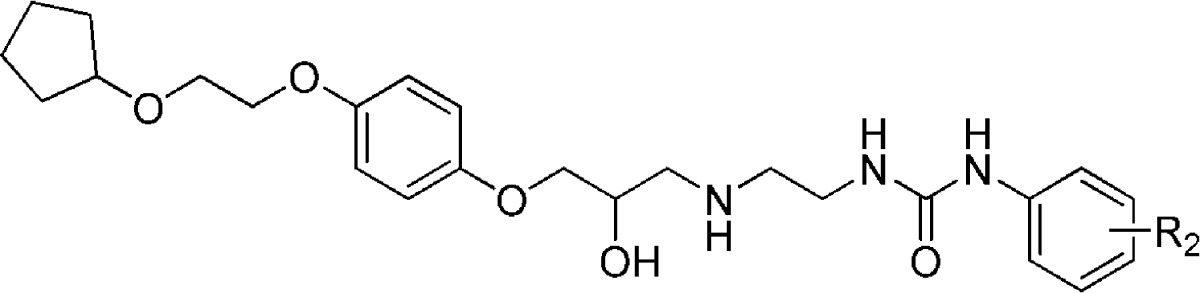
Informed from previously established SARs,36 we explored three variants of the p-alkoxyalkoxy side chain present in 1 and 19, namely a para-positioned 2-(cyclopentyloxy)ethoxy- (29a), a 2-(4-fluorophenethyloxy)ethoxy- (29b), and an 2-(ethyloxy)ethoxy- (29c) moiety. Because the 2-(cyclopentyloxy)ethoxy side chain displayed the best combination of an increase in both binding affinity and β1/β2-selectivity when compared to 19, we retained this moiety to undertake a SAR evaluation of the phenylurea portion of the molecule (Table 5). The unsubstituted phenylurea (37a) displayed a loss in affinity at both the β1- and β2-adrenoceptors yet maintained good β1-selectivity. Insertion onto the phenyl ring of individual electron-withdrawing or electron-donating groups at the ortho-, meta-, or para-positions displayed a set of measurable trends. In general, the ligand binding affinity remained within an order of magnitude for the respective adrenoceptor subtypes (β1 log KD between −6.94 and −8.17 M; β2 log KD between −5.52 and −6.54 M) with all ligands showing selectivity for the β1-subtype. In most cases (37b, 37e, 37k, 37n, 37q, and 37t) an ortho-positioned substituent resulted in the weakest β1-adrenoceptor binding affinity within each three-ligand subset alongside the smallest β1/β2-selectivity. The only exception to this observation was that of the o-fluoro derivative (37h), which, while displaying the second highest affinity of the three ligands (37h–j) for the β1-adrenoceptor, did possess the greatest, albeit modest, β1/β2-selectivity (76-fold).
Table 5. Human β1- and β2-Adrenoceptor Binding Affinities and Receptor Selectivity of Substituted Phenylureas (37a–37u)a.
| compd | R2 | log KD β1 | n | log KD β2 | n | selectivity ratio (β1/β2) |
|---|---|---|---|---|---|---|
| 37a | H | –7.91 ± 0.03 | 10 | –5.58 ± 0.03 | 9 | 213 |
| 37b | o-CH3 | –7.33 ± 0.03 | 6 | –6.27 ± 0.02 | 6 | 11 |
| 37c | m-CH3 | –8.04 ± 0.04 | 6 | –6.06 ± 0.03 | 6 | 96 |
| 37d | p-CH3 | –7.76 ± 0.04 | 6 | –5.80 ± 0.03 | 6 | 91 |
| 37e | o-OCH3 | –7.02 ± 0.04 | 7 | –5.93 ± 0.02 | 8 | 12 |
| 37f | m-OCH3 | –7.76 ± 0.03 | 7 | –6.05 ± 0.03 | 10 | 51 |
| 37g | p-OCH3 | –7.80 ± 0.04 | 7 | –5.86 ± 0.04 | 5 | 87 |
| 37h | o-F | –7.82 ± 0.03 | 6 | –5.94 ± 0.04 | 6 | 76 |
| 37i | m-F | –8.17 ± 0.03 | 6 | –6.54 ± 0.02 | 6 | 43 |
| 37j | p-F | –7.70 ± 0.04 | 5 | –5.92 ± 0.09 | 6 | 60 |
| 37k | o-Cl | –7.11 ± 0.03 | 7 | –5.54 ± 0.03 | 5 | 37 |
| 37l | m-Cl | –8.21 ± 0.06 | 10 | –5.69 ± 0.07 | 12 | 331 |
| 37m | p-Cl | –7.95 ± 0.05 | 7 | –5.91 ± 0.06 | 9 | 110 |
| 37n | o-Br | –7.11 ± 0.01 | 5 | –6.10 ± 0.03 | 6 | 10 |
| 37o | m-Br | –7.92 ± 0.05 | 6 | –5.99 ± 0.04 | 6 | 85 |
| 37p | p-Br | –7.77 ± 0.04 | 6 | –5.86 ± 0.04 | 6 | 81 |
| 37q | o-CF3 | –6.94 ± 0.01 | 6 | –5.84 ± 0.02 | 6 | 13 |
| 37r | m-CF3 | –7.77 ± 0.06 | 6 | –5.86 ± 0.06 | 6 | 81 |
| 37s | p-CF3 | –7.70 ± 0.04 | 6 | –5.76 ± 0.04 | 6 | 87 |
| 37t | o–OH | –6.99 ± 0.08 | 6 | –5.96 ± 0.05 | 6 | 11 |
| 37u | m–OH | –7.89 ± 0.06 | 6 | –5.85 ± 0.06 | 6 | 110 |
Values represent the mean ± SEM of n separate experiments. Log KD values were obtained from 3H-CGP 12177 whole cell binding studies in CHO cells stably expressing the human β1- or β2-adrenoceptors.
The optimal ligands in terms of both affinity and selectivity were substituted in the meta- or para-positions with either a chlorine atom (37l and 37m) or a hydroxyl group (29a and 37u) or, as mentioned above, were unsubstituted (37a). To cross-validate these results and attempt to determine β3-adrenoceptor affinity, we repeated the alternative assay for determining affinity whereby the shift of a cimaterol-mediated dose–response curve is measured, focusing upon the best ligands from the phenylurea SAR study and re-evaluating the original alkoxyalkoxy compounds (Table 6).
Table 6. Antagonist Affinities for Second- and Third-Generation Ligands at the Human β1-, β2-, and β3-Adrenoceptorsa.
| compd | log KD β1 | n | log KDβ2 | n | log KDβ3 | n | selectivity β1 vs β2 |
|---|---|---|---|---|---|---|---|
| 19 | –7.75 ± 0.06 | 21 | –5.15 ± 0.06 | 7 | >10 μM | 7 | 398 |
| 29a | –8.55 ± 0.04 | 12 | –5.84 ± 0.06 | 4 | >10 μM | 4 | 513 |
| 29b | –9.00 ± 0.04 | 9 | –6.05 ± 0.05 | 4 | –5.58 ± 0.09 | 4 | 891 |
| 29c | –7.57 ± 0.05 | 12 | >10 μM | 4 | >10 μM | 4 | >372 |
| 37a | –8.46 ± 0.05 | 12 | –5.92 ± 0.02 | 4 | >10 μM | 4 | 347 |
| 37l | –8.83 ± 0.06 | 9 | –6.10 ± 0.10 | 4 | –5.22 ± 0.08 | 4 | 537 |
Values represent the mean ± SEM of n separate experiments. Log KD values were obtained from 3H-cAMP accumulation assays following inhibition of the cimaterol agonist response in CHO cells stably expressing the human β1-, β2-, or β3-adrenoceptors.
