

Abstract
Multiple sclerosis (MS) is the most common autoimmune disease of the central nervous system (CNS) in the Western world. The disease is characterized histologically by the infiltration of encephalitogenic TH1/TH17-polarized CD4+ T cells, B cells, and a plethora of myeloid cells, resulting in severe demyelination ultimately leading to a degeneration of neuronal structures. These pathological processes are substantially modulated by microglia, the resident immune competent cells of the CNS. In this overview, we summarize the current knowledge regarding the highly diverse and complex function of microglia during CNS autoimmunity in either promoting tissue injury or tissue repair. Hence, understanding microglia involvement in MS offers new exciting paths for therapeutic intervention.
1. Multiple Sclerosis: The Most Frequent Autoimmune Disease of the CNS
An autoimmune disease is characterized by the loss of self-tolerance of the immune system, which can be caused by either genetic or environmental factors or a combination of both [1]. As a consequence of this malfunction, an immune response is initiated against certain cell types or even entire organs of the body. For the central nervous system (CNS) several autoimmune diseases are described of which multiple sclerosis (MS) is the most common form, affecting approximately 2.5 million people worldwide, mainly in the third and fourth decades of live. While the exact etiology of MS is still unknown, much progress has been made in understanding its pathology. MS comprises a blood-brain-barrier (BBB) disruption accompanied by an activation of macrophages/microglia as well as T- and B-cell infiltration into the CNS, ultimately resulting in demyelination and degeneration of neuronal structures [2]. MS can be clinically divided into different forms. Most patients experience relapsing-remitting stage (RRMS) of the disease, which in many cases results in continuous disease progression called secondary progressive MS (SPMS). On the other hand, some patients suffer from primarily progressive MS (PPMS), characterized by a continuously progressing disease course [2]. To date no cure for any form of MS exists, but several treatment options which might reduce the symptoms are available [3]. One of these approaches compromises the application of IFN-β, which is thought to be anti-inflammatory in RRMS and thereby reduces the relapse rate [3–5]. Unfortunately, even though IFN-β is well tolerated by patients, approximately 50% of them respond to and benefit from the treatment [5]. The effects of IFN-β are complex and far from being fully understood. Profound insights into the pathogenic mechanisms involved in MS as well as possible therapeutic interventions were gained through the use of experimental autoimmune encephalitis (EAE), the most used animal model for CNS autoimmunity [6, 7]. Several key features of MS, such as paralysis, weight loss, demyelination, and inflammation, observed in human patients, are recapitulated during EAE in rodents [7]. Depending on the strain, EAE can be induced by active immunization with myelin derived proteins such as myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), or proteolipid protein (PLP) in combination with an adjuvant, usually complete Freund's adjuvant (CFA) [7]. CFA contains inactivated mycobacteria and is thought to break peripheral tolerance, which results in the induction of CNS autoimmunity. CFA is recognized by pattern recognition receptors such as Toll-like receptors (TLRs). Especially, myeloid differentiation primary response gene (88) (MyD88), TLR7, and TLR9 have been found to be essential disease modifiers. In addition to these surface and endosomal receptors, newly discovered endosomal molecules such as retinoic acid inducible gene- (RIG-) I and melanoma differentiation-associated protein- (MDA-) 5 also have been shown to be crucial for EAE induction [8, 9]. Some of these disease modifying recognition receptors release type I interferons (IFNs) upon activation that in turn robustly change both the innate and adaptive arms of autoimmunity in mice [9–12]. However, for C57BL/6 mice the EAE model induced by MOG35-55 peptide is thought to be a monophasic chronically active disease without significant recovery and relapse phases, thereby only partially reflecting the clinical course found in MS patients [13]. Nevertheless, this EAE model in addition to the increasing availability of (cell type-)specific knock-out mice has greatly expanded our understanding of MS pathology and might open new avenues for specific treatment options in the future.
