

Abstract
Detailed studies of ribosomal proteins, essential components of the protein biosynthetic machinery, have been hampered by the lack of readily accessible chromosomal deletions of the corresponding genes. Here, we report the systematic genomic deletion of 41 individual ribosomal protein genes in Escherichia coli, which are not included in the Keio collection. Chromosomal copies of these genes were replaced by an antibiotic resistance gene in the presence of an inducible, easy-to-exchange plasmid-born allele. Using this knockout collection, 9 ribosomal proteins (L15, L21, L24, L27, L29, L30, L34, S9 and S17) were found nonessential for survival under induction conditions at various temperatures. Taken together with previous results, this analysis revealed that 22 of the 54 E. coli ribosomal protein genes can be individually deleted from the genome. These strains also allow expression of truncated protein variants to probe the importance of RNA-protein interactions in functional sites of the ribosome. This set of strains should enhance in vivo studies of ribosome assembly/function, and may ultimately allow systematic substitution of ribosomal proteins with RNA.
Keywords: ribosome, translation, recombineering, ribonucleoprotein complex, protein engineering
Introduction
Ribosomes are large ribonucleoprotein particles that iteratively translate the triplet codons of mRNA into the corresponding amino acids in the growing polypeptide chain. Upon each round of translational elongation, an mRNA codon at the decoding center in the A (Aminoacyl) site is decoded by its cognate aminoacyl-tRNA. A peptidyl moiety attached to tRNA in the P (Peptidyl) site is transferred to the α-amino group of the A-site aminoacyl-tRNA by nucleophilic attack on the ester bond linkage of the peptidyl-tRNA, and tRNAs in the A and P sites then translocate to their adjacent P and E (Exit) sites, respectively. The functional ribosome (70S in prokaryotes and 80S in eukaryotes) contains 2 subunits: one large subunit and one small subunit. In E. coli, the large (50S) subunit consists of 2 ribosomal RNAs (rRNA; 23S and 5S) and 33 different ribosomal proteins (L1-L7, L9-L36), while the small (30S) subunit consists of 1 rRNA and 21 ribosomal proteins (S1-S21).
Ribosomal proteins are highly specialized, RNA-interacting proteins—approximately half of them are conserved among the three domains of life, while others are domain-specific. Moreover, the composition of ribosomal proteins is highly conserved within each domain 1. Crystal structures have revealed intricate protein-RNA interactions both in the peripheral and internal regions of the ribosome 2; 3. In addition, several ribosomal proteins are suggested to be involved in essential ribosomal functions 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14. Over the last several decades, there have been significant efforts to elucidate the roles of ribosomal proteins in translation, and numerous mutations that confer diverse phenotypes and antibiotic resistance in E. coli have been isolated 15; 16; 17; 18. For instance, restrictive mutations in S12, originally identified in streptomycin-resistant/dependent strains, confer a hyperaccurate phenotype (where frequency of miscoding and stop-codon readthrough are decreased) to the ribosome 19; 20, while ram mutations in S4 and S5, identified in revertants of streptomycin-dependence, give rise to an error-prone phenotype (where frequency of miscoding and stop-codon readthrough are increased) 21; 22. Additionally, some restrictive mutations in S12 (R86S and P91Q), which are known to slow the rate of ribosome translation, can give rise to increased folding and expression of aggregation-prone eukaryotic proteins in E. coli 23. However, the functions of most ribosomal proteins have not been fully deciphered, primarily due to a lack of readily accessible mutant lines containing genetic deletions of individual essential ribosomal proteins.
λ-Red-mediated recombination is a powerful tool to precisely alter bacterial genomes, providing a simple method to study protein function, genetic networks, and genome evolution in living cells 24. Previous efforts to generate a complete set of gene deletion strains (of non-essential genes) in E. coli have resulted in a collection of 3985 individual mutants covering ~90% of known E. coli genes (Keio collection 25). However, only 13 of the 54 ribosomal protein genes (encoding L1, L9, L11, L25, L31, L32, L33, L35, L36, S6, S15, S20, or S21) could be deleted in this way. Here, we employed a one-step E. coli genome recombineering technique 26, to create a series of genetic deletion mutants covering 41 ribosomal protein genes (excluding those that have already been covered by the publically-available Keio collection). Deleterious effects of the deletions are minimized by the presence of a complementing plasmid carrying the gene of interest, and by precise genomic replacement using an antibiotic marker. We have begun to use these deletion strains to probe the functions of individual ribosomal proteins in translation.
Results
Deletion of ribosomal protein genes in the E. coli chromosome
Because many ribosomal proteins are thought to be essential, we deleted each chromosomal gene in the presence of a plasmid-born, inducible copy of the same gene (see Supplemental Experimental Procedures). We first individually cloned each of the 41 targeted ribosomal protein (RP) genes (amplified from E. coli DH10B genomic DNA) into a target plasmid (pCDSSara) under an arabinose-inducible araBAD promoter (Fig. 1). E. coli strain DH10B was used as the parental strain due to its ability to stably maintain these complementing plasmids. A selective toxic marker sacB, encoding levansucrase from Bacillus subtilis, was also included within each target plasmid to facilitate subsequent exchange of the plasmid by a vector of interest. The toxic effects conferred by sacB and successful exchange of pCDSSara-RP with another RP expression plasmid were confirmed in the presence of 5% sucrose (Fig. S1). pCDSSara-RP plasmids were then transformed into E. coli DH10B, and the corresponding genes on the chromosome were replaced with the chloramphenicol acetyltransferase (CAT) gene by λ Red-mediated recombination in the presence of arabinose 26. The CAT gene was amplified by polymerase chain reaction such that it included 40-100 bp extensions homologous to the genomic regions flanking each target gene. These PCR fragments were purified and transformed into cells expressing the λ-Red recombination system in the presence of the complementing plasmid.
