

Abstract
The central nervous system (CNS) is immune privileged with access to leukocytes being limited. In several neurological diseases, however, infiltration of immune cells from the periphery into the CNS is largely observed and accounts for the increased representation of macrophages within the CNS. In addition to extensive leukocyte infiltration, the activation of microglia is frequently observed. The functions of activated macrophages/microglia within the CNS are complex. In three animal models of multiple sclerosis (MS), namely, experimental autoimmune encephalomyelitis (EAE) and cuprizone- and lysolecithin-induced demyelination, there have been many reported detrimental roles associated with the involvement of macrophages and microglia. Such detriments include toxicity to neurons and oligodendrocyte precursor cells, release of proteases, release of inflammatory cytokines and free radicals, and recruitment and reactivation of T lymphocytes in the CNS. Many studies, however, have also reported beneficial roles of macrophages/microglia, including axon regenerative roles, assistance in promoting remyelination, clearance of inhibitory myelin debris, and the release of neurotrophic factors. This review will discuss the evidence supporting the detrimental and beneficial aspects of macrophages/microglia in models of MS, provide a discussion of the mechanisms underlying the dichotomous roles, and describe a few therapies in clinical use in MS that impinge on the activity of macrophages/microglia.
1. Introduction
The central nervous system (CNS), consisting of the brain and spinal cord, is immune-privileged with access to leukocytes being limited. In several neurological diseases including multiple sclerosis (MS), however, significant infiltration of immune cells from the periphery into the CNS is observed. Demyelination and axonal degeneration are common consequences of CNS inflammation [1]. In addition to extensive accumulation of macrophages, the activation of microglia, the phagocytic cells of the CNS, is a common occurrence following neurological injury [2–6]. This review will discuss the roles of macrophages and microglia as evidenced in the common immune-mediated animal model of MS, experimental autoimmune encephalomyelitis (EAE), as well as in the two prominent demyelinating models of MS, cuprizone and lysolecithin injury.
2. Microglia and Macrophages
Microglia and bone marrow-derived macrophages are two genetically distinct myeloid populations [7, 8]. Microglia are the resident immune cells of the CNS and originate from erythromyeloid precursors in the embryonic yolk sac. In early gestation, these precursor cells differentiate into microglia and invade the developing neural tube [7, 9, 10]. In contrast, macrophages are derived from hematopoietic stem cells in the bone marrow. These cells differentiate into blood monocytes which circulate the peripheral vasculature and populate tissues such as the liver, lungs, and nonparenchymal areas of the CNS, including the meninges, choroid plexus, and perivascular space [11, 12]. In the healthy CNS, resting microglia are characterized by many ramified processes, surveying the parenchyma for any possible threats to neurons and macroglia. Under physiological conditions, bone marrow-derived monocytes do not contribute to the local microglia pool [13, 14]. These observations suggest that microglia are sustained by local progenitors. Upon CNS injury, these cells become activated and take on an amoeboid shape, characterized by retracted processes. It is during this state of the CNS when bone marrow-derived macrophages also infiltrate the CNS and accumulate at the injury site, contributing to both further damage and tissue repair [11]. Macrophages within CNS lesion sites are difficult to distinguish from activated microglia, as both are amoeboid-shaped and express many of the same antigenic markers [15]. Due to difficulty in distinguishing these phagocytic cells, many authors refer to these cells collectively as macrophages/microglia.
Although there seems to be a spectrum of different types of macrophages/microglia, there are two main phenotypes that occur prominently in inflammatory lesions. These phenotypes are the classically activated “M1” cells and the alternatively activated “M2” cells [16, 17]. The following discussion is a simplified description as a more sophisticated discussion of these different subsets is beyond the scope of this review (refer to [2, 18]). The M1 macrophages/microglia are generally considered proinflammatory, as they are associated with the secretion of many proinflammatory cytokines including interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α). M1 cells also express the cell surface markers CD86 and CD16/32 and have inducible nitric oxide synthase (iNOS) activity [11]. The M2 macrophages/microglia on the other hand are anti-inflammatory in nature, as they are associated with the secretion of anti-inflammatory cytokines, such as IL-10. This subclass of macrophages/microglia can be identified by the expression of the mannose receptor (CD206) as well as the enzyme arginase 1 (Arg1). Most cells in the injured spinal cord were found to be of the M1 subset particularly at early stages of injury [19].
3. Roles of Macrophages/Microglia in CNS Injury
In the CNS of patients with Alzheimer's disease and MS patients, as well as in their models, a substantial infiltration of macrophages as well as an activation of resident microglia is prominent. This infiltration has been associated with disease severity and represents the predominant immune cell type present in MS autopsy studies [20, 21]. In addition, targeted depletion of CD95 ligand (CD95L), a protein thought to be important in the survival and migration of these cells into the injured CNS, reduced the representation of macrophages in an experimental rodent spinal cord injury model, correlating with enhanced locomotor recovery [22]. Furthermore, treatment with minocycline, an antibiotic that has been thought to reduce the activation of macrophages/microglia and the expression of inflammatory cytokines and matrix metalloproteinases (MMPs), reduced the amount of phagocytic cells in the lesion site, leading to a greater neurological recovery [23–25]. Popovich et al. [26] found that the depletion of macrophages using clodronate liposomes improved neural and motor recovery after experimental spinal cord injury. In addition to these detrimental effects observed in vivo, Kigerl et al. [19] showed the ability of M1-polarized macrophages to induce neuronal death. The mechanisms by which activated microglia kill neurons have been amply summarized [27] and include the elaboration of free radicals, proteases, and glutamate.
Despite these observations of detrimental roles of macrophages/microglia within the injured CNS, many studies have also reported beneficial roles of these innate immune cells. For instance, transplantation of macrophages into the injured CNS promoted the survival of neurons, functional recovery, and nerve regeneration [28]. In the study reporting enhanced nerve regeneration, activated macrophages in the crushed optic nerve were found to secrete a calcium-binding protein, called oncomodulin, which promoted neuron survival and axonal regeneration [29]. Macrophages/microglia have been demonstrated to produce other prorepair molecules, including brain-derived neurotropic factor (BDNF), IL10, and ferritin [30–33]. The latter molecule has been demonstrated to increase the proliferation and differentiation of oligodendrocyte precursor cells (OPCs). In mice expressing a mutated HSV-1 thymidine kinase (TK) gene controlled by the myeloid-specific CD11b promoter, administration of ganciclovir depletes cells of myeloid origin. In a study employing this technique, Barrette et al. [34] was able to show decreased axonal regeneration, increased myelin debris, and severely compromised locomotor recovery in CD11b-TK mice administered ganciclovir. This study demonstrates the reparative role that macrophages play in nerve injury. In mice deficient of CCR2, a receptor involved in recruiting macrophages to the lesion site, accelerated progression of Alzheimer's-like disease was demonstrated, with increasing cognitive impairments and amyloid deposition becoming evident [35, 36]. Another study depleted microglia and subsequently observed the formation and maintenance of amyloid plaques, suggesting that plaques can form independent of microglia [37]. In another study of Alzheimer's-like disease, CD11b-TK mice were again used to show that macrophages play an important role in clearing amyloid deposits [38].
From these studies, it is clear that macrophages/microglia can be both detrimental and beneficial in many different diseases affecting the CNS (Figure 1). To further highlight the dichotomy present in the roles of macrophages/microglia, the remainder of this review will discuss the functions of these cells in three animal models of MS. The reader should bear in mind that although these animal models have yielded a wealth of information concerning the biology of multiple sclerosis, such models represent an artificial mechanism of disease induction.
Figure 1.