2. Microglia-Resident Macrophages of the CNS
Microglia cells are resident tissue macrophages located in the CNS and are considered to be a patrolling immune competent cell type within the parenchyma [14–17]. Microglia make up around 10% of the cells in CNS and are evenly distributed in the parenchyma of a healthy brain [14]. In contrast to neurons and macroglia (oligodendrocytes and astrocytes), microglia originate from the primitive hematopoiesis within the yolk sac (YS) and migrate to the neuronal tube during embryogenesis [17, 18]. Recently, we could identify lin−-erythromyeloid precursors as the genuine microglia progenitors on a stem cell level [19]. These pioneer cells continue their physiological and morphological development on their way from the YS to the developing brain, where the mature cells finally reside and build the ultimate pool of microglia [17, 19, 20]. By using parabiotic mice, a contribution of bone marrow-derived phagocytes (BMDPs) to the pool of existing microglia throughout life could be ruled out [20–22]. In fact, BMDP engraftment from the circulation can only be achieved after significant “priming” of the host CNS, for example, by irradiation-induced changes of the BBB or alterations of the tissue micromilieu [19, 23–25].
Due to their morphology, microglia cells are ascribed to be in a “resting” state under healthy conditions. This term is somewhat misleading, since in vivo imaging has revealed that microglia actively scavenge and monitor with their ramified branches the environment of the CNS for pathogens [26, 27]. Upon tissue damage, or inflammation, viral or bacterial insult, microglia change their morphology towards an amoeboid shape by retracting these branches. In addition to the morphological differentiation, several surface markers, such as F4/80 or Mac-1, which is typical for macrophages, are upregulated [28, 29]. The status of activation can be further subdivided into “classically” (M1) and “alternatively” (M2) activated microglia [15, 30] (Figure 1). This subdivision is based on function that while M1 microglia are often associated with acute infection, M2 cells play a role in tissue remodelling, repair, and healing [30]. However, M1 macrophages are also vital and important for the defence against microorganisms. The functional classification relies on the microenvironment of chemo- or cytokines as a result of microbial products or damaged cells [30]. IFN-γ and lipopolysaccharide (LPS) polarize microglia towards the M1 state and induce the release or expression of interleukin- (IL-) 1, IL-6, IL-12, IL-23, and inducible nitric oxide synthase (iNOS). On the other hand, the presence of IL-4, IL-10, and IL-13 turns microglia into M2 cells, which produce IL-10 and express arginase 1 [30, 31]. However, one has to keep in mind that this distinction into M1/M2 is a simplification and represents the extreme states. During disease both of these extremes as well as intermediate states may be present. However, the described subdivision of M1 and M2 cells nicely reflects the Janus-like behavior of microglia regarding their promotion of either tissue injury or repair. However, more and more evidence has accumulated that microglia do not only take part in immunological processes but also play a role in nonpathological conditions by, for example, eliminating or remodelling synapses and support of myelin turnover [32–34]. Among all the described functions of microglia, this review concentrates specifically on the involvement of microglia in autoimmune diseases of the CNS. In particular, we elucidate the mechanisms of microglia activation and the subsequent results of activation. Finally, we highlight current ideas to interfere in microglia activation as promising therapeutic strategies for MS.
Figure 1.
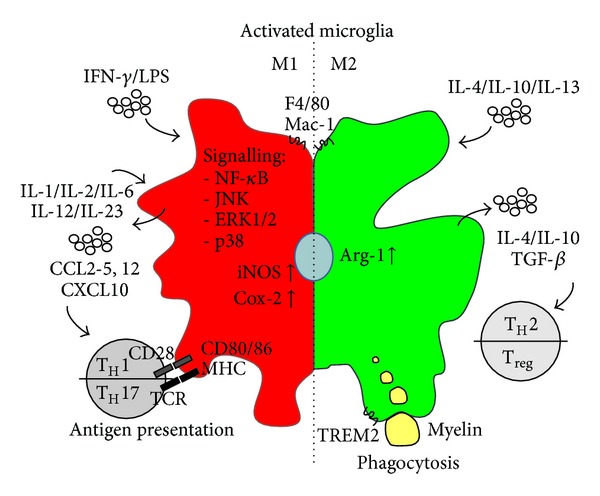
Polarization and function of activated microglia in CNS autoimmune inflammation. Microglia are activated by diverse stimuli, which define the polarization status of the cell. While IFN-γ/LPS promote the proinflammatory M1 status, IL-4/IL-10 or IL-13 induce the anti-inflammatory M2 status. M1 microglia take part in the attraction and differentiation of pathogenic TH1/TH17 T-cells, whereas M2 microglia promote phagocytosis of myelin debris, which is important for remyelination.