Interestingly, this assay gave values for ligand affinity (and selectivity) slightly higher than that of the binding assay, for example, with 29a β1-selectivity 478-fold (over β2) in the binding assay but 513-fold in the cAMP assay and with 37l 331-fold in the binding assay and 537-fold in the cAMP assay, similar to what was observed with previous ligands (Tables 1 and 2). This method therefore provides a similar pattern of β1- versus β2-selectivity, allowing comparison between different chemical analogues. Even using this method, the affinity at the human β3-adrenoceptor once again could not be assessed, because the maximum concentration of test compounds did not cause a sufficient shift of the cimaterol concentration response curve (e.g., Tables 2 and 6; Figure 2).
Partial Agonism of Second- and Third-Generation Ligands
Although the affinity and selectivity profiles for many of the ligands were extremely encouraging, we were also aware that the original lead compounds (19 and 1) also displayed significant partial agonism at the β-adrenoceptors. We therefore investigated the full concentration response relationships for the best ligands (Table 7).
Table 7. Agonist Responses of Second- and Third-Generation Ligands at the Human β1-, β2-, and β3-Adrenoceptorsa.
| β1 |
β2 |
β3 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | log EC50 β1 | % isop max | n | β2b | % isop max at 10 μM | n | β3b | % isop max at 10 μM | n |
| 1 | –8.64 ± 0.04 | 37.1 ± 2.2 | 5 | 10 μM | 15.2 ± 0.7 | 4 | 10 μM | 8.6 ± 0.4 | 4 |
| 19 | –8.01 ± 0.04 | 33.6 ± 2.4 | 6 | 10 μM | 8.0 ± 1.1 | 7 | 10 μM | 12.5 ± 2.3 | 7 |
| 29a | –8.44 ± 0.04 | 25.5 ± 1.1 | 5 | 10 μM | 15.3 ± 1.1 | 4 | 10 μM | 4.0 ± 0.6 | 3 |
| 29b | –8.64 ± 0.04 | 23.8 ± 1.5 | 5 | 10 μM | 16.0 ± 1.3 | 4 | 10 μM | 7.9 ± 0.1 | 4 |
| 29c | –7.64 ± 0.04 | 35.9 ± 1.7 | 5 | 10 μM | 27.8 ± 2.3 | 3 | 10 μM | 1.3 ± 0.1 | 3 |
| 37a | –8.35 ± 0.10 | 28.3 ± 1.9 | 5 | 10 μM | 15.3 ± 1.0 | 4 | 10 μM | 4.0 ± 0.3 | 4 |
| 37l | –8.52 ± 0.08 | 29.1 ± 1.7 | 4 | 10 μM | 12.7 ± 0.7 | 3 | 10 μM | 2.0 ± 0.1 | 3 |
Values represent the mean ± SEM of n separate experiments. Log EC50 values and % isoprenaline maximal responses were obtained from 3H-cAMP accumulation for cells expressing the human β1-adrenoceptor.
For the human β2- and β3-adrenoceptors, the top of the agonist concentration response curve was not obtained even with the maximum concentration of ligand. In these instances, the percentage of isoprenaline response is given for the response at 10 μM of ligand.
Whereas the affinities of the second- and third-generation ligands were comparable to those of 1 and 19, there was a modest drop in efficacy at the β1-adrenoceptor, suggesting a marginal attenuation of partial agonism with these compounds. Interestingly, the one exception to this observation was 29c, which contained the linear 2-(ethyloxy)ethoxy side chain. In this example, the affinity at the β1-adrenoceptor was an order of magnitude less than that of 1 and the 2-(4-fluorophenethyloxy)ethoxy derivative (29b), yet this compound displayed a similar efficacy profile.
Assessing the Site of Agonist Action at the Human β1-Adrenoceptor
As the β1-adrenoceptor is now accepted to exist in at least two agonist conformational states,45−48 it was important to assess at which state of the β1-adrenoceptor these novel β-adrenoceptor ligands were exerting their partial agonist actions. CGP 20712A (neutral high-affinity β1-adrenoceptor antagonist, capable of inhibiting both conformational states of the β1-adrenoceptor but with different affinities)42 measured in the presence of cimaterol yielded a high affinity value of log KD = −9.48 (Supporting Information Table 1) compatible with the agonist activity occurring via the primary catecholamine site of the receptor. CGP 12177 (log EC50 = −7.91 ± 0.04, 58.2 ± 2.6% isoprenaline maximum, n = 5) was antagonized by CGP 20712A to yield a much lower affinity (log KD = −7.22 ± 0.03, Supporting Information Table 1), demonstrating that a far higher concentration of CGP 20712A is required to block the CGP 12177 agonist response. The agonist activity of CGP 12177 was therefore occurring via the secondary site of the β1-adrenocptor (Supporting Information Figure 1).42 The agonist response of our compounds was therefore inhibited by CGP 20712A to determine the site of their agonist actions. The affinity values for CGP 20712A were all high (log KD values between −9.51 and −9.09, Supporting Information Table 1). suggesting that, like the partial agonist xamoterol and ICI 89406 (and unlike CGP 12177), our new partial agonists were indeed achieving their agonist actions via the catecholamine conformation of the receptor. The human β3-adrenoceptor, similar to the human β1-adrenoceptor, has also been demonstrated to exist in two conformational states.49 However, given the low affinity and efficacy of our novel compounds for the human β3-adrenoceptor, it was not possible to determine at which receptor conformational state the β3-agonist responses were occurring.
Conclusions
Despite 70 years of experience and the availability of many adrenoceptor ligands, there remain very few truly selective ligands for the β1-adrenoceptor. To date, CGP 20712A is a widely known and truly selective β1-antagonist, whereas ICI 118551 is a β2-selective antagonist.18−20 However, although several very highly β2-selective agonists exist (e.g., salmeterol and formoterol), there are no selective β1-agonist compounds – noradrenaline, dobutamine, xamoterol, and ICI 89406 all having relatively poor selectivity.18−20 Here we have characterized the pharmacological properties of 1 and some novel derivatives and thus present the most selective β1-partial agonists reported to date.
1 was confirmed as being a truly selective β1-adrenoceptor ligand with a β1 over β2 selectivity of 562-fold as measured by whole-cell binding. The first analogue of 1 made was 19, a decyanated version of the molecule, and the pharmacological effect of this maneuver was to reduce affinity at both human β1- and β2-adrenoceptors.