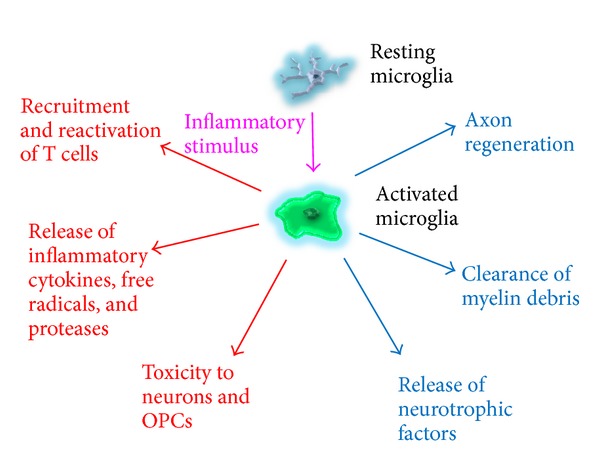
The dichotomy of macrophages/microglia is displayed. Macrophages/microglia have been shown to recruit and reactivate T cells in the CNS and release many detrimental molecules such as proteases, inflammatory cytokines, and free radicals. Through the latter molecules and other mechanisms, macrophages/microglia have been reported to contribute to toxicity to neurons as well as oligodendrocyte precursor cells. Conversely, they have also been observed to aid in axonal regeneration and remyelination as well as assist in the clearance of inhibitory myelin debris. In addition, macrophages/microglia have been shown to release a variety of neurotrophic factors. It can therefore be seen that macrophages/microglia possess an array of detrimental and beneficial functions, with the balance being dictated by the temporal and spatial specifications following CNS injury.
4. MS and Its Inflammatory Model, EAE
MS is a chronic, inflammatory neurodegenerative disease characterized by demyelination and remyelination in the majority of patients in the early phase of the disease. Clinical relapses are thought to be due to multifocal infiltration of immune cells, leading to loss of oligodendrocytes, demyelination, and axonal injury/loss. After a course of the relapsing-remitting part of the disease, most patients enter a phase of the disease characterized by progressive neurodegeneration, manifesting in irreversible disability in MS patients [39]. Demyelinating lesions are often found in the white matter of the brain stem, spinal cord, optic nerve, and cerebellum [39, 40]. Activated microglia and macrophages are frequently observed in active MS plaques [1, 20, 21, 41].
A common animal model of MS is EAE. The disease is induced by immunization with myelin components, including myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), and proteolipid protein (PLP) [42]. Most studies are carried out with mice immunized with a 35 to 55 residue MOG peptide emulsified in Freund's adjuvant supplemented with Mycobacterium tuberculosis extract. Mice may then be injected with pertussis toxin on the day of immunization and then two days later [43]. Along with EAE lesions resembling plaques in MS autopsies, EAE is advantageous in that its myelin-reactive CD4+ T-cell inflammation provides an ample platform for studying the T-cell inflammatory components of MS [43].
Substantial evidence exists for the involvement of macrophages/microglia in EAE. However, as mentioned in other animal models of CNS injury, there is a clear dichotomy in the roles of these phagocytic cells in EAE.
5. Detriments and Benefits of Macrophages/Microglia in EAE
The majority of studies demonstrating the detrimental or beneficial involvement of macrophages/microglia in EAE used loss-of-function approaches, in which macrophages/microglia were inhibited or depleted and the subsequent histopathology and behavioural symptoms monitored. For example, in a parabiosis study in which hematopoietic cells were ablated in the recipients but not the donor animals, it was shown that EAE progression was correlated with macrophage infiltration in recipient mice [14]. In other studies, inhibitors to macrophages or their depletion resulted in an attenuation of EAE progression [44–46]. Heppner et al. [47] used CD11b-HSVTK transgenic mice with systemic administration of ganciclovir to reduce macrophage/microglial density. In this study, it was found that ganciclovir administration corresponded to a suppression in the development of EAE, suggesting that macrophage/microglia activation is detrimental in EAE. Agrawal et al. [48] showed that ablation of both matrix metalloproteinase-2 (MMP-2) and MMP-9, which reduced leukocyte penetration across the BBB, suppressed EAE. In addition, Bartholomäus et al. [49] reported the reactivation of CNS-infiltrating pathogenic T cells by MHC class II-presenting macrophages in EAE. In another study, Nikić et al. [50] observed mitochondrial pathology and focal axonal degeneration being initiated by the macrophage-mediated production of reactive oxygen and nitrogen species (ROS and RNS). A regression in axonal degeneration was noticed when this group neutralized the ROS and RNS. In another study providing evidence for a detrimental role of microglia, Rasmussen et al. [51] found that the activation of these cells correlated with degradation of synaptic proteins as well as with atypical phosphorylation of neurofilaments in the cerebral cortex. Another study deleted galectin-1, a critical deactivator of M1 microglia activation secreted by astrocytes, and observed pronounced inflammation-induced neurodegeneration, demonstrating the detrimental effects of M1 polarized microglia [52].
In contrast to these studies reporting detrimental roles of macrophages/microglia in EAE, three studies demonstrated beneficial roles for these cells, particularly those of the M2 variety. In this regard, the transfer of M2-polarized monocytes resulted in a suppression of EAE [53, 54]. Finally, Denney et al. [55] reported that activation of invariant NKT cells increased the proportion of M2 macrophages in the CNS of EAE mice and this led to the attenuation of clinical signs.
In these studies, investigators employed gain-of-function or loss-of-function approaches to determine the roles that macrophages/microglia perform in EAE. When macrophages of the M2 subset were present, EAE severity was dampened, demonstrating a beneficial role for these cells. However, when macrophage activity was depleted, an improvement in EAE resulted, implying a detrimental role for these cells. To further study the role of macrophages/microglia in the processes of demyelination and remyelination, other animal models of MS must be used. In EAE, demyelination and remyelination are difficult to quantitate as the severity, lesion location, and temporal specifications of the pathophysiology vary from animal to animal. Models which can reproducibly induce a focal demyelinating lesion in a white matter tract would offer a significantly better approach to studying the effect that these cells have on the de-/remyelination process. In addition, the latter demyelinating models normally do not have extensive T-cell representation, allowing effects of drugs on macrophages/microglia to be determined. The reproducibility of these models allows for the examination of the effect that loss-of-function, gain-of-function, or pharmacological treatment experiments have on the demyelination/remyelination process [56]. The following sections discuss the roles that have been elucidated for macrophages/microglia in focal demyelinating models, namely, the cuprizone and lysolecithin models.
6. The Cuprizone Demyelinating Model
The cuprizone model is the more commonly used of the two main focal demyelinating models, partly due to its ease of administration in the diet to produce injury.
Cuprizone is a copper chelator that is normally fed to mice for 4 to 6 weeks to induce significant, reproducible demyelinating lesions in the corpus callosum, hippocampus, anterior commissure, olfactory bulb, optic chiasm, brain stem, cerebellum, caudate putamen, cerebral cortex, and the cingulum [57–64]. The mechanism of demyelination is thought to be due to selective toxicity to oligodendrocytes, specifically through disruption in the mitochondrial complex IV of these myelin-forming cells. Discontinuation of cuprizone results in remyelination in the corpus callosum, usually through recruitment and differentiation of OPCs [43]. Significant M/M accumulation has been reported in lesions in this model, allowing the specific involvement of these cells in demyelination and remyelination to be examined [59, 61, 65–68]. The cuprizone model is advantageous to other focal demyelinating models in that the insult is easy to administer. However, a longer period is normally required for demyelination to occur, typically taking a few weeks. In addition, once cuprizone is removed, remyelination occurs very quickly, posing a difficulty in examining remyelination-specific events, such as the effect of remyelinating drugs on white matter tracts.
In the last fifteen years, there has been a large amount of research using this model, yielding many reports of the detrimental and beneficial roles of macrophages/microglia in de-/remyelination. The observed detriments will be first discussed followed by an overview of the beneficial aspects associated with these cells.