3. Function of Microglia in CNS Autoimmunity
3.1. Activation of Microglia as Hallmark of Disease
For several decades, the activation of microglia has been described in the damaged CNS during the pathology of MS/EAE, reflecting an initial event in MS pathology [35]. Even in early stages of MS, activated microglia clusters, so-called microglia nodules, are found in preactive lesions in the white matter of MS patients [36, 37]. In response of becoming activated, microglia also start to proliferate during MOG35-55-induced EAE [22]. In order to gain further insights into the involvement of activated microglia, a CD11b-HSVTK transgenic mouse line was generated [38]. In this mouse model the HSVTK gene is driven by the CD11b promoter, which is only expressed in cells with myeloid origin including microglia and macrophages. Importantly, HSVTK acts suicidal upon ganciclovir treatment. Thus, CD11b-HSVTK represents a pharmacogenetically inducible in vivo model of microglia depletion. MOG immunization of ganciclovir-treated CD11b-HSVTK mice significantly repressed disease onset and the severity of clinical EAE signs. The importance of microglia activation during EAE pathology was confirmed by application of macrophage inhibitory factor (TKP) or minocycline, which also attenuated EAE symptoms [39, 40]. Recently, the involvement of the complement system, which usually plays a part in defence and elimination of microorganisms in the adaptive immune response [41], in microglia activation in EAE was shown [42]. Under pathological conditions, like MS, a first disease-causing stimulus primes microglia, which upon a second stimulus result in overactivated microglia [42]. The presence of primed microglia was observed in MS lesions and in a knock-out of complement receptor-1-related protein y (Crry) [42]. During EAE primed microglia led to overactivated microglia resulting in an enhanced clinical severity in Crry-deficient mice [42]. Thus, microglia activation presents a histological hallmark in MS pathogenesis. However, during the disease, activated microglia might fill two positions depending on their polarization, M1 cells are involved in the pathogenic T-cell response (TH1 and TH17), while M2 microglia are designated as being protective in MS [43, 44]. Therefore, it is important to understand the processes and mechanisms of microglia activation and polarization.
Recently, miRNA124 (miR-124) was identified to be a key regulator and modulator of microglia/monocyte activation [29]. miR-124, which is highly expressed in ramified microglia, belongs to the family of noncoding microRNAs, which are known to have regulatory functions such as promoting mRNA degeneration or interfering with mRNA translation [29, 45]. Interestingly, while during onset and peak of EAE expression of the surface markers CD45 and MHC class II was upregulated in microglia, the expression of miR-124 was repressed [29]. miR-124 targets C/EBP-α and thereby decreases the expression of C/EBP-α and indirectly Pu.1, both important myeloid regulatory transcription factors responsible for expression of activation markers [29]. Finally, intravenous treatment of miR-124 in the preclinical phase as well as after onset of the disease inhibited or substantially ameliorated the clinical course of EAE. Importantly, application of miR-124 turns microglia towards an M2 polarization.
In addition, suppressor of cytokine signalling (SOCS-) 3 was identified as another key player of microglia activation [31]. SOCS proteins are known to interfere with JAK/STAT signalling. SOCS3 in particular inhibits IL-6 signalling by limiting STAT3 activation [46]. Induction of EAE in the absence of SOCS3 on myeloid cells, by using LysMCre-SOCS3fl/fl mice, resulted in an early onset and more severe disease course, with an overly activated STAT3/4 signalling [31]. Myeloid-specific SOCS3 deficiency polarized the microglia/monocytes towards the M1 state and induced neuronal death. Thus, SOCS3 limits activation of macrophages/microglia and controls their polarization status. Taken together, both, miR-124 as well as SOCS3, are important factors critical for microglia activation. The hereby activated microglia then take over several responsibilities depending on their polarization.
3.2. Microglia as a Source of Cytokines during Autoimmune Inflammation
Cytokines and chemokines are small secreted signalling molecules important for inter- and intracellular communication during inflammation. While both are mostly expressed at very low, basal levels in healthy conditions, the expression and secretion, primarily by microglia but also by astrocytes, are markedly increased upon CNS insult [47–50]. Microglial secretion of diverse cytokines can modulate cells in a paracrine manner but can also affect microglia in an autocrine fashion by either positive or negative feedback loops [47, 50]. Finally, the amount and composition of cytokines and chemokines in the microenvironment determines the function of microglia as indicated earlier in description of M1/M2 microglia polarization.