It was clear from the antagonist studies that 1 and its derivative 19 had substantial agonist activity at the human β-adrenoceptors. Given this observation, we investigated the potential clinical impact of this degree of partial agonism in a fully conscious rat model. Infusion of the hydrochloride salt of 19 resulted in a substantial increase in the basal (β1-mediated) heart rate, in keeping with its β1-partial agonist effects, followed by inhibition of the β1-heart rate response to the isoprenaline (compare Figure 2a in CHO β1 cells and Figure 3a in conscious rats). No effect on basal hindquarters vascular conductance or on the β2-mediated hindquarters response to isoprenaline was observed, consistent with high β1-selectivity, thereby demonstrating the β1-selective partial agonist effects in vivo. This agonist effect is significant because β-ligands with agonist properties (e.g., xamoterol) have been shown to be less beneficial and, at times, even detrimental if used long-term in cardiovascular disease.44,50,51 Thus, although 19 maintained its β1-selectivity in vivo, its partial agonist properties were too great for it to be pursued as a potential clinical compound.
Given 19 had reduced affinity and selectivity compared to 1, we designed a series of analogues of the molecule looking for evidence of structure–activity relationships related to affinity, selectivity, and partial agonism. Substitution of the alkoxyalkoxy side chain afforded ligands with increased β1-adrenoceptor affinity (29a and 29b). Because all molecules remained partial agonists at the human β1-adrenoceptor (Table 7), we therefore examined the phenylurea portion of the molecule in an attempt to attenuate this pharmacology. Generally, regardless of the substituent, substitution in the meta- and para-positions of the phenyl ring afforded higher β1-affinity (and therefore higher β1-selectivity) than substitutions at the ortho-position. Once again, however, these ligands retained their β1-selective partial agonist activity, and the agonist actions were demonstrated to occur via the catecholamine conformation.
In summary, 1 and the new family of ligands that we have generated are β1-selective partial agonists. Compared with currently published ligands, for example, xamoterol and ICI 89406, 1 and some of our novel analogues are the most β1-selective agonists to date and will prove to be useful pharmacological tools; however, the observed clinically deleterious effects of significant partial agonism in β-blockers44,50,51 preclude any of these ligands from further clinical development. However, this preliminary body of work clearly demonstrates that it is possible to generate more β1-selective molecules than those currently available and in clinical use. Molecules of higher selectivity, but devoid of partial agonist effects, should prove to be a very powerful treatment for people with concomitant cardiovascular and respiratory disease and is the focus of ongoing work within our laboratories.
Experimental Section
General Chemistry Methods
Chemicals and solvents were purchased from standard suppliers and used without further purification. Merck Kieselgel 60, 230–400 mesh, for flash column chromatography (FCC), was supplied by Merck KgaA (Darmstadt, Germany), and deuterated solvents were purchased from Goss International Limited (England) and Sigma-Aldrich Co. Ltd. (England).
Unless otherwise stated, reactions were carried out at ambient temperature. Reactions were monitored by thin layer chromatography on commercially available precoated aluminum-backed plates (Merck Kieselgel 60 F254). Visualization was by examination under UV light (254 and 366 nm). General staining was carried out with KMnO4 or phosphomolybdic acid. A solution of ninhydrin (in ethanol) was used to visualize primary and secondary amines. All organic extracts collected after aqueous workup procedures were dried over anhydrous MgSO4 or Na2SO4 before gravity filtration and evaporation to dryness. Organic solvents were evaporated in vacuo at ≤40 °C (water bath temperature). Purification using preparative layer chromatography (PLC) was carried out using Fluka silica gel 60 PF254 containing gypsum (200 mm × 200 mm × 1 mm). Flash chromatography was performed using Merck Kieselgel 60 (0.040–0.063 mm).
Melting points (mp) were recorded on a Reichert 7905 apparatus or Perkin-Elmer Pyris 1 differential scanning calorimeter and were uncorrected. Fourier transform infrared (FT-IR) spectra were recorded as thin films or KBr disks in the range of 4000–500 cm–1 using an Avatar 360 Nicolet FT-IR spectrophotometer. Hig- resolution mass spectra (HRMS)–time-of-flight electrospray (TOF ES ±) were recorded on a Waters 2795 separation module/micromass LCT platform. 1H NMR spectra were recorded on a Bruker-AV 400 at 400.13 MHz. 13C NMR spectra were recorded at 101.62 MHz. Chemical shifts (δ) are recorded in parts per million (ppm) with reference to the chemical shift of the deuterated solvent/an internal tetramethylsilane (TMS) standard. Coupling constants (J) and carbon–fluorine coupling constants (JCF) are recorded in hertz and the significant multiplicities described by singlet (s), doublet (d), triplet (t), quadruplet (q), broad (br), multiplet (m), doublet of doublets (dd), and doublet of triplets (dt). Spectra were assigned using appropriate COSY, distortionless enhanced polarization transfer (DEPT), HSQC, and HMBC sequences. Unless otherwise stated, all spectra were recorded in CDCl3.
Analytical reverse-phase high-performance liquid chromatography (RP-HPLC) was performed on a Waters Millenium 995 LC using both system 1 and system 2 described below and was used to confirm that all final products were ≥95% pure.
System 1:
Phenomenex Onyx Monolithic reverse phase C18 column (100 × 4.6 mm), a flow rate of 3.00 mL/min and UV detection at 287 nm. Linear gradient 5–95% solvent B over 10 min. Solvent A, 0.1% formic acid (FA) in water; solvent B, 0.1% FA in MeCN.
System 2:
Waters symmetry reverse phase C18 column (75 × 4.6 mm), a flow rate of 1.00 mL/min, and UV detection at 287 nm. Linear gradient 5–95% solvent B over 20 min. Solvent A, 0.1% FA in water; solvent B, 0.1% FA in MeOH. Preparative HPLC was performed using a Phenomenex Onyx Monolithic reverse phase C18 column (100 × 10 mm), a flow rate of 14.10 mL/min, and UV detection at 287 nm. Samples were run in 5–95% solvent B over 10 min. Solvent A, 0.1% FA in water; solvent B, 0.1% FA in MeCN. All retention times (Rt) are quoted in minutes.
1-(2-Aminoethyl)-3-(4-benzyloxy)phenylurea (12)
A solution of 4-(benzyloxy) phenylisocyanate (3.739 g, 16.61 mmol) in anhydrous DCM (30 mL) was dripped into a flask containing vigorously stirred 1,2-ethanediamine (11) (6 mL, 89.80 mmol, 5.4 equiv) under nitrogen. Instant precipitation of a white solid was noted, and the reaction was allowed to stir for a further 3 h after addition of isocyanate solution was complete. After removal of all volatiles under reduced pressure, the crude solid was washed with Et2O, before drying to give 4.472 g (94%) of white solid: mp 147–149 °C; FT-IR 3300 (primary (1°) amine N–H, str), 2932, 2864 (alkyl C–H, str), 1642 (urea C=O, str), 1604 (1° amine N–H, bend), 1111 (C–O, str), 830 (aryl C–H, bend, para-disubstituted ring), 741, 697 (aryl C–H, bend, phenyl ring); 1H NMR (DMSO-d6) δ 8.48 (s, 1H, NHAr), 7.31–7.44 (m, 5H, aromatic benzyl CH), 7.28, 6.88 (d, J = 9.0 Hz, 2 × 2H, para-disubstituted ring), 6.24 (t, J = 5.2 Hz, 1H, NHCONHAr), 5.02 (s, 2H, PhCH2O), 4.27 (br s, 2H, NH2), 3.10–3.17 (m, 2H, CH2NH), 2.67 (t, J = 6 Hz, CH2NH2); 13C NMR (DMSO-d6) δ 155.72 (C=O), 152.88, 137.37, 133.97 (4 °C), 128.37, 127.71, 127.63 (benzyl CH), 119.29, 114.87 (aryl CH), 69.37 (benzyl CH2), 40.87 (CH2NH2), 40.38 (CHNH); m/z HRMS (TOF ES+) C16H20N3O2 [MH]+ calcd 286.1550; found 286.1547.