7. Detriments of Macrophages/Microglia in the Cuprizone Model
Wergeland et al. [69] used the cuprizone model on mice being supplemented with vitamin D3 in their diet. With high and very high doses of this vitamin, the amount of microglia activation was decreased, correlating with a reduction in white matter demyelination. Although these results suggest that activated microglia and infiltrating macrophages may be aiding the demyelination process, one cannot rule out the possibility of vitamin D3 having a protective effect on its own and thereby reducing macrophage/microglia activity. Another study found that administration of erythropoietin to mice fed cuprizone reduced vestibulomotor impairment, an observation that was associated with a decrease in microglia activation in the corpus callosum [70]. Yoshikawa et al. [71] found that pharmacological inhibition of 5-lipoxygenase (5-LO), an enzyme involved in the biosynthesis of leukotrienes, reduced cuprizone-induced axonal damage and motor deficits. Cuprizone-induced demyelination, however, was not attenuated. This observed reduction in axonal damage was associated with a decrease in microglial activation, suggesting that the 5-LO pathway contributes to microglial activation and neurotoxicity. Another study examined the expression of iNOS in the M1 proinflammatory subclass of macrophages/microglia. In that study, the authors found that the administration of fumaric acid esters, compounds which significantly inhibited LPS-induced nitric oxide production by microglia, slightly accelerated remyelination in the corpus callosum [63]. These findings suggest a negative role for iNOS in demyelinating lesions. When cuprizone-treated mice were administered the microglia inhibitor minocycline, a decrease in demyelination and an improvement in motor coordination behaviour was observed [72–74].
17 beta-estradiol (E2) is a form of estrogen that has been shown to reduce symptoms in EAE [75]. When this compound was administered to cuprizone-treated male mice, there was a reduction in demyelination accompanied by a delay in microglia activation. This correlation implies a protective effect corresponding to decreased microglia activation. Millet et al. [76] demonstrated that the injection of a proteasome inhibitor, lactacystin, into the corpus callosum during the remyelination process in cuprizone-treated mice resulted in a large improvement in remyelination, corresponding with attenuation of the recruitment of macrophages/microglia. Macrophage inflammatory protein-1α (MIP-1α) is a protein that has been associated with the recruitment of macrophages/microglia to demyelinating lesions in cuprizone treatment. A study conducted by McMahon et al. [77] found a significant decrease in demyelination in mice deficient in MIP-1α, indicating a role of macrophages/microglia in promoting demyelination.
These studies point to a harmful role for macrophages/microglia in cuprizone-induced demyelination. Decreased microglia activation corresponding with decreased demyelination, motor impairment, and increased remyelination, suggests a negative role for microglia in the demyelination/remyelination process. However, there is evidence demonstrating a beneficial role for these cells as well.
8. Benefits of Macrophages/Microglia in the Cuprizone Model
In contrast to the studies mentioned above, there are several studies that have noted beneficial roles for macrophages/microglia in cuprizone-induced demyelination. Olah et al. [78] isolated microglia from the corpus callosum of mice in a cuprizone experiment and performed a genome-wide gene expression investigating the upregulated genes associated with remyelination or demyelination. In that study, they found that microglia displayed a phenotype associated with the phagocytosis of myelin debris as well as with the recruitment of OPCs through the expression of cytokines and chemokines. The study provided transcriptomic evidence for the ability of microglia to support remyelination. Jurevics et al. [79] found that genes related to macrophages/microglia appeared in a temporal fashion corresponding to phagocytosis of myelin debris and repair of lesions, suggesting beneficial roles of these cells in clearing the inhibitory environment for repair. Another study looked at the role astrocytes played in providing prorepair signals to microglia in a demyelinated environment. Skripuletz et al. [80] ablated astrocytes in the cuprizone model using a TK transgene under the control of the astrocyte-specific promoter, glial fibrillary acidic protein (GFAP). These mice showed a failure to remove collapsed myelin debris, which was associated with a reduction in microglial activation, suggesting the importance of astrocyte signalling to macrophages/microglia in myelin debris clearance.
The major histocompatibility complex class II (MHC II) is predominantly present on microglia to present antigens to lymphocytes crossing the BBB. In MHC II null mice, there was delay in remyelination and differentiation of OPCs, pointing to the potential beneficial role played by microglia in remyelination [81]. The same group also found that in mice lacking the microglial enzyme, iNOS, and in mice lacking the microglial cytokine, tumor necrosis factor-α (TNF-α), there was a significant delay in remyelination and more severe demyelination, indicating the importance of the macrophages/microglial expression of iNOS and TNF-α in remyelination [82, 83]. Morell et al. [84] suggested that macrophages/microglia may be important in recruiting OPCs and stimulating their differentiation into mature myelinating oligodendrocytes. This was postulated when they observed that upregulation of mRNA transcripts for myelin-associated glycoprotein (MAG) and myelin basic protein (MBP) coincided with the accumulation of macrophages/microglia, before any remyelination was observed. Mason et al. [85] induced demyelination with cuprizone in mice lacking interleukin-1β (IL-1β), a cytokine normally secreted by macrophages/microglia. In that study, they found an impairment in remyelination in the homozygous IL-1β null mice, pointing to the macrophage/microglial-derived secretion of IL-1β as a potentially beneficial molecule for remyelination.
From these studies, it is evident that the cuprizone model of demyelination has been able to provide a large body of experimental evidence concerning the beneficial roles played by macrophages/microglia in de-/remyelination. These studies have demonstrated that macrophages/microglia have the potential to clear myelin debris, promote the recruitment and differentiation of OPCs, and release cytokines which may be beneficial for the remyelination process. As to when these cells are detrimental or beneficial will most likely depend on other mediators in the microenvironment, the extent of injury, and the temporal coordination between signalling molecules from other sources such as astrocytes. The following sections will now examine the body of evidence accumulated in the other commonly used focal demyelination model, the lysolecithin injury.
9. The Lysolecithin Demyelinating Model
Lysolecithin (lysophosphatidylcholine) is a demyelinating chemical that is administered through a stereotactic injection into white matter tracts in the CNS [43]. The dorsal and ventral funiculi of the thoracic and lumbar spinal cord are the most common injection targets, although the corpus callosum is sometimes used as well. Noticeable demyelination occurs hours after injection of the chemical, with significant demyelination lasting about seven to ten days. Substantial remyelination is normally evident twenty-one days after lesion formation [86]. Considerable macrophage infiltration and activated microglia are observed in the lesions with minimal T-cell involvement [87, 88]. Imai et al. [88] found that transplanted GFP-positive, bone marrow-derived macrophages represented the major cell population in lysolecithin-induced mice at days 2, 4, and 7-time points that are early in the demyelination/remyelination process. This model is advantageous in that it is quick to induce with a prolonged remyelination period, thus allowing processes promoting or interfering with remyelination to be studied more comprehensively than with the cuprizone model.
10. Detriments of Macrophages/Microglia in the Lysolecithin Model
The lysolecithin model has provided considerable insight into the biology of macrophages/microglia in the de-/remyelination process. Although not as frequently used as the cuprizone model, due largely to its greater challenge in producing injury as it involves a stereotaxic surgery, it has significant potential in elucidating the impact of specific treatment and intervention on demyelination and remyelination. In particular, its rapid induction of demyelination as well as its drawn out course in remyelination offers significant advantages over the cuprizone model, characteristics which may draw in more researchers into employing this model in the study of remyelination.
The detriment of macrophages/microglia in the lysolecithin model was suggested by the result that corticosteroid-treated animals had an enhancement in remyelination associated with a reduction in the number of macrophages/microglia at the lesion site [89]. Another study administered progesterone to adult male mice with lysolecithin demyelination [90] and found that on day 7, there was reduced representation of macrophages/microglia whilst remyelination, and numbers of OPCs and oligodendrocytes were elevated. Furthermore, Schonberg et al. [91] activated macrophages/microglia using zymosan, a toll-like receptor-2 agonist, following lysolecithin injury and observed a loss of OPCs and oligodendrocytes; the results suggest that macrophage/microglia activation can hinder remyelination through an inhibition of OPC recruitment and differentiation.