IFN-γ, tumour necrosis factor- (TNF-) α, IL-1β, and IL-6 are potent proinflammatory cytokines activating microglia and shifting them towards the cytotoxic M1 phenotype, thereby potentiating the inflammatory response [49–52] (Figure 1). However, to induce the complete cytotoxic M1 state, which leads to the expression of TNF-α, IL-1β, IL-2, IL-6, and IL-12, as well as iNOS and cyclooxygenase-2 (COX-2), binding of more than one cytokine is necessary [50]. Successful stimulation then triggers the expression of target genes via various signalling cascades including NF-κB, JNK, ERK1/2, and p38 [50, 53, 54]. Subsequently, constitutive activation of, for example, the NF-κB signalling cascade in microglia/macrophages in LysMCreIκBα fl/fl mice worsens clinical symptoms of EAE and increases the expression of, for example, IL-1β and IL-6 [55]. Interestingly, NF-κB signalling is not only important in microglia, but also plays an important role in neuroectodermal-derived astrocytes during autoimmune inflammation of the CNS [56, 57].
The roles of the proinflammatory cytokines IL-12, IL-23, and IL-17 produced by classically activated M1 microglia are more sophisticated [58–60] (Figure 1). Interestingly, IL-12 and IL-23 belong to the IL-12 family of cytokines and share a similar β-chain p40 but differ in their α-chain, which is p19 for IL-23 and p35 for IL-12 [60]. Their contribution to the pathogenesis of EAE lies in the regulation, proliferation, and differentiation of naïve CD4+ T cells. While IL-12 facilitates TH1 effector cell differentiation, IL-23 is critical for stable IL-17 expression which is ultimately important for the differentiation of pathogenic TH17 cells [50, 60, 61]. Although TH1 and TH17 both hold central and distinct roles in the development and pathogenesis in CNS autoimmune inflammation, their exact functions remain to be fully resolved [61]. In contrast, alternatively activated M2 microglia express and release the cytokines IL-4 and IL-10, as well as transforming growth factor- (TGF-) β through which the differentiation of anti-inflammatory and protective TH2 and T regulatory (Treg) cells is induced [61].
In addition to cytokines, microglia are also activated by and are able to release diverse chemoattractant chemokines, such as CCL2, CCL3, CCL4, CCL5, CXCL10, and/or CCL12 [47, 48, 52, 62, 63] (Figure 1). The importance of CCL2, also known as monocyte chemoattractant protein- (MCP-) 1, and its receptor CCR2 has been investigated for various CNS pathologies such as neurodegeneration [23] and autoimmune inflammation [63–65]. Many different proinflammatory stimuli, such as IFN-γ, TNF-α, and IL-1β, can induce the expression of CCL2 in the brain [63]. Until now, two major functions have been allocated to CCL2 during CNS autoimmunity. First, it participates in the disruption of blood brain barrier integrity. Secondly it is involved in the recruitment of CCR2+CD11b+Ly-6Chi mononuclear leukocytes into the CNS [63, 64, 66]. Consequently, deletion or blockade of CCL2 synthesis by the chemical inhibitors significantly reduced the clinical symptoms of EAE [67–70]. In addition, therapeutic interference with the function of CCL5/Rantes reduced leukocyte activation and trafficking to the CNS [68]. In summary, by arranging the cyto-/chemokine milieu, microglia play a key role both in regulating and recruiting leukocytes into the CNS as well as macrophage polarization during autoimmune inflammation [71].
3.3. Phagocytosis Mediated by Microglia: Pathfinding for Reengineering
Phagocytosis can be seen as a double-edged sword; on the one hand, it is beneficial by clearing cellular debris, but on other the hand it can also be destructive by inducing an oxidative burst [72]. Microglia, as macrophages, are per definition phagocytes of either invading pathogens or dead cells in the CNS [72, 73]. During EAE, activated microglia are known to phagocytize myelin debris and oligodendrocytes in lesions, which in turn leads to the release of proinflammatory cytokines [71, 73] (Figure 1). One of the key receptors involved in this process during EAE is the triggering receptor expressed on myeloid cells (TREM-) 2 together with its associated signalling molecule DNAX-activating protein- (DAP-) 12, both expressed in microglia and macrophages [74, 75]. Virus-mediated overexpression of TREM2 in myeloid cells increased the clearance of myelin debris, thereby improving the tissue regeneration and consequently reducing the severity of clinical symptoms [75]. In addition to TREM2, several other receptors are important for myelin debris phagocytosis, such as complement receptor (CR) 3, signal regulatory protein (SIRP)-α, or Fcγ receptor [72].