1-(2-Aminoethyl)-3-(4-hydroxy)phenylurea (13)
1-(2-Aminoethyl)-3-(4-(benzyloxy)phenyl) urea (12) (113 mg, 0.40 mmol) was stirred overnight in a solution of concd HCl (10 mL). The mixture was concentrated under reduced pressure and the residue redissolved in water (10 mL) before neutralization with aqueous 0.5 M NaOH. After reconcentration, the residue was dissolved in the minimum amount of MeOH and filtered (gravity), before purification by PLC (eluent 37% aqueous NH3/MeOH/DCM 2:25:73). This gave 56 mg (73%) of a brown semisolid: FT-IR 3339 (1° amine N–H, str), 3118 (br, O–H, str), 1643 (urea C=O, str), 1615 (1° amine N–H, bend), 1570 (aryl, str), 836 (aryl C–H, bend, para-disubstituted ring); 1H NMR (MeOD-d4) δ 7.17 (d, J = 8.7 Hz, 2H, aryl 3-H and 5-H), 6.73 (d, J = 8.7 Hz, 2H, aryl 2-H and 6-H), 3.45 (t, J = 5.6 Hz, 2H, CH2NH(C=O)NH), 3.05 (t, J = 6.0 Hz, 2H, CH2NH2); 13C NMR (MeOD-d4) δ 159.38 (4° aryl 4-C), 154.70 (C=O), 132.02 (4° aryl 1-C), 123.61, 116.32 (aryl CH), 41.73 (CH2NH2), 38.79 (CH2NH(C=O)NH); m/z HRMS (TOF ES+) C9H14N3O2 [MH]+ calcd 196.1081; found 196.1081.
1-(Benzyloxy)-4-[2-(cyclopentyloxy)ethoxy]benzene (26a)
2-(Cyclopentyloxy)ethanol (21) (3.751 g, 28.81 mmol), triphenylphosphine (9.448 g, 36.02 mmol, 1.25 equiv), and 4-(benzyloxy)phenol (14) (5.769 g, 28.81 mmol, 1 equiv) were dissolved in DCM (70 mL). Di-tert-butyl azodicarboxylate (8.294 g, 36.02 mmol, 1.25 equiv) in DCM (20 mL) was added dropwise to the reaction mixture, which was allowed to stir overnight. After removal of approximately half of the solvent from the reaction mixture under reduced pressure, the resulting slurry was diluted with hexanes (100 mL) and washed with aqueous 1 M HCl (2 × 50 mL), aqueous 1 M NaOH (2 × 50 mL), water (2 × 50 mL), and brine (1 × 50 mL). The organic layer was concentrated and redissolved in DCM (30 mL). On addition of hexanes a precipitate of triphenylphosphine oxide began to form. The flask was left in the freezer for 1 h before filtration of the precipitate and washing with hexanes and Et2O. After concentration of the filtrate, purification was achieved via FCC (eluent Et2O/hexanes 10:90) to give 6.75 g (75%) of a clear colorless oil: FT-IR 2870, 2954 (alkyl C–H, str), 1507 (aryl, str), 1109 (C–O–C, str), 824 (aryl C–H, bend, para-disubstituted ring), 738, 696 (aryl C–H bend, phenyl ring); 1H NMR δ 7.34 – 7.48 (m, 5H, aromatic benzyl CH), 6.91, 6.96 (d, J = 9.2 Hz, 2 × 2H, aryl-dioxy ring), 5.04 (s, 2H, PhCH2O), 4.09 (t, J = 4.9 Hz, 2H, CH2OArOBn), 4.02–4.06 (m, 1H, cPe CH), 3.76 (t, J = 5.3 Hz, 2H, CH2CH2OArOBn), 1.73–1.84 (m, 6H, cPe CH2), 1.56–1.63 (m, 2H, cPe CH2); 13C NMR δ 153.02, 153.26 (4 °C, aryl-dioxy ring), 137.29 (4° benzyl C), 127.44, 127.83, 128.50 (benzyl CH), 115.62, 115.68 (CH aryl-dioxy ring), 81.89 (cPe CH), 70.55 (benzyl CH2), 68.16 (CH2OArOBn), 67.28 (CH2CH2OArOBn), 32.27 (2-C and 5-C cPe ring), 23.55 (3-C and 4-C cPe ring); m/z HRMS (TOF ES+) C20H25O3 [MH]+ calcd 313.1798; found 313.1766.
4-(2-[Cyclopentyloxy]ethoxy)phenol (27a)
1-(2-(Cyclopentyloxy)ethoxy)-4-(benzyloxy)benzene (26a) (6.326 g, 20.25 mmol) was hydrogenated according to the general procedure for O-benzyl deprotection to give the title compound in quantitative yield as a clear colorless oil: FT-IR 3381 (br, O–H, str), 2960, 2871 (alkyl C–H, str), 1510 (aryl, str), 1104 (C–O–C, str), 827 (aryl C–H, bend, para-disubstituted ring); 1H NMR δ 7.60 (br s, 1H, OH), 6.69, 6.73 (d, J = 9.2 Hz, 2 × 2H, para-disubstituted phenol), 3.96–3.99 (m, 3H, CH, CH2OAr), 3.70 (t, J = 5.0 Hz, 2H, cPeOCH2), 1.62–1.78 (m, 6H, cPe CH2), 1.45–1.53 (m, 2H, cPe CH2); 13C NMR δ 150.05, 152.01 (4 °C), 115.53, 115.88 (CH phenolic ring), 82.05 (cPe CH), 67.82 (CH2OAr), 67.11 (CH2CH2OAr), 32.87 (2-C and 5-C cPe ring), 23.20 (3-C and 4-C cPe ring); m/z HRMS (TOF ES–) C13H17O3 [M – H]− calcd 221.1183; found 221.1191.