In summary, there is good data suggesting the negative roles of macrophages/microglia in demyelination/remyelination. As demonstrated in the aforementioned studies, when macrophages and microglia are diminished, the recruitment and differentiation of OPCs are increased, resulting in a better outcome for remyelination. This macrophage/microglia-mediated impairment of OPC recruitment and differentiation may be due to a variety of factors, including the release of toxic molecules. The next section will outline evidence for the positive roles of these cells in lysolecithin-induced demyelination.
11. Benefits of Macrophages/Microglia in the Lysolecithin Model
The benefits of macrophages/microglia in the lysolecithin model was first highlighted by Triarhou and Herndon [92, 93] who showed that depleting macrophages with silica quartz dust and dexamethasone resulted in an impairment in the clearance of collapsed myelin debris as well as a delay in the remyelination process. Using the clodronate liposome method to deplete macrophages, Kotter et al. [94] described that the depletion of macrophages soon after injury (day 1) significantly reduced remyelination 21 days after lysolecithin demyelination. When the authors administered clodronate liposomes later in the remyelination phase (day 8), the outcome of remyelination did not change, indicating that infiltrating macrophages were important in the early stages of remyelination. A later study carried out by the same group [95] found that depletion of macrophages resulted in delayed recruitment of OPCs to the lesion site; the mechanism of the requirement of macrophages was likely related to their clearance of myelin debris that otherwise was an inhibitory milieu for the differentiation of OPCs [96]. A recent study paired the circulatory systems of a GFP-expressing young mouse with a lysolecithin-induced demyelinated old mouse and found macrophages from the young mouse infiltrating the lesion in the old mouse and stimulating an increase in OPC differentiation as well as remyelination [97]. This study very clearly demonstrated the beneficial role macrophages play in promoting remyelination in the lesioned CNS as well as served to demonstrate the ability of young macrophages to enhance the recovery of an aged CNS, an environment in which remyelination and macrophage/microglia accumulation were significantly delayed. In support of the benefits of macrophages/microglia, the early administration of minocycline to demyelinated rodents inhibited macrophage/microglia activation examined at 1 and 3 days postlesion and reduced oligodendrocyte repopulation and remyelination [98].
This model of demyelination has accumulated a significant amount of knowledge demonstrating the benefits and detriments of macrophages/microglia in remyelination. While there is data suggesting that these cells may be inhibiting the recruitment of OPCs to the lesion, there is evidence suggesting the alternative. The recurrent observation of these cells being critical in the clearance of inhibitory myelin debris seems to be a requisite for the migration and subsequent differentiation of OPCs into the lesion. Certainly, the temporal coordination of cytokines and signalling molecules, the size and extent of the lesion, and the inhibitory properties of the extracellular environment likely dictate whether these cells will be beneficial or detrimental to the remyelination process. More studies using this model will inevitably yield more information on the complex roles characterizing these cells, thus allowing therapies to be developed which take advantage of the beneficial aspects and suppress the detrimental aspects.
12. Basis for Detriments of Macrophages/Microglia
There is a large accumulation of evidence demonstrating the harmful aspects of macrophages/microglia in the demyelinated lesion. Mechanisms underlying these detrimental roles may include the recruitment and reactivation of T cells in the CNS through the release of proteases, the production of proinflammatory cytokines, and the release of reactive oxygen species to induce neurotoxicity and OPC toxicity through excitatory amino acids. This section will discuss some of the evidence supporting these possible mechanisms.
In order for pathogenic T cells to enter the CNS parenchyma, they must be able to cross the parenchymal basement membrane. This is a laminin-containing basement membrane which lies in direct apposition to the CNS parenchyma and venules within the CNS. For these cells to successfully transmigrate this membrane, proteases such as the MMPs must be present. Macrophages/microglia are a major source of MMPs, and when selective MMPs are blocked, the amelioration of EAE clinical signs is observed [48, 99–101]. These observations suggest that the secretion of MMPs by macrophages/microglia aid in the recruitment of pathogenic T cells to the CNS.
Macrophages/microglia also assist in the reactivation of T cells once they enter the CNS parenchyma. In order for a T cell to contribute to CNS pathogenesis, it must be reactivated with antigen presenting cells within the CNS parenchyma. It has been observed that macrophages/microglia upregulate MHC II molecules when they are activated. MHC II molecules are required for antigen presentation to T cells [102]. These observations suggest the capability of activated macrophages/microglia to reactivate primed T cells entering the CNS. It should also be noted that the expression of MHC II has been correlated with the infiltration of T cells as well as with the progression of EAE [103].
In addition to recruitment and reactivating T cells, macrophages/microglia have also been observed to strip myelin, as well as kill neurons and OPCs [104–106]. One of the mechanisms explaining this macrophage/microglia-mediated toxicity is through the production of cytokines, glutamate, and reactive oxygen species [27]. When macrophages/microglia are activated, they release an array of inflammatory cytokines, such as TNF-α and IFN-γ. These cytokines induce the release of glutamate. Excessive glutamate stimulation on N-methyl-D-aspartate (NMDA) receptors results in mitochondrial death and ultimately excitotoxic neuronal and oligodendrocyte death [106]. When glutamate release is blocked, EAE progression is attenuated, providing evidence for glutamate excitotoxicity as a plausible mechanism for macrophage/microglia-mediated toxicity [107]. In addition to stimulating the release of glutamate, proinflammatory cytokines and chemokines promote inflammation and antigen presentation, thereby mediating the recruitment and reactivation of T cells to the lesion [108]. Finally, the release of free radicals, such as nitric oxide (NO), has been shown to induce oxidative damage to neurons and oligodendrocyte precursor cells [109].
It is evident that the array of detriments associated with macrophage/microglial activity in MS and its animal models function through many mechanisms of action. Such modes of action include the expression of proteases to aid in the recruitment of T cells into the CNS, the upregulation of molecules associated with antigen presentation, thereby aiding in the reactivation of pathogenic T cells, and through the release of inflammatory cytokines, free radicals, and glutamate. Developing therapies which target these mechanisms will certainly offer hope in the quest for improving the outcome of remyelination.
13. Basis for Benefits of Macrophages/Microglia
From the array of studies conducted with the different animal models of MS, it is clear that macrophages/microglia, although being shown to be predominantly detrimental, have a variety of beneficial effects. The mechanisms of these beneficial effects are thought to be due to the production of growth factors, the removal of inhibitory debris and toxic products, and through the removal of inhibitory extracellular matrix (ECM) molecules. This section entails a discussion of some of the evidence supporting these plausible mechanisms.
Macrophages/microglia have been reported to secrete many well-known neurotrophic factors, which may help in the recruitment and differentiation of OPCs, as well as in the regeneration of axons. Herx et al. [110] showed that the production of IL-1β by microglia regulated the production of ciliary neurotrophic factor (CNTF), an important growth factor for the survival of oligodendrocytes. Studies have also demonstrated the production by macrophages/microglia of other neurotrophic factors, such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin-3 (NT-3) [111, 112]. As mentioned above, Yin et al. [29] reported that axonal regeneration after optic nerve injury was partially due to oncomodulin, a growth factor secreted by macrophages. Overall, there is evidence to support the claim that macrophages/microglia promote remyelination and axonal regeneration through the secretion of beneficial growth factors. A challenge remains, though, in determining the temporal and spatial characteristics in which these benefits are most likely to occur.