Interestingly, remaining myelin debris inhibits the regenerative remyelination, indicated by a loss of PDGFRα+ oligodendrocyte precursor recruitment [76, 77]. A detailed analysis, making use of gene arrays, could determine a “single remyelination-supportive microglia” phenotype, which gradually develops [78]. Additionally, during remyelination microglia express several genes, which are known to promote oligodendrocyte differentiation [78]. Thus, efficient resolution of inflammation, by phagocytosis of myelin debris, is important for tissue reengineering.
3.4. Antigen Presentation and T Cell Priming by Microglia
MS/EAE pathology is characterized by harmful T cell infiltration [5]. For T cell activation by an antigen presenting cell (APC), two signals are prerequisites: first, antigen peptide presentation at the cell surface via major histocompatibility complex (MHC) to the specific T cell receptor [79, 80] and, second, a costimulatory interaction of CD80/86 or CD40 located on APCs with CD28 or CD40L present on T cells [71, 81]. Typically, dendritic cells represent the most prominent cell type for antigen presentation [82]. However, in addition to T cell attraction, by expressing CCL2 and CCL5, microglia can also serve as APCs by presenting myelin [83]. During MS/EAE pathology, microglia highly express MHC class I and II proteins as well as CD40 and CD80/86 [48, 84] (Figure 1). Microglia “reprime” or reactivate T cells in lesion sites and thereby exacerbate the disease by epitope spreading [71, 85, 86]. Following the T cell-APC interaction, differentiation into mature proinflammatory encephalitogenic T cells (TH17, TH1) or anti-inflammatory T cells (TH2 or Treg) takes place, depending on the cytokine environment [71].
3.5. Microglia as Therapeutic Targets
With the increased understanding of microglia function in MS/EAE pathology gained thus far, it might be possible to find and establish efficient therapies by targeting microglial responses. Since M1 microglia seem to be harmful during CNS autoimmune inflammation, most therapeutic ideas aim to either deactivate M1 microglia or turn them into M2 cells. Recently, galectin- (gal-) 1 was identified as a potent microglia regulator [87]. Gal-1 preferentially binds to M1 microglia, thereby modulating M1 key features, such as CCL2 and iNOS expression, by controlling NF-κB and p38 signalling [87]. Finally, gal-1 application ameliorated disease course of EAE, making it a potentially attractive therapeutic option [87]. Similar approaches were employed using tuftsin, a naturally occurring tetrapeptide known to induce phagocytosis, or ghrelin, which acts as an anti-inflammatory hormone [88, 89]. Treatment with tuftsin or ghrelin shifted microglia into M2 polarization in vitro and reduced EAE severity in vivo [88, 89]. In addition, application of compound A, a plant-derived phenyl aziridine precursor, ameliorated the clinical course of EAE, and subsequent in vitro analyses revealed an inhibition of NF-κB signalling by compound A in microglia [90]. β-Lapachone treatment, a natural substance from the bark of the lapacho tree, significantly reduced the expression of IL-12 family cytokines in microglia and thereby suppressed clinical severity [91].
During the termination of the acute inflammatory reaction, microglia become deactivated [15]. Therefore, the deactivation of microglia also presents a promising therapeutic concept. Several molecules, including chemokines and steroid hormones, are known as ligands regulating microglia activation status and mediate signalling via different nuclear receptors [15]. In this respect, stimulation of the oestrogen receptor- (ER-) β with synthetic ligands efficiently turned down microglia activation and thereby reduced EAE symptoms [92, 93].
Recently, the use of reprogrammed embryonic stem (ES) cell-derived microglia-like cells (ESdM) for MS was suggested [94]. Intravenously applied ESdM populate lesion sites in the spinal cord of EAE mice but do not influence the clinical course [94]. Application of lentiviral transduced ESdM with NT-3, a neurotrophic factor, significantly reduced clinical disease severity as well as the degree of demyelination and axonal damage [94].