2-{4-[2-(Cyclopentyloxy)ethoxy]phenoxymethyl}oxirane (28a)
NaH 60% suspension in mineral oil (863 mg, equivalent to 518 mg of NaH, 21.58 mmol, 1.1 equiv) was suspended in anhydrous DMF (20 mL) with stirring under a nitrogen atmosphere. After 5 min, 4-(2-(cyclopentyloxy)ethoxy)phenol (27a) (4.360 g, 19.61 mmol) in anhydrous DMF (20 mL) was added dropwise, with the vessel cooled over an ice bath. This mixture was then allowed to stir at rt for 20 min before addition of rac-epichlorohydrin (15.34 mL, 196.10 mmol, 10 equiv). The mixture was stirred for 7 h and then quenched cautiously with MeOH. After removal of all volatiles under reduced pressure, the crude residue was partitioned between water (30 mL) and Et2O (30 mL) and the aqueous layer washed again with Et2O (3 × 30 mL). The combined organic extracts were concentrated before purification through a silica plug (initial wash with hexanes, followed by EtOH/DCM 5:95) to give 4.558 g (84%) of a clear yellow oil: FT-IR 3052 (epoxide C–H, str, weak), 2961, 2870 (alkyl C–H, str), 1508 (aryl, str), 1110 (C–O–C, str), 828 (aryl C–H, bend, para-disubstituted ring); 1H NMR δ 6.80 (s, 4H, aryl C–H), 4.11 (dd, J = 11.1/3.1 Hz, 1H, ArOCH2CH), 3.99 (t, J = 4.9 Hz, 2H, CH2OAr), 3.92–3.96 (m, 1H, cPe CH), 3.82 (dd, J = 11.1/5.7, 1H, ArOCH2CH), 3.67 (t, J = 5.3 Hz, 2H, cPeOCH2), 3.25–3.29 (m, 1H, epoxide CH), 2.82 (d, J = 4.9/4.9 Hz, 1H, epoxide CH2), 2.67 (dd, J = 5.0/2.7 Hz, 1H, epoxide CH2), 1.58 – 1.77 (m, 6H, cPe CH2), 1.41 – 1.56 (m, 2H, cPe CH2); 13C NMR δ 152.65, 153.36 (4 °C), 115.46, 115.53 (aryl CH), 69.35 (ArOCH2CH), 68.08 (CH2OAr), 67.18 (CH2CH2OAr), 50.13 (epoxide CH), 44.49 (epoxide CH2), 32.17 (2-C and 5-C cPe ring), 23.45 (3-C and 4-C cPe ring); m/z HRMS (TOF ES+) C16H22NaO4 [MNa]+ calcd 301.1410; found 301.1414.
1-{2-[(3-{4-[2-(Cyclopentyloxy)ethoxy]phenoxy}-2-hydroxypropyl)amino]ethyl}-3-(4-hydroxyphenyl)urea (29a)
2-((4-(2-(Cyclopentyloxy)ethoxy)phenoxy)methyl)oxirane (28a) was opened with 1-(2-aminoethyl)-3-(4-hydroxyphenyl)urea (13) as described in the general procedure for aminolysis of oxiranes (cf. Supporting Information). Yield: 29%, white amorphous solid; mp 135–138 °C; FT-IR 3311 (br, O–H, str), 2929, 2869 (alkyl C–H, str), 1636 (urea C=O, str), 1509, 1568 (aryl, str), 1111 (C–O–C, str), 820 (aryl C–H, bend, para-disubstituted ring); 1H NMR (DMSO-d6) δ 8.91 (br s, 1H, phenol), 8.20 (s, 1H, NH(C=O)NHAr), 7.13 (d, J = 8.8 Hz, 2H, aryl C–H ortho to urea), 6.84 (s, 4H, aryl-dioxy ring), 6.62 (d, J = 8.8 Hz, 2H, aryl C–H ortho to phenol), 6.02 (t, J = 5.4 Hz, 1H, NH(C=O)NHAr), 4.96 (br s, 1H, NH), 3.79–3.97 (m, 6H, CH2OAr, cPe CH, CH(OH), ArOCH2), 3.61 (t, J = 4.8 Hz, 2H, cPeOCH2), 3.12–3.16 (m, 2H, NHCH2CH2), 2.56 – 2.71 (m, 4H, CH(OH)CH2NH, NHCH2CH2), 1.54–1.69 (m, 6H, cPe CH2), 1.42–1.51 (m, 2H, cPe CH2); 13C NMR (DMSO-d6) δ 152.74, 152.49, 151.85, 132.14 (aryl 4 °C), 155.65 (C=O), 119.72 (aryl C–H ortho to urea), 115.33 (CH aryl-dioxy ring), 115.04 (aryl C–H ortho to phenol), 80.84 (cPe CH), 71.23 (ArOCH2), 68.08 (CH(OH)), 67.71 (CH2OAr), 66.72 (cPeOCH2), 52.20 (CH(OH)CH2NH), 49.38 (NHCH2CH2), 39.15 (NHCH2CH2), 31.78 (2-C and 5-C cPe ring), 23.12 (3-C and 4-C cPe ring); m/z HRMS (TOF ES+) C25H36N3O6 [MH]+ calcd 474.2599; found 474.2578; RP-HPLC Rt 3.17 (system 1), 10.30 (system 3).
General Pharmacology Methods
(A) Pharmacology Materials
Fetal calf serum was from PAA Laboratories (Teddington, Middlesex, UK). White-sided view plates were supplied by Thermo Fisher Scientific (Basingstoke, UK). Microscint 20 scintillation fluid and Ultima Gold XR scintillation fluid were from PerkinElmer. Radioligands (3H-CGP 12177, 3H-adenine, and 14C-cAMP) were from Amersham International (Buckinghamshire, UK). ICI 89406, cimaterol, and CGP 20712A were from Tocris Life Sciences (Avonmouth, UK). All other reagents were from Sigma Chemicals (Poole, Dorset, UK).
(B) In Vitro Experiments
(1) Cell Culture
CHO-K1 stably expressing either the human β1 (1146fmol/mg protein), the human β2 (466fmol/mg protein), or the human β3-adrenoceptor (790fmol/mg protein) was used throughout this study.20 Cells were grown in Dulbecco’s modified Eagle’s medium nutrient mix F12 (DMEM/F12) containing 10% fetal calf serum and 2 mM l-glutamine in a 37 °C humidified 5% CO2/95% air atmosphere.
(2) 3H-CGP 12177 Whole Cell Binding
Cells were grown to confluence in white-sided, flat-bottom 96-well view plates. 3H-CGP 12177 whole cell binding was performed as described.20 Briefly, the medium was removed from each well, and 100 μL of serum-free medium containing the competing ligand at twice the final required concentration was added to each well. A fixed concentration of 100 μL 3H-CGP 12177 was then immediately added to each well (1:2 dilution in well), and the cells were incubated for 2 h at 37 °C, 5% CO2. The cells were washed twice by the addition and removal of 200 μL of 4 °C phosphate-buffered saline per well. One hundred microliters of Microscint 20 was added to each well, a white sticky base was applied to the bottom of the plate, and a sealant top was applied to the top of the plate. The plates were left at room temperature overnight in the dark and then counted on a Topcount at 21 °C for 2 min/well.