Another beneficial mechanism of macrophages/microglia may be through the removal of inhibitory debris or toxic products. As mentioned previously, Kotter et al. [95] found that macrophages were important for the clearance of myelin debris and in enabling subsequent remyelination to occur. These authors found in a later study that myelin debris acted to inhibit OPC differentiation into mature, myelinating oligodendrocytes [96], thus supporting the ability of macrophages/microglia to clear debris as a beneficial role in remyelination. In another study, the depletion of macrophages with silica dust resulted in a hindered clearance of myelin debris and significantly delayed remyelination [92]. In an Alzheimer's-like disease model, it was found that macrophages recruited to the CNS cleared intracerebral Aβ deposits [38]. Altogether, it can be seen that there is evidence supporting the capability of macrophages/microglia in clearing inhibitory myelin debris as well as toxic products such as Aβ deposits, ultimately associating with a decrease in the severity of the disease.
Finally, our group has shown that macrophages/microglia act beneficially to remove inhibitory extracellular matrix molecules. As mentioned previously, CSPGs are normally deposited around a CNS lesion. We have shown that NG2, a type of proteoglycan, hinders the differentiation of OPCs [113]. It was found that the production of MMP-9 by macrophages/microglia cleared the accumulation of NG2 within a demyelinating lesion, leading to subsequent oligodendrocyte maturation and remyelination.
Although the mechanisms discussed above serve to explain some of the benefits observed with macrophages/microglia, there are likely other positive mechanisms that aid in the remyelination process. One such mechanism may be through the stimulation of astrocytes to secrete trophic factors as well as to reseal the blood-brain barrier (BBB) [114]. By determining the mechanisms involved in the beneficial aspects of these immune cells, we will be able to better target these endogenous processes with therapies that may stimulate remyelination and axon regeneration. This can already be seen by some of the therapies already in use for the treatment of multiple sclerosis. The next section will discuss some of these therapies and briefly describe the mechanisms in which the macrophage/microglia population is targeted.
14. Therapies Involving Macrophage/Microglia Activity
There are currently seven therapies approved by the US Food and Drug Administration (FDA) for treatment of multiple sclerosis. All of these therapies function by modulating the immune system, with many having broad effects affecting multiple components of both innate and adaptive immunity. While T cells are most commonly targeted by these medications, there are reports describing the involvement of macrophages/microglia in these therapies. This section will briefly describe five of the seven licensed therapies in which significant involvement of macrophages/microglia has been documented.
Glatiramer acetate (Copaxone) is a first-line therapy that has been shown to reduce the relapse rate and progression of disability in patients with relapsing remitting MS [115]. This compound is a random polypeptide composed of four amino acids designed to mimic myelin basic protein (MBP), a critical component of the myelin sheath [116]. The major mechanism of action is to induce T cells of the T helper type 2 subset (Th2). These cells secrete the anti-inflammatory cytokines interleukin-4 (IL-4), IL-5, IL-6, IL-10, and IL-13 and transforming growth factor-β (TGF-β). In addition to the induction of Th2 cells, glatiramer acetate has been shown to have an effect of macrophages/microglia, with many studies suggesting a role in promoting an anti-inflammatory M2 phenotype. It was found that glatiramer acetate increased the expression of the anti-inflammatory cytokine IL-10 and reduced the expression of the proinflammatory cytokine tumor necrosis factor-α (TNF-α) in macrophages/microglia [117–119]. Another study found that this compound inhibited the production of NO, suggesting the polarization away from an M1 phenotype [120]. Finally, this compound has been demonstrated to increase the phagocytic activity of microglia and monocytes, possibly contributing to increased myelin debris clearance [121]. Altogether, it appears as though glatiramer acetate has many effects on the macrophage/microglia population, while at the same time polarizing the T-cell response to one of an anti-inflammatory nature.
Interferon-β (Avonex, Betaseron, Extavia, and Rebif) is another first line therapy which has demonstrated success in reducing the rate and severity of relapses, as well in slowing the progression of disability [121]. The main mechanisms of action seem to be in the inhibition of T-cell recruitment and activation, as well as in the modulation of cytokines [116, 122, 123]. Though the reduction in the T-cell response is quite evident, aberrations in the macrophage/microglia response may also contribute. For instance, interferon-β has been demonstrated to reduce the proliferation of both macrophages and monocytes as well as reduce the expression of MHC II on these cells [124]. This reduced expression of MHC II may be a reason for the decreased activation of T cells, as the antigen-presentation capabilities of macrophages/microglia would be hindered. Another example of macrophage/microglia involvement is demonstrated in the study by Hall et al. [124], which highlighted the reduction of toxic microglia-derived respiratory bursts as a result of interferon-β interaction. As interferon-β is a cytokine, many other unknown mechanisms may be occurring, possibly involving other aspects of the macrophage/microglia response.
Fingolimod (Gilenya) has demonstrated success in reducing the relapse rate as well as the rate of disability progression [116, 125]. It acts by modulating the sphingosine 1-phosphate receptors, thereby preventing lymphocyte recruitment, as these receptors are required for the exit of lymphocytes from secondary lymphoid organs to the CNS [126]. In addition, fingolimod induces an anti-inflammatory phenotype in activated macrophages. This is supported by the observation of decreased production of proinflammatory cytokines and free radicals by macrophages upon application of this drug [127].
Mitoxantrone (Novatrone) is a cytotoxic agent that functions through the intercalation of DNA [128]. Due to this broad mechanism of action, it affects many of the different types of immune cells, resulting in widespread immunosuppressive activity [129]. This immunosuppression results in the inhibition of monocyte and lymphocyte migration into the CNS [130]. Reductions in the proinflammatory cytokines, TNF-α, IL-2, and IFN-γ, have also been observed [131]. This reduction in monocyte activity may explain an observation made in which macrophage-mediated myelin degradation was inhibited [132].
Dimethyl fumarate (Tecfidera) is a recently approved first-line therapy for multiple sclerosis. It is an oral tablet thought to act by decreasing the expression of NF-κB dependent genes [133, 134]. These genes regulate the expression of inflammatory cytokines, and as such, studies have reported decreased production of TNF-α, IL-1β, IL-6, and NO in microglia [135]. Adding to these effects, when dimethyl fumarate is administered to mice with EAE, a significant reduction in infiltrating macrophages is observed in the lesions [136]. As is the case with the aforementioned medications, there may be many modes of action that are still unknown. Nonetheless, involvement of macrophage/microglia seems to be consistent across the approved medications used for multiple sclerosis.
15. Conclusion
Macrophages/microglia are implicated in promoting demyelination; yet remyelination in MS lesions appears to require these cells as repair occurs in the presence of macrophages/microglia [137, 138]. From the research conducted on macrophages/microglia in models of CNS injury, including EAE, cuprizone-, and lysolecithin-induced demyelination, it is evident that a complex dichotomy exists in the roles of these cells in the demyelination/remyelination process. More detailed studies analyzing the spectrum of activities of macrophages/microglia at different time points in the focal demyelination models, as well as studies depleting and activating these cells at different temporal points will only serve to dissect the dichotomy presented to us. A thorough understanding of when macrophages/microglia are beneficial or detrimental in the demyelination/remyelination process will allow us to develop therapeutic interventions which take advantage of the respective characteristics, with the ultimate goal to enhance remyelination and suppress demyelination. In addition to the importance of the cuprizone and lysolecithin demyelination models in being critical tools for dissecting the dichotomy of macrophages/microglia, studies using EAE will serve to complement what we learn from the focal demyelinating models, altogether serving to further our understanding of the complex processes of de-/remyelination in injuries to the CNS.
Acknowledgments
The authors thank the Multiple Sclerosis Scientific Research Foundation and the Multiple Sclerosis Society of Canada for support of operating funds. K. S. Rawji is supported by studentships from the University of Calgary Faculty of Medicine, Multiple Sclerosis Society of Canada, and the Hotchkiss Brain Institute. V. W. Yong acknowledges salary support from the Canada Research Chair (Tier 1) Program.