4. Conclusion
Activation of microglia is a hallmark of MS/EAE pathology. During recent years more and more knowledge about how and why microglia become activated has accumulated. Subsequently, several investigations have already proven the feasibility of beneficially modulating microglia function in animal models of MS. Nevertheless, this path has to be taken further. There is a need for comprehensive and detailed analysis to further illuminate the triggering and signalling mechanisms of microglia in inflammation. Thoroughly understanding microglia action in the context of a disease will help us identify new targets for therapeutic approaches, which may ultimately be translated to the clinics.
Acknowledgments
The authors would like to thank Dr. Stefanie Brendecke and Dr. Katrin Kierdorf for critical reading and editing. M. Prinz is supported by the DFG-funded research unit (FOR) 1336, the BMBF-funded competence network of multiple sclerosis (KKNMS), the competence network of neurodegenerative disorders (KNDD), and the DFG (SFB 620, PR 577/8-2).
References
- 1.Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. Journal of Autoimmunity. 2009;33(1):3–11. doi: 10.1016/j.jaut.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature Reviews Neurology. 2012;8(11):647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- 3.Luessi F, Siffrin V, Zipp F. Neurodegeneration in multiple sclerosis: novel treatment strategies. Expert Reviews. 2012;12(9):1061–1077. doi: 10.1586/ern.12.59. [DOI] [PubMed] [Google Scholar]
- 4.Brendecke SM, Prinz M. How type I interferons shape myeloid cell function in CNS autoimmunity. Journal of Leukocyte Biology. 2012;92(3):479–488. doi: 10.1189/jlb.0112043. [DOI] [PubMed] [Google Scholar]
- 5.Axtell RC, Raman C. Janus-like effects of type I interferon in autoimmune diseases. Immunological Reviews. 248:23–35. doi: 10.1111/j.1600-065X.2012.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nature Neuroscience. 2012;15(8):1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Z, Tsirka SE. Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurology Research International. 2011;2011:1–9. doi: 10.1155/2011/383087.383087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuerten S, Angelov DN. Comparing the CNS morphology and immunobiology of different EAE models in C57BL/6 mice—a step towards understanding the complexity of multiple sclerosis. Annals of Anatomy. 2008;190(1):1–15. doi: 10.1016/j.aanat.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Dann A, Poeck H, Croxford AL, et al. Cytosolic RIG-I-like helicases act as negative regulators of sterile inflammation in the CNS. Nature Neuroscience. 2012;15(1):98–106. doi: 10.1038/nn.2964. [DOI] [PubMed] [Google Scholar]
- 10.Prinz M, Schmidt H, Mildner A, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells Limit autoimmunity in the central nervous system. Immunity. 2008;28(5):675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Prinz M, Kalinke U. New lessons about old molecules: how type I interferons shape Th1/Th17-mediated autoimmunity in the CNS. Trends in Molecular Medicine. 2010;16(8):379–386. doi: 10.1016/j.molmed.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Teige I, Liu Y, Issazadeh-Navikas S. IFN-β inhibits T cell activation capacity of central nervous system APCs. The Journal of Immunology. 2006;177(6):3542–3553. doi: 10.4049/jimmunol.177.6.3542. [DOI] [PubMed] [Google Scholar]
- 13.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. The Journal of Clinical Investigation. 2008;118(5):1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 15.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 16.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulz C, Perdiguero EG, Chorro L, et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science. 2012;335(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 19.Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu. 1- and Irf8-dependent pathways. Nature Neuroscience. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 20.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nature Neuroscience. 2011;14(10):1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 21.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 22.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature Neuroscience. 2011;14(9):1142–1150. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 23.Mildner A, Schlevogt B, Kierdorf K, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. The Journal of Neuroscience. 2011;31(31):11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mildner A, Schmidt H, Nitsche M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nature Neuroscience. 2007;10(12):1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 25.Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 26.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 27.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 28.Djukic M, Mildner A, Schmidt H, et al. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain. 2006;129(9):2394–2403. doi: 10.1093/brain/awl206. [DOI] [PubMed] [Google Scholar]
- 29.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nature Medicine. 2011;17(1):64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. The Journal of Pathology. 2013;229(2):176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 31.Qin H, Yeh W-I, De Sarno P, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(13):5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tremblay MÈ. The role of microglia at synapses in the healthy CNS: novel insights from recent imaging studies. Neuron Glia Biology. 2011;7(1):67–76. doi: 10.1017/S1740925X12000038. [DOI] [PubMed] [Google Scholar]