(3) 3H-cAMP Accumulation
Cells were grown to confluence in 24-well plates. The cells were prelabeled with 3H-adenine by incubation for 2 h with 2 μCi/mL 3H-adenine in serum-free medium (0.5 mL per well). The 3H-adenine was removed, and each well washed by the addition and removal of 1 mL of serum-free medium. One milliliter of serum-free medium containing 1 mM IBMX was added to each well, and the cells were incubated for 15 min. When used, antagonists were added at a final concentration at this stage and thus had 15 min of incubation. Agonist (in 10 μL of serum-free medium) was added to each well, and the plates were incubated for 5 h. The reaction was terminated by the addition of 50 μL of concentrated HCl per well. The plates were then frozen and thawed, and 3H-cAMP was separated from other 3H-nucleotides by sequential Dowex and alumina column chromatography, as previously described.52
As measurements of partial agonism depend on many factors, including particularly the receptor expression level in the cells being examined, all cAMP experiments were performed in cells at the same passage throughout this entire study. In addition, whenever possible, experiments were performed with all ligands being investigated in parallel studies on the same day. Thus, every ligand was examined on the same split of cells on the same day, with each separate n number being performed on different days.
(C) Pharmacological Data Analysis
(1) Whole Cell Binding
All data points on each binding curve were performed in triplicate, and each 96-well plate also contained three to six determinations of total and nonspecific binding. Nonspecific binding was determined in the presence of 10 μM propranolol.
A one-site sigmoidal binding curve (eq 1) was then fitted to the data using Graphpad Prism 2.01, and the IC50 was then determined as the concentration required to inhibit 50% of the specific binding.
| 1 |
In eq 1, A is the concentration of the competing ligand, IC50 is the concentration at which half of the specific binding of 3H-CGP 12177 has been inhibited, and NS is the nonspecific binding.
From the IC50 value and the known concentration of radioligand [3H-CGP 12177], a KD (concentration at which half the receptors are bound by the competing ligand) value was calculated using eq 2:
| 2 |
(2) 3H-cAMP Accumulation
Agonist responses were best described by a one-site sigmoidal concentration response curve (eq 3)
| 3 |
where Emax is the maximum response, [A] is the agonist concentration, and EC50 is the concentration of agonist that produces 50% of the maximal response
As several ligands have too low affinity at the human β2 and β3 receptors to assess accurately in the whole cell binding assay, the affinity (log KD value) of these antagonists was calculated in functional assays from the shift of the agonist concentration responses in the presence of a fixed concentration of antagonist using eq 4
| 4 |
where DR (dose ratio) is the ratio of the agonist concentration required to stimulate an identical response in the presence and absence of a fixed concentration of antagonist [B].
Several of the compounds, however, displaced clear partial agonist activities; that is, they antagonized the more efficacious agonist cimaterol while stimulating an agonist response of their own. When clear partial agonism was seen, the affinity (log KD) was calculated according to the method of Stephenson using eq 5:43
| 5 |
In eq 5 [P] is the concentration of the partial agonist, [A1] is the concentration of the agonist at the point where the partial agonist alone causes the same response, [A2] is the concentration of agonist causing a given response above that achieved by the partial agonist, and [A3] is the concentration of the agonist in the presence the partial agonist causing the same stimulation as [A2].
Isoprenaline (10 μM) was included in all experiments, and therefore all maximal responses are expressed as a percentage of this maximum.
(D) In Vivo Experiments
(1) Animals and Surgery
Adult male Sprague–Dawley rats (Charles River, Margate, Kent, UK), weighing 300–350 g, were housed in groups in a temperature-controlled (21–23 °C) environment with a 12 h light–dark cycle (lights on at 6:00 a.m.) and free access to food (Teklad Global 18% Protein Rodent Diet, Bicester, Oxon, U.K.) and water for at least 7 days after arrival from the supplier before any surgical intervention.
Surgery was performed in two stages under general anesthesia (fentanyl and medetomidine, 300 μg/kg of each ip, supplemented as required), with reversal of anesthesia and postoperative analgesia provided by atipamezole (1 mg/kg sc) and buprenorphine (0.02 mg/kg sc). At the first surgical stage, a miniature pulsed Doppler flow probe was sutured around the distal abdominal aorta to monitor hindquarters hemodynamics. The wires from the probe were taped and sutured at the nape of the neck, and the animals were returned to the holding room. At the second surgical stage, which took place at least 10 days after the surgery for probe implantation, and following a satisfactory inspection from the Named Veterinary Surgeon, catheters were implanted in the distal abdominal aorta via the caudal artery (for arterial blood pressure (BP) monitoring and the derivation of heart rate (HR)) and in the right jugular vein (for drug administration). Three separate intravenous catheters were placed in the jugular vein to enable concurrent administration of different substances. At this stage, the wires from the probe were soldered into a miniature plug (Microtech Inc., Boothwyn, PA, USA), which was mounted onto a custom-designed harness worn by the rat. The catheters emerged from the same point as the probe wires and were fed through a protective spring secured to the harness and attached to a counterbalanced pivot system. The arterial catheter was connected to a fluid-filled swivel for overnight infusion of heparinized (15 units/mL) saline to maintain patency.
Experiments began 24 h after surgery for catheter implantation, with animals fully conscious and unrestrained in home cages, with free access to food and water. All procedures were carried out with approval of the University of Nottingham Local Ethical Review Committee, under Home Office Project and Personal License Authority.
(2) Cardiovascular Recordings
Cardiovascular variables were recorded using a customized, computer-based system (Instrument Development Engineering Evaluation (IDEEQ), Maastrich Instruments Bv, The Netherlands) connected to a transducer amplifier (Gould, USA; model 13-4615-50) and a Doppler flowmeter (Crystal Biotech (Holliston, USA) VF-1 mainframe (pulse repetition frequency = 125 kHz) fitted with high-velocity (HVPD-20) modules). Raw data were sampled by IDEEQ every 2 ms, averaged, and stored to disc every cardiac cycle. Hindquarters vascular conductance (HVC) changes were calculated from the changes in BP and Doppler shift.
(3) Experimental Protocol
In all experiments, atropine methyl nitrate (1 mg/kg/h; 0.4 mL/h) was infused continuously to remove any parasympathetic influence on the control of heart rate. Starting 2 h after the onset of the atropine infusion, rats were given 3 min infusions (0.15 mL/min) of isoprenaline (12, 40, and 120 ng/kg/min) in ascending order separated by at least 20 min. At least 45 min after the last infusion of isoprenaline, saline or 19 was given as an iv bolus (0.1 mL) maintained by continuous infusion (0.4 mL/h), and the isoprenaline infusions were repeated, starting 30 min thereafter.
(4) In Vivo Data Analysis
Data were analyzed offline using IDEEQ software. Responses to isoprenaline were measured as the difference between steady-state values immediately before the isoprenaline infusion and during the third minute of infusion. Changes in baseline were measured as the difference between the control values for the last dose of isoprenaline before, and the first dose of isoprenaline after, saline or 19 administration. Data were analyzed by t test or ANOVA with Bonferroni correction as appropriate; P < 0.05 was taken as significant (GraphPad Prism version 5.02).
Acknowledgments
This work was supported by the BBSRC and the University of Nottingham. J.G.B. is a Wellcome Trust Clinician Scientist (Grant 073377/Z/.3/Z). We thank June McCulloch for technical assistance in running the cAMP chromatography columns and Philip Kemp and Julie March for technical assistance in running the in vivo experiments.