References
- 1.Shechter R, Schwartz M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer if ‘but how’. Journal of Pathology. 2013;229(2):332–346. doi: 10.1002/path.4106. [DOI] [PubMed] [Google Scholar]
- 2.David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nature Reviews Neuroscience. 2011;12(7):388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- 3.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nature Neuroscience. 2011;14(10):1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 4.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468(7321):253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 5.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 6.Graeber MB. Changing face of microglia. Science. 2010;330(6005):783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- 7.Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nature Neuroscience. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 8.Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science. 2012;335(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Developmental Brain Research. 1999;117(2):145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 11.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nature Neuroscience. 2012;15(8):1096–1101. doi: 10.1038/nn.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 14.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature Neuroscience. 2011;14(9):1142–1150. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 15.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1(5):301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 16.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annual Review of Immunology. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 18.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. Journal of Pathology. 2013;229(2):176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 19.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. Journal of Neuroscience. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Annals of Neurology. 2000;47(6):707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 21.Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Laboratory Investigation. 1978;38(4):409–421. [PubMed] [Google Scholar]
- 22.Letellier E, Kumar S, Sancho-Martinez I, et al. CD95-ligand on peripheral myeloid cells activates Syk kinase to trigger their recruitment to the inflammatory site. Immunity. 2010;32(2):240–252. doi: 10.1016/j.immuni.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 23.Wells JEA, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126(7):1628–1637. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48(6):1393–1401. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 25.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Annals of Neurology. 2002;52(1):54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 26.Popovich PG, Guan Z, Wei P, Huitinga I, Van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Experimental Neurology. 1999;158(2):351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi H. Neurotoxicity by microglia: mechanisms and potential therapeutic strategy. Clinical and Experimental Neuroimmunology. 2010;1(1):12–21. [Google Scholar]
- 28.Rapalino O, Lazarov-Spiegler O, Agranov E, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nature Medicine. 1998;4(7):814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- 29.Yin Y, Henzl MT, Lorber B, et al. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nature Neuroscience. 2006;9(6):843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- 30.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 31.Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiology of Disease. 2000;7(6):574–585. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- 32.Shechter R, London A, Varol C, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Medicine. 2009;6(7, article e1000113) doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schonberg DL, Goldstein EZ, Sahinkaya FR, Wei P, Popovich PG, McTigue DM. Ferritin stimulates oligodendrocyte genesis in the adult spinal cord and can be transferred from macrophages to NG2 cells in vivo. The Journal of Neuroscience. 2012;32(16):5374–5384. doi: 10.1523/JNEUROSCI.3517-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrette B, Hébert MA, Filali M, et al. Requirement of myeloid cells for axon regeneration. Journal of Neuroscience. 2008;28(38):9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Medicine. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 36.Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Journal of Neuroscience. 2011;31(16):6208–6220. doi: 10.1523/JNEUROSCI.0299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grathwohl SA, Kälin RE, Bolmont T, et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Neuroforum. 2009;12(11):1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 39.Lassmann H, Brück W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends in Molecular Medicine. 2001;7(3):115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- 40.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. The New England Journal of Medicine. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 41.Sriram S. Role of glial cells in innate immunity and their role in CNS demyelination. Journal of Neuroimmunology. 2011;239:13–20. doi: 10.1016/j.jneuroim.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 42.Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nature Reviews Neuroscience. 2002;3(4):291–301. doi: 10.1038/nrn784. [DOI] [PubMed] [Google Scholar]
- 43.Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nature Neuroscience. 2012;15(8):1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martiney JA, Rajan AJ, Charles PC, et al. Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage-deactivating agent. Journal of Immunology. 1998;160(11):5588–5595. [PubMed] [Google Scholar]
- 45.Bhasin M, Wu M, Tsirka SE. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunology. 2007;8, article 10 doi: 10.1186/1471-2172-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran EH, Hoekstra K, Van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. Journal of Immunology. 1998;161(7):3767–3775. [PubMed] [Google Scholar]
- 47.Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nature Medicine. 2005;11(2):146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 48.Agrawal S, Anderson P, Durbeej M, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. Journal of Experimental Medicine. 2006;203(4):1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartholomäus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462(7269):94–98. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- 50.Nikić I, Merkler D, Sorbara C, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nature Medicine. 2011;17(4):495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- 51.Rasmussen S, Wang Y, Kivisäkk P, et al. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing-remitting experimental autoimmune encephalomyelitis. Brain. 2007;130(11):2816–2829. doi: 10.1093/brain/awm219. [DOI] [PubMed] [Google Scholar]
- 52.Starossom SC, Mascanfroni ID, Imitola J, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37(2):249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mikita J, Dubourdieu-Cassagno N, Deloire MS, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Multiple Sclerosis. 2011;17(1):2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 54.Vaknin I, Kunis G, Miller O, et al. Excess circulating alternatively activated myeloid (M2) cells accelerate ALS progression while inhibiting experimental autoimmune encephalomyelitis. PLoS One. 2011;6(11, article e26921) doi: 10.1371/journal.pone.0026921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denney L, Kok WL, Cole SL, Sanderson S, McMichael AJ, Ho LP. Activation of invariant NKT cells in early phase of experimental autoimmune encephalomyelitis results in differentiation of Ly6Chi inflammatory monocyte to M2 macrophages and improved outcome. The Journal of Immunology. 2012;189(2):551–557. doi: 10.4049/jimmunol.1103608. [DOI] [PubMed] [Google Scholar]
- 56.Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathology. 2001;11(1):107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silvestroff L, Bartucci S, Soto E, Gallo V, Pasquini J, Franco P. Cuprizone-Induced demyelination in CNP::GFP transgenic mice. Journal of Comparative Neurology. 2010;518(12):2261–2283. doi: 10.1002/cne.22330. [DOI] [PubMed] [Google Scholar]
- 58.Hoffmann K, Lindner M, Gröticke I, Stangel M, Löscher W. Epileptic seizures and hippocampal damage after cuprizone-induced demyelination in C57BL/6 mice. Experimental Neurology. 2008;210(2):308–321. doi: 10.1016/j.expneurol.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 59.Koutsoudaki PN, Skripuletz T, Gudi V, et al. Demyelination of the hippocampus is prominent in the cuprizone model. Neuroscience Letters. 2009;451(1):83–88. doi: 10.1016/j.neulet.2008.11.058. [DOI] [PubMed] [Google Scholar]