- 33.Blank T, Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia. 2013;61(1):62–70. doi: 10.1002/glia.22372. [DOI] [PubMed] [Google Scholar]
- 34.Fitzner D, Schnaars M, Van Rossum D, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. Journal of Cell Science. 2011;124(3):447–458. doi: 10.1242/jcs.074088. [DOI] [PubMed] [Google Scholar]
- 35.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. Journal of Molecular Medicine. 1997;75(3):165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 36.Van Der Valk P, Amor S. Preactive lesions in multiple sclerosis. Current Opinion in Neurology. 2009;22(3):207–213. doi: 10.1097/WCO.0b013e32832b4c76. [DOI] [PubMed] [Google Scholar]
- 37.Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathologica. 2013;125(4):595–608. doi: 10.1007/s00401-013-1082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nature Medicine. 2005;11(2):146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 39.Popovic N, Schubart A, Goetz BD, Zhang S-C, Linington C, Duncan D. Inhibition of autoimmune encephalomyelitis by a tetracycline. Annals of Neurology. 2002;51(2):215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- 40.Bhasin M, Wu M, Tsirka SE. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunology. 2007;8, article 10 doi: 10.1186/1471-2172-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nature Reviews Immunology. 2009;9(10):729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 42.Ramaglia V, Hughes TR, Donev RM, et al. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(3):965–970. doi: 10.1073/pnas.1111924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. The Journal of Neuroscience. 2007;27(40):10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature Immunology. 2011;12(3):231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 45.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nature Reviews Immunology. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 47.Hanisch U-K. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 48.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. The Journal of Immunology. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 49.Schmitz T, Chew L-J. Cytokines and myelination in the central nervous system. The Scientific World Journal. 2008;8:1119–1147. doi: 10.1100/tsw.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merson TD, Binder MD, Kilpatrick TJ. Role of cytokines as mediators and regulators of microglial activity in inflammatory demyelination of the CNS. NeuroMolecular Medicine. 2010;12(2):99–132. doi: 10.1007/s12017-010-8112-z. [DOI] [PubMed] [Google Scholar]
- 51.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Science Signaling. 2010;3(105) doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 52.Juedes AE, Hjelmström P, Bergman CM, Neild AL, Ruddle NH. Kinetics and cellular origin of cytokines in the central nervous system: insight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. The Journal of Immunology. 2000;164(1):419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- 53.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, et al. Transcription factor NF-κB is activated in microglia during experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 1994;55(1):99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 54.Waetzig V, Czeloth K, Hidding U, et al. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50(3):235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- 55.Ellrichmann G, Thöne J, Lee D-H, Rupec RA, Gold R, Linker RA. Constitutive activity of NF-κB in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. Journal of Neuroinflammation. 2012;9, article 15 doi: 10.1186/1742-2094-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Loo G, De Lorenzi R, Schmidt H, et al. Inhibition of transcription factor NF-κB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nature Immunology. 2006;7(9):954–961. doi: 10.1038/ni1372. [DOI] [PubMed] [Google Scholar]
- 57.Raasch J, Zeller N, Van Loo G, et al. IκB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-κB in the central nervous system. Brain. 2011;134(4):1184–1198. doi: 10.1093/brain/awq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. The Journal of Experimental Medicine. 2003;198(12):1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. The Journal of Experimental Medicine. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nature Immunology. 2012;13(8):722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KHG. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clinical and Experimental Immunology. 2010;162(1):1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-γ but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55(16):1728–1739. doi: 10.1002/glia.20587. [DOI] [PubMed] [Google Scholar]