Glossary
Abbreviations Used
- aq
aqueous solution
- β-AR
beta-adrenoceptor
- Bn
benzyl
- Boc
tert-butoxycarbonyl
- Bz
benzoyl
- calcd
calculated
- cAMP
cyclic adenosine monophosphate
- CAN
ceric(IV) ammonium nitrate
- CGP 20712A
2-hydroxy-5-(2-[{hydroxy-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)propyl}amino]ethoxy)benzamide
- CHO
Chinese hamster ovary
- concd
concentrated
- COPD
chronic obstructive pulmonary disease
- cPr
cyclopropyl
- CRE-SPAP
cAMP response element-secreted placental alkaline phosphatase
- CV
column volume
- DCM
dichloromethane
- DIAD
diisopropyl azodicarboxylate
- DMF
N,N-dimethylformamide
- FCC
flash column chromatography
- FSK
forskolin
- GPCR
G protein-coupled receptor
- ICI 118551
(−)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol
- LABA
long-acting β-2 agonist
- lit.
literature
- LK 204-545
1-(2-(3-(2-cyano-4-(2-(cyclopropylmethoxy)ethoxy)phenoxy)-2-hydroxypropyl-amino)ethyl)-3-(4-hydroxyphenyl)urea
- m-CPBA
meta-chloroperbenzoic acid
- mp
melting point
- MW
microwave
- PE
petroleum ether 40–60
- Phth
phthalimide
- PLC
preparative layer chromatography
- PMB
para-methoxybenzyl
- rac
racemic
- RP-HPLC
reverse phase high-performance liquid chromatography
- rt
room temperature
- SAR
structure–activity relationship
- TEA
triethylamine
Supporting Information Available
Full experimental procedures and compound characterization data for compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ S.N.M. and J.G.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Louis S. N. S.; Nero T. L.; Iakovidis D.; Jackman G. P.; Louis W. J. LK 204-545, a highly selective β1-adrenoceptor antagonist at human β-adrenoceptors. Eur. J. Pharmacol. 1999, 367, 431–435. [DOI] [PubMed] [Google Scholar]
- Segal M. S.; Beakey J. F. The use of isuprel for the management of bronchial asthma. Bull. New Engl. Med. Cent. 1947, 9, 62–67. [PubMed] [Google Scholar]
- Lowell F. C.; Curry J. J.; Schiller I. W. A clinical and experimental study of isuprel in spontaneous and induced asthma. N. Engl. J. Med. 1948, 239, 45–51. [DOI] [PubMed] [Google Scholar]
- Black J. W.; Stephenson J. S. Pharmacology of a new adrenergic β-receptor-blocking compound (Nethalide). Lancet 1962, 2, 311–314. [DOI] [PubMed] [Google Scholar]
- Black J. W.; Duncan W. A.; Shanks R. G. Comparison of some properties of pronethalol and propranolol. Br. J. Pharmacol. Chemother. 1965, 25, 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. A life in new drug research. Br. J. Clin. Pharmacol. 2010, 70, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen T. R. Six-year follow-up of the Norwegian Multicenter Study on Timolol after Acute Myocardial Infarction. N. Engl. J. Med. 1985, 313, 1055–1058. [DOI] [PubMed] [Google Scholar]
- Cuculi F.; Radovanovic D.; Pedrazzini G.; Regli M.; Urban P.; Stauffer J. C.; Erne P. AMIS Plus Investigators: Is pretreatment with B-blockers beneficial in patients with acute coronary syndrome?. Cardiology 2010, 115, 91–97. [DOI] [PubMed] [Google Scholar]
- Waagstein F.; Hjalmarson A.; Varnauskas E.; Wallentin I. Effect of chronic β-adrenergic receptor blockade in congestive cardiomyopathy. Br. Heart J. 1975, 37, 1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg K.; Hjalmarson A.; Waagstein F.; Wallentin I. Prolongation of survival in congestive cardiomyopathy by β-receptor blockade. Lancet 1979, 1, 1374–1376. [DOI] [PubMed] [Google Scholar]
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999, 353, 9–13. [PubMed] [Google Scholar]
- Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in-congestive heart failure (MERIT-HF). Lancet 1999, 353, 2001–2007. [PubMed] [Google Scholar]
- Packer M.; Fowler M. B.; Roecker E. B.; Coats A. J. S.; Katus H. A.; Krum H.; Mohacsi P.; Rouleau J. L.; Tendera M.; Staiger C.; Holcslaw T. L.; Amann-Zalan I.; DeMets D. L. Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) study. Circulation 2002, 106, 2194–2199. [DOI] [PubMed] [Google Scholar]
- Wild D. M.; Kukin M. β-Blockers to prevent symptomatic heart failure in patients with stage A and B heart failure. Curr. Heart Fail. Rep. 2007, 4, 99–102. [DOI] [PubMed] [Google Scholar]
- Baker J. G.; Hill S. J.; Summers R. J. Evolution of β-blockers: from anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 2011, 32, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M.; Thomson N. C. (R)-Salbutamol in the treatment of asthma and chronic obstructive airways disease. Expert Opin. Pharmacother. 2011, 12, 1133–1141. [DOI] [PubMed] [Google Scholar]
- Smith C.; Teitler M. β-Blocker selectivity at cloned human β1- and β2-adrenergic receptors. Cardiovasc. Drugs Ther. 1999, 13, 123–126. [DOI] [PubMed] [Google Scholar]
- Schnabel P.; Maack C.; Mies F.; Tyroller S.; Scheer A.; Böhm M. Binding properties of β-blockers at recombinant β1-, β2-, and β3-adrenoceptors. J. Cardiovasc. Pharm. 2000, 36, 466–471. [DOI] [PubMed] [Google Scholar]
- Hoffmann C.; Leitz M. R.; Oberdorf-Maass S.; Lohse M. J.; Klotz K. N. Comparative pharmacology of human β-adrenergic receptor subtypes – characterization of stably transfected receptors in CHO cells. N. S. Arch. Pharmacol. 2004, 369, 151–159. [DOI] [PubMed] [Google Scholar]
- Baker J. G. The selectivity of β-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br. J. Pharmacol. 2005, 144, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G. The selectivity of β-adrenoceptor agonists at human β1-, β2- and β3-adrenoceptors. Br. J. Pharmacol. 2010, 160, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. P.; Beek D. Is prenalterol (H133/80) really a selective β1-adrenoceptor agonist? Tissue selectivity resulting from differences in stimulus-response relationships. J. Pharmacol. Exp. Ther. 1980, 213, 406–413. [PubMed] [Google Scholar]
- Berthold R.; Louis W. J. 3-Aminopropoxyphenyl derivatives, their preparation and pharmaceutical compositions containing them. Patent 4,661,513, 1987.