- 60.Norkute A, Hieble A, Braun A, et al. Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. Journal of Neuroscience Research. 2009;87(6):1343–1355. doi: 10.1002/jnr.21946. [DOI] [PubMed] [Google Scholar]
- 61.Groebe A, Clarner T, Baumgartner W, Dang J, Beyer C, Kipp M. Cuprizone treatment induces distinct demyelination, astrocytosis, and microglia cell invasion or proliferation in the mouse cerebellum. Cerebellum. 2009;8(3):163–174. doi: 10.1007/s12311-009-0099-3. [DOI] [PubMed] [Google Scholar]
- 62.Pott F, Gingele S, Clarner T, et al. Cuprizone effect on myelination, astrogliosis and microglia attraction in the mouse basal ganglia. Brain Research. 2009;1305:137–149. doi: 10.1016/j.brainres.2009.09.084. [DOI] [PubMed] [Google Scholar]
- 63.Moharregh-Khiabani D, Blank A, Skripuletz T, et al. Effects of fumaric acids on cuprizone induced central nervous system de- and remyelination in the mouse. PLoS ONE. 2010;5(7, article e11769) doi: 10.1371/journal.pone.0011769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skripuletz T, Lindner M, Kotsiari A, et al. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. American Journal of Pathology. 2008;172(4):1053–1061. doi: 10.2353/ajpath.2008.070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hiremath MM, Saito Y, Knapp GW, Ting JPY, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. Journal of Neuroimmunology. 1998;92(1-2):38–49. doi: 10.1016/s0165-5728(98)00168-4. [DOI] [PubMed] [Google Scholar]
- 66.Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. Journal of Molecular Neuroscience. 2012;48(1):66–76. doi: 10.1007/s12031-012-9773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kipp M, Norkus A, Krauspe B, et al. The hippocampal fimbria of cuprizone-treated animals as a structure for studying neuroprotection in multiple sclerosis. Inflammation Research. 2011;60(8):723–726. doi: 10.1007/s00011-011-0339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gudi V, Moharregh-Khiabani D, Skripuletz T, et al. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Research. 2009;1283:127–138. doi: 10.1016/j.brainres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 69.Wergeland S, Torkildsen O, Myhr KM, Aksnes L, Mork SJ, Bo L. Dietary vitamin D3 supplements reduce demyelination in the cuprizone model. PLoS One. 2011;6(10, article e26262) doi: 10.1371/journal.pone.0026262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hagemeyer N, Boretius S, Ott C, et al. Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Molecular Medicine. 2012;18:628–635. doi: 10.2119/molmed.2011.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshikawa K, Palumbo S, Toscano CD, Bosetti F. Inhibition of 5-lipoxygenase activity in mice during cuprizone-induced demyelination attenuates neuroinflammation, motor dysfunction and axonal damage. Prostaglandins Leukotrienes and Essential Fatty Acids. 2011;85(1):43–52. doi: 10.1016/j.plefa.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan R, Xu F, Previti ML, et al. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. Journal of Neuroscience. 2007;27(12):3057–3063. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skripuletz T, Miller E, Moharregh-Khiabani D, et al. Beneficial effects of minocycline on cuprizone induced cortical demyelination. Neurochemical Research. 2010;35(9):1422–1433. doi: 10.1007/s11064-010-0202-7. [DOI] [PubMed] [Google Scholar]
- 74.Pasquini LA, Calatayud CA, Bertone Uña AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochemical Research. 2007;32(2):279–292. doi: 10.1007/s11064-006-9165-0. [DOI] [PubMed] [Google Scholar]
- 75.Bebo BF, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. Journal of Immunology. 2001;166(3):2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 76.Millet V, Moiola CP, Pasquini JM, Soto EF, Pasquini LA. Partial inhibition of proteasome activity enhances remyelination after cuprizone-induced demyelination. Experimental Neurology. 2009;217(2):282–296. doi: 10.1016/j.expneurol.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 77.McMahon EJ, Cook DN, Suzuki K, Matsushima GK. Absence of macrophage-inflammatory protein-1α delays central nervous system demyelination in the presence of an intact blood-brain barrier. Journal of Immunology. 2001;167(5):2964–2971. doi: 10.4049/jimmunol.167.5.2964. [DOI] [PubMed] [Google Scholar]
- 78.Olah M, Amor S, Brouwer N, et al. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60(2):306–321. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- 79.Jurevics H, Largent C, Hostettler J, et al. Alterations in metabolism and gene expression in brain regions during cuprizone-induced demyelination and remyelination. Journal of Neurochemistry. 2002;82(1):126–136. doi: 10.1046/j.1471-4159.2002.00954.x. [DOI] [PubMed] [Google Scholar]
- 80.Skripuletz T, Hackstette D, Bauer K, et al. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 2013;136, part 1:147–167. doi: 10.1093/brain/aws262. [DOI] [PubMed] [Google Scholar]
- 81.Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JPY. Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. Journal of Neuroscience. 2003;23(30):9824–9832. doi: 10.1523/JNEUROSCI.23-30-09824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arnett HA, Hellendall RP, Matsushima GK, et al. The protective role of nitric oxide in a neurotoxicant-induced demyelinating model. Journal of Immunology. 2002;168(1):427–433. doi: 10.4049/jimmunol.168.1.427. [DOI] [PubMed] [Google Scholar]
- 83.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JPY. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nature Neuroscience. 2001;4(11):1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 84.Morell P, Barrett CV, Mason JL, et al. Gene expression in brain during cuprizone-induced demyelination and remyelination. Molecular and Cellular Neurosciences. 1998;12(4-5):220–227. doi: 10.1006/mcne.1998.0715. [DOI] [PubMed] [Google Scholar]
- 85.Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1β promotes repair of the CNS. Journal of Neuroscience. 2001;21(18):7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lau LW, Keough MB, Haylock-Jacobs S, et al. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Annals of Neurology. 2012;72(3):419–432. doi: 10.1002/ana.23599. [DOI] [PubMed] [Google Scholar]
- 87.Waxman SG, Kocsis JD, Nitta KC. Lysophosphatidyl choline-induced focal demyelination in the rabbit corpus callosum. Light-microscopic observations. Journal of the Neurological Sciences. 1979;44(1):45–53. doi: 10.1016/0022-510x(79)90221-1. [DOI] [PubMed] [Google Scholar]
- 88.Imai M, Watanabe M, Suyama K, et al. Delayed accumulation of activated macrophages and inhibition of remyelination after spinal cord injury in an adult rodent model: laboratory investigation. Journal of Neurosurgery. 2008;8(1):58–66. doi: 10.3171/SPI-08/01/058. [DOI] [PubMed] [Google Scholar]
- 89.Pavelko KD, Van Engelen BGM, Rodriguez M. Acceleration in the rate of CNS remyelination in lysolecithin-induced demyelination. Journal of Neuroscience. 1998;18(7):2498–2505. doi: 10.1523/JNEUROSCI.18-07-02498.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garay L, Tungler V, Deniselle MC, Lima A, Roig P, De Nicola AF. Progesterone attenuates demyelination and microglial reaction in the lysolecithin-injured spinal cord. Neuroscience. 2011;192:588–597. doi: 10.1016/j.neuroscience.2011.06.065. [DOI] [PubMed] [Google Scholar]
- 91.Schonberg DL, Popovich PG, McTigue DM. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. Journal of Neuropathology and Experimental Neurology. 2007;66(12):1124–1135. doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- 92.Triarhou LC, Herndon RM. Effect of macrophage inactivation on the neuropathology of lysolecithin-induced demyelination. British Journal of Experimental Pathology. 1985;66(3):293–301. [PMC free article] [PubMed] [Google Scholar]
- 93.Triarhou LC, Herndon RM. The effect of dexamethasone on L-α-lysophosphatidyl choline (lysolecithin)-induced demyelination of the rat spinal cord. Archives of Neurology. 1986;43(2):121–125. doi: 10.1001/archneur.1986.00520020015008. [DOI] [PubMed] [Google Scholar]
- 94.Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJM. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 2001;35(3):204–212. doi: 10.1002/glia.1085. [DOI] [PubMed] [Google Scholar]