- 63.Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. Journal of Cerebral Blood Flow and Metabolism. 2010;30(3):459–473. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mildner A, MacK M, Schmidt H, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132(9):2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- 65.Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. Journal of Neuroimmunology. 2010;224(1-2):80–84. doi: 10.1016/j.jneuroim.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 66.King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113(14):3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang D, Tani M, Wang J, et al. Pertussis toxin-induced reversible encephalopathy dependent on monocyte chemoattractant protein-1 overexpression in mice. The Journal of Neuroscience. 2002;22(24):10633–10642. doi: 10.1523/JNEUROSCI.22-24-10633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dos Santos AC, Barsante MM, Esteves Arantes RM, Bernard CCA, Teixeira MM, Carvalho-Tavares J. CCL2 and CCL5 mediate leukocyte adhesion in experimental autoimmune encephalomyelitis—an intravital microscopy study. Journal of Neuroimmunology. 2005;162(1-2):122–129. doi: 10.1016/j.jneuroim.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 69.Ge S, Shrestha B, Paul D, et al. The CCL2 synthesis inhibitor bindarit targets cells of the neurovascular unit, and suppresses experimental autoimmune encephalomyelitis. Journal of Neuroinflammation. 2012;9(1):p. 171. doi: 10.1186/1742-2094-9-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo X, Nakamura K, Kohyama K, et al. Inhibition of glial cell activation ameliorates the severity of experimental autoimmune encephalomyelitis. Neuroscience Research. 2007;59(4):457–466. doi: 10.1016/j.neures.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 71.Chastain EML, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochimica et Biophysica Acta. 2011;1812(2):265–274. doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Frontiers in Cellular Neuroscience. 2013;7:p. 6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Napoli I, Neumann H. Protective effects of microglia in multiple sclerosis. Experimental Neurology. 2010;225(1):24–28. doi: 10.1016/j.expneurol.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 74.Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. The Journal of Experimental Medicine. 2005;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Medicine. 2007;4(4):675–689. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kotter MR, Li W-W, Zhao C, Franklin RJM. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. The Journal of Neuroscience. 2006;26(1):328–332. doi: 10.1523/JNEUROSCI.2615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132(2):288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olah M, Amor S, Brouwer N, et al. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60(2):306–321. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- 79.Vyas JM, Van Der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nature Reviews Immunology. 2008;8(8):607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class i and MHC class II antigen presentation. Nature Reviews Immunology. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 81.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nature Reviews Immunology. 2003;3(12):939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 82.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nature Reviews Immunology. 2007;7(7):543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 83.Mack CL, Vanderlugt-Castaneda CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. Journal of Neuroimmunology. 2003;144(1-2):68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 84.Satoh J, Lee YB, Kim SU. T-cell costimulatory molecules B7-1 (CD80) and B7-2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Research. 1995;704(1):92–96. doi: 10.1016/0006-8993(95)01177-3. [DOI] [PubMed] [Google Scholar]
- 85.Tompkins SM, Padilla J, Dal Canto MC, Ting JP-Y, Van Kaer L, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. The Journal of Immunology. 2002;168(8):4173–4183. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- 86.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nature Medicine. 2005;11(3):335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 87.Starossom SC, Mascanfroni ID, Imitola J, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37(2):249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Theil M-M, Miyake S, Mizuno M, et al. Suppression of experimental autoimmune encephalomyelitis by ghrelin. The Journal of Immunology. 2009;183(4):2859–2866. doi: 10.4049/jimmunol.0803362. [DOI] [PubMed] [Google Scholar]
- 89.Wu M, Nissen JC, Chen EI, Tsirka SE. Tuftsin promotes an anti-inflammatory switch and attenuates symptoms in experimental autoimmune encephalomyelitis. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034933.e34933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Van Loo G, Sze M, Bougarne N, et al. Antiinflammatory properties of a plant-derived nonsteroidal, dissociated glucocorticoid receptor modulator in experimental autoimmune encephalomyelitis. Molecular Endocrinology. 2010;24(2):310–322. doi: 10.1210/me.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu J, Wagoner G, Douglas JC, Drew PD. β-Lapachone ameliorization of experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 2013;254(1-2):46–54. doi: 10.1016/j.jneuroim.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145(4):584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JÅ. Targeting estrogen receptor β in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):3543–3548. doi: 10.1073/pnas.1300313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beutner C, Lepperhof V, Dann A, et al. Engineered stem cell-derived microglia as therapeutic vehicle for experimental autoimmune encephalomyelitis. Gene Therapy. 2013;2013 doi: 10.1038/gt.2012.100. [DOI] [PubMed] [Google Scholar]