- Talukdar S.; Hsu J.-L.; Chou T.-C.; Fang J.-M. Direct transformation of aldehydes to nitriles using iodine in ammonia water. Tetrahedron Lett. 2001, 42, 1103–1105. [Google Scholar]
- Chen S.-W.; Kim J. H.; Song C. E.; Lee S.-G. Self-supported oligomeric Grubbs/Hoveyda-type Ru-carbene complexes for ring-closing metathesis. Org. Lett. 2007, 9, 3845–3848. [DOI] [PubMed] [Google Scholar]
- Nowak I.; Robins M. J. Synthesis of 3′-deoxynucleosides with 2-oxabicyclo[3.1.0]hexane sugar moieties: addition of difluorocarbene to a 3′,4′-unsaturated uridine derivative and 1,2-dihydrofurans derived from d- and l-xylose1. J. Org. Chem. 2007, 72, 3319–3325. [DOI] [PubMed] [Google Scholar]
- Okimoto M.; Chiba T. Electrochemical transformations of aldehydes into methyl carboxylates and nitriles. J. Org. Chem. 1988, 53, 218–219. [Google Scholar]
- McBriar M. D.; Guzik H.; Shapiro S.; Paruchova J.; Xu R.; Palani A.; Clader J. W.; Cox K.; Greenlee W. J.; Hawes B. E.; Kowalski T. J.; O’Neill K.; Spar B. D.; Weig B.; Weston D. J.; Farley C.; Cook J. Discovery of orally efficacious melanin-concentrating hormone receptor-1 antagonists as antiobesity agents. Synthesis, SAR, and biological evaluation of bicyclo[3.1.0]hexyl ureas. J. Med. Chem. 2006, 49, 2294–2310. [DOI] [PubMed] [Google Scholar]
- Brimble M. A.; Duncalf L. J.; Phythian S. J. Synthesis of the monomeric unit of γ-actinorhodin. J. Chem. Soc., Perkin Trans. 1 1997, 1399–1404. [Google Scholar]
- Hoefle M. L.; Hastings S. G.; Meyer R. F.; Corey R. M.; Holmes A.; Stratton C. D. Cardioselective β-adrenergic blocking agents. 1. 1-((3,4-Dimethoxyphenethyl)amino)-3-aryloxy-2-propanols. J. Med. Chem. 1975, 18, 148–152. [DOI] [PubMed] [Google Scholar]
- Crowther A. F.; Gilman D. J.; McLoughlin B. J.; Smith L. H.; Turner R. W.; Wood T. M. β-Adrenergic blocking agents V. 1-Amino-3-(substituted phenoxy)-2-propanols. J. Med. Chem. 1969, 12, 638–642. [DOI] [PubMed] [Google Scholar]
- Smith L. H. β-Adrenergic blocking agents 13. (3-Amino-2-hydroxypropoxy)benzamides. J. Med. Chem. 1976, 19, 1119–1123. [DOI] [PubMed] [Google Scholar]
- Smith L. H. β-Adrenergic blocking agents 15. 1-Substituted ureidophenoxy-3-amino-2-propanols. J. Med. Chem. 1977, 20, 705–708. [DOI] [PubMed] [Google Scholar]
- Smith L. H. β-Adrenergic blocking agents. 16. 1-(Acylaminomethyl-, ureidomethyl-, and ureidoethylphenoxy)-3-amino-2-propanols. J. Med. Chem. 1977, 20, 1254–1258. [DOI] [PubMed] [Google Scholar]
- Large M. S.; Smith L. H. β-Adrenergic blocking agents. 23. 1-[Substituted-amido)phenoxy]-3-[[(substituted-amido)alkyl]amino] propan-2-ols. J. Med. Chem. 1983, 26, 352–357. [DOI] [PubMed] [Google Scholar]
- Louis S.; Nero T.; Iakovidis D.; Colagrande F.; Jackman G.; Louis W. β1- and β2-adrenoceptor antagonist activity of a series of para-substituted N-isopropylphenoxypropanolamines. Eur. J. Med. Chem. 1999, 34, 919–937. [DOI] [PubMed] [Google Scholar]
- Chary K. P.; Laxmi Y. R. S.; Iyengar D. S. Reductive cleavage of acetals/ketals with ZrCl4/NaBH4. Synth. Commun. 1999, 29, 1257–1261. [Google Scholar]
- Machin P. J.; Hurst D. N.; Bradshaw R. M.; Blaber L. C.; Burden D. T.; Fryer A. D.; Melarange R. A.; Shivdasani C. β1-Selective adrenoceptor antagonists. 2. 4-Ether-linked phenoxypropanolamines. J. Med. Chem. 1983, 26, 1570–1576. [DOI] [PubMed] [Google Scholar]
- Jensen K. B.; Braxmeier T. M.; Demarcus M.; Frey J. G.; Kilburn J. D. Synthesis of guanidinium-derived receptor libraries and screening for selective peptide receptors in water. Chem.–Eur. J. 2002, 8, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Englund E. A.; Gopi H. N.; Appella D. H. An efficient synthesis of a probe for protein function: 2,3-diaminopropionic acid with orthogonal protecting groups. Org. Lett. 2004, 6, 213–215. [DOI] [PubMed] [Google Scholar]
- Lelais G.; Micuch P.; Josien-Lefebvre D.; Rossi F.; Seebach D. Preparation of protected β2- and β3-homocysteine, β2- and β3-homohistidine, and β2-homoserine for solid-phase syntheses. Helv. Chim. Acta 2004, 87, 3131–3159. [Google Scholar]
- Baker J. G. Site of action of β-ligands at the human β1-adrenoceptor. J. Pharmacol. Exp. Ther. 2005, 313, 1163–1171. [DOI] [PubMed] [Google Scholar]
- Stephenson R. P. A modification of receptor theory. Br. J. Pharmacol. Chemother. 1956, 11, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G.; Kemp P.; March J.; Fretwell L.; Hill S. J.; Gardiner S. M. Predicting in vivo cardiovascular properties of β-blockers from cellular assays: a quantitative comparison of cellular and cardiovascular pharmacological responses. FASEB J. 2011, 25, 4486–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arch J. R. S. Do low-affinity states of β-adrenoceptors have roles in physiology and medicine?. Br. J. Pharmacol. 2004, 143, 517–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G.; Hall I. P.; Hill S. J. Agonist actions of “β-blockers” provide evidence for two agonist activation sites or conformations of the human β1-adrenoceptor. Mol. Pharmacol. 2003, 63, 1312–1321. [DOI] [PubMed] [Google Scholar]
- Granneman J. G. The putative β4-adrenergic receptor is a novel state of the β1-adrenergic receptor. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E199–E202. [DOI] [PubMed] [Google Scholar]
- Kaumann A. J.; Molenaar P. The low-affinity site of the β1-adrenoceptor and its relevance to cardiovascular pharmacology. Pharmacol. Ther. 2008, 118, 303–336. [DOI] [PubMed] [Google Scholar]
- Baker J. G. Evidence for a secondary state of the human β3-adrenoceptor. Mol. Pharmacol. 2005, 68, 1645–1655. [DOI] [PubMed] [Google Scholar]
- A trial of the β-blocker bucindolol in patients with advanced chronic heart failure. N. Engl. J. Med. 2012, 344, 1659–1667. [DOI] [PubMed] [Google Scholar]
- Xamoterol in severe heart failure. The Xamoterol in Severe Heart Failure Study Group. Lancet 1990, 336, 1–6. [PubMed] [Google Scholar]
- Donaldson J.; Brown A. M.; Hill S. J. Influence of rolipram on the cyclic-3′,5′-adenosine monophosphate response to histamine and adenosine in slices of guinea-pig cerebral cortex. Biochem. Pharmacol. 1988, 37, 715–723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.