- 95.Kotter MR, Zhao C, Van Rooijen N, Franklin RJM. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiology of Disease. 2005;18(1):166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 96.Kotter MR, Li WW, Zhao C, Franklin RJM. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. Journal of Neuroscience. 2006;26(1):328–332. doi: 10.1523/JNEUROSCI.2615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruckh JM, Zhao JW, Shadrach JL, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10(1):96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li WW, Setzu A, Zhao C, Franklin RJM. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. Journal of Neuroimmunology. 2005;158(1-2):58–66. doi: 10.1016/j.jneuroim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 99.Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nature Reviews Neuroscience. 2001;2(7):502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Toft-Hansen H, Nuttall RK, Edwards DR, Owens T. Key metalloproteinases are expressed by specific cell types in experimental autoimmune encephalomyelitis. Journal of Immunology. 2004;173(8):5209–5218. doi: 10.4049/jimmunol.173.8.5209. [DOI] [PubMed] [Google Scholar]
- 101.Agrawal SM, Silva C, Tourtellotte WW, Yong VW. EMMPRIN: a novel regulator of leukocyte transmigration into the CNS in multiple sclerosis and experimental autoimmune encephalomyelitis. Journal of Neuroscience. 2011;31(2):669–677. doi: 10.1523/JNEUROSCI.3659-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Research Reviews. 2004;46(3):261–281. doi: 10.1016/j.brainresrev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 103.Murphy AC, Lalor SJ, Lynch MA, Mills KHG. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain, Behavior, and Immunity. 2010;24(4):641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 104.Peterson JW, Bö L, Mörk S, Chang A, Ransohoff RM, Trapp BD. VCAM-1-positive microglia target oligodendrocytes at the border of multiple sclerosis lesions. Journal of Neuropathology and Experimental Neurology. 2002;61(6):539–546. doi: 10.1093/jnen/61.6.539. [DOI] [PubMed] [Google Scholar]
- 105.Merrill JE, Zimmerman RP. Natural and induced cytotoxicity of oligodendrocytes by microglia is inhibitable by TGF beta. Glia. 1991;4(3):327–331. doi: 10.1002/glia.440040311. [DOI] [PubMed] [Google Scholar]
- 106.Takeuchi H, Mizuno T, Zhang G, et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. Journal of Biological Chemistry. 2005;280(11):10444–10454. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- 107.Shijie J, Takeuchi H, Yawata I, et al. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku Journal of Experimental Medicine. 2009;217(2):87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
- 108.Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkstra CD. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia. 1995;15(4):437–446. doi: 10.1002/glia.440150407. [DOI] [PubMed] [Google Scholar]
- 109.Reynolds A, Laurie C, Lee Mosley R, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. International Review of Neurobiology. 2007;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- 110.Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1β is required for the production of ciliary neurotrophic factor. Journal of Immunology. 2000;165(4):2232–2239. doi: 10.4049/jimmunol.165.4.2232. [DOI] [PubMed] [Google Scholar]
- 111.Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. Journal of Neuroscience. 1996;16(8):2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Batchelor PE, Liberatore GT, Wong JYF, et al. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. Journal of Neuroscience. 1999;19(5):1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. Journal of Neuroscience. 2003;23(35):11127–11135. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wee Yong V. Inflammation in neurological disorders: a help or a hindrance? Neuroscientist. 2010;16(4):408–420. doi: 10.1177/1073858410371379. [DOI] [PubMed] [Google Scholar]
- 115.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing- remitting multiple sclerosis: results of a phase III multicenter, double- blind, placebo-controlled trial. Neurology. 1995;45(7):1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 116.Luessi F, Siffrin V, Zipp F. Neurodegeneration in multiple sclerosis: novel treatment strategies. Expert Review of Neurotherapeutics. 2012;12(9):1061–1076. doi: 10.1586/ern.12.59. [DOI] [PubMed] [Google Scholar]
- 117.Jung S, Siglienti I, Grauer O, Magnus T, Scarlato G, Toyka K. Induction of IL-10 in rat peritoneal macrophages and dendritic cells by glatiramer acetate. Journal of Neuroimmunology. 2004;148(1-2):63–73. doi: 10.1016/j.jneuroim.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 118.Weber MS, Starck M, Wagenpfeil S, Meinl E, Hohlfeld R, Farina C. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain. 2004;127(6):1370–1378. doi: 10.1093/brain/awh163. [DOI] [PubMed] [Google Scholar]
- 119.Weber MS, Prod’homme T, Youssef S, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nature Medicine. 2007;13(8):935–943. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 120.Iarlori C, Gambi D, Lugaresi A, et al. Reduction of free radicals in multiple sclerosis: effect of glatiramer acetate (Copaxone) Multiple Sclerosis. 2008;14(6):739–748. doi: 10.1177/1352458508088918. [DOI] [PubMed] [Google Scholar]
- 121.Trojano M, Pellegrini F, Fuiani A, et al. New natural history of interferon-β-treated relapsing multiple sclerosis. Annals of Neurology. 2007;61(4):300–306. doi: 10.1002/ana.21102. [DOI] [PubMed] [Google Scholar]
- 122.Yong VW. Differential mechanisms of action of interferon-β and glatiramer acetate in MS. Neurology. 2002;59(6):802–808. doi: 10.1212/wnl.59.6.802. [DOI] [PubMed] [Google Scholar]
- 123.Kieseier BC, Hartung HP. Interferon-β and neuroprotection in multiple sclerosis-Facts, hopes and phantasies. Experimental Neurology. 2007;203(1):1–4. doi: 10.1016/j.expneurol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 124.Hall GL, Wing MG, Compston DAS, Scolding NJ. β-interferon regulates the immunomodulatory activity of neonatal rodent microglia. Journal of Neuroimmunology. 1997;72(1):11–19. doi: 10.1016/s0165-5728(96)00128-2. [DOI] [PubMed] [Google Scholar]
- 125.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. The New England Journal of Medicine. 2010;362(5):387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 126.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nature Reviews Immunology. 2005;5(7):560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 127.Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circulation Research. 2008;102(8):950–958. doi: 10.1161/CIRCRESAHA.107.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loma I, Heyman R. Multiple sclerosis: pathogenesis and treatment. Current Neuropharmacology. 2011;9(3):409–416. doi: 10.2174/157015911796557911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fox EJ. Mechanism of action of mitoxantrone. Neurology. 2004;63(12):S15–S18. doi: 10.1212/wnl.63.12_suppl_6.s15. [DOI] [PubMed] [Google Scholar]
- 130.Kopadze T, Dehmel T, Hartung HP, Stüve O, Kieseier BC. Inhibition by mitoxantrone of in vitro migration of immunocompetent cells: a possible mechanism for therapeutic efficacy in the treatment of multiple sclerosis. Archives of Neurology. 2006;63(11):1572–1578. doi: 10.1001/archneur.63.11.1572. [DOI] [PubMed] [Google Scholar]
- 131.Vollmer T, Stewart T, Baxter N. Mitoxantrone and cytotoxic drugs’ mechanisms of action. Neurology. 2010;74, supplement 1:S41–S46. doi: 10.1212/WNL.0b013e3181c97f5a. [DOI] [PubMed] [Google Scholar]
- 132.Watson CM, Davison AN, Baker D, O’Neill JK, Turk JL. Suppression of demyelination by mitoxantrone. International Journal of Immunopharmacology. 1991;13(7):923–930. doi: 10.1016/0192-0561(91)90045-9. [DOI] [PubMed] [Google Scholar]
- 133.Stoof TJ, Flier J, Sampat S, Nieboer C, Tensen CP, Boorsma DM. The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. British Journal of Dermatology. 2001;144(6):1114–1120. doi: 10.1046/j.1365-2133.2001.04220.x. [DOI] [PubMed] [Google Scholar]
- 134.Moharregh-Khiabani D, Linker RA, Gold R, Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis. Current Neuropharmacology. 2009;7(1):60–64. doi: 10.2174/157015909787602788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wierinckx A, Brevé J, Mercier D, Schultzberg M, Drukarch B, Van Dam AM. Detoxication enzyme inducers modify cytokine production in rat mixed glial cells. Journal of Neuroimmunology. 2005;166(1-2):132–143. doi: 10.1016/j.jneuroim.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 136.Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clinical and Experimental Immunology. 2006;145(1):101–107. doi: 10.1111/j.1365-2249.2006.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Patani R, Balaratnam M, Vora A, Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathology and Applied Neurobiology. 2007;33(3):277–287. doi: 10.1111/j.1365-2990.2007.00805.x. [DOI] [PubMed] [Google Scholar]
- 138.Kipp M, Victor M, Martino G, Franklin RJ. Endogeneous remyelination: findings in human studies. CNS & Neurological Disorders. 2012;11(5):598–609. doi: 10.2174/187152712801661257. [DOI] [PubMed] [Google Scholar]