Fig. 1. Complementing plasmids which rescue genomic deletions of ribosomal protein genes.
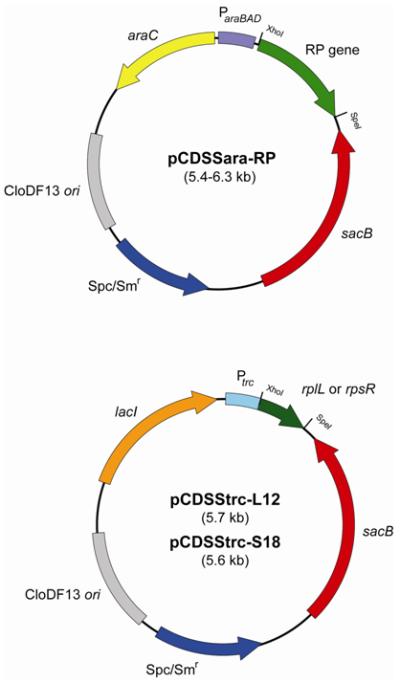
All of the genes encoding the 41 targeted ribosomal proteins in the E. coli genome were disrupted in the presence of a complementing plasmid. Of these, 39 genes were disrupted in the presence of pCDSSara-RP (RP corresponds to the protein name encoded by the gene) which expresses the ribosomal protein in the presence of arabinose induction. rplL and rpsR (encoding L12 and S18, respectively) were disrupted in the presence of pCDSStrc-L12 or S18, respectively, which expresses the protein in the presence of IPTG induction. Spc, specitinomycin; Sm, streptomycin.
Roughly 70% of the targeted genes (29 genes) were precisely replaced with the CAT gene; the remainder required partial replacement after our initial attempts failed (Tables 1; Fig. S2). For rplD (encoding L4), rplO (L15), rpmD (L30), and rpsE (S5) which contain an internal Shin-Dalgarno (SD) sequence for the 3′-downstream gene, the stop codon in the CAT gene was introduced immediately 5′ to the SD sequence, preserving translation of the downstream gene. For rplE (encoding L5), rplM (L13), rplW (L23), rpmB (L28), rpsB (S2), and rpsC (S3), the 5′ region of the gene was left intact, and the 3′ portion was replaced by an in-frame stop codon followed by a SD sequence and the CAT gene. Likewise, for rpsD (encoding S4) and rpsJ (S10), the 3′ region of the gene was left intact, and the 5′ portion was replaced by the CAT gene, followed by a SD sequence and a start codon to minimize possible polar effects on downstream genes within the same operon. Probable reasons for the difficulties associated with complete replacement of some of the RP genes are the presence of an important regulatory element within the polycistronic mRNAs and/or stable secondary structures in the recombination sites. Unexpectedly, replacement of the rplL and rpsR genes, encoding L7/L12 and S18, respectively, occurred only when the plasmid copy of the gene was expressed under control of an IPTG (isopropyl β-D-1-thiogalactopyranoside)-inducible trc promoter, perhaps due to a requirement for higher expression levels (Fig. 1). All recombination events were verified by genomic PCR with primer pairs targeting the CAT gene and flanking regions in the genome (Fig. S3).
Table 1.
Summary of deletion strains of ribosomal protein genesa.
| Protein | Gene | Operon | Replaced (bp)b |
Remaining (bp)c |
Plasmidd | Essentialitye |
|---|---|---|---|---|---|---|
| L2 | rplB | S10 | All | None | pCDSSara-L2 | Essential |
| L3 | rplC | S10 | All | None | pCDSSara-L3 | Essential |
| L4 | rplD | S10 | 1-585 | [586-606] | pCDSSara-L4 | Essential |
| L5 | rplE | spc | 130-540 | 1-129 | pCDSSara-L5 | Essential |
| L6 | rplF | spc | All | None | pCDSSara-L6 | Essential |
| L10 | rplJ | rif | All | None | pCDSSara-L10 | Essential |
| L7/L12 | rplL | rif | All | None | pCDSStrc-L12 | Essential |
| L13 | rplM | L13 | 31-429 | 1-30 | pCDSSara-L13 | Essential |
| L14 | rplN | spc | All | None | pCDSSara-L14 | Essential |
| L15 | rplO | spc | 1-424 | [425-435] | pCDSSara-L15 | Nonessential |
| L16 | rplP | S10 | All | None | pCDSSara-L16 | Essential |
| L17 | rplQ | alpha | All | None | pCDSSara-L17 | Essential |
| L18 | rplR | spc | All | None | pCDSSara-L18 | Essential |
| L19 | rplS | S16 | All | None | pCDSSara-L19 | Essential |
| L20 | rplT | L20 | All | None | pCDSSara-L20 | Essential |
| L21 | rplU | L21 | All | None | pCDSSara-L21 | Nonessential |
| L22 | rplV | S10 | All | None | pCDSSara-L22 | Essential |
| L23 | rplW | S10 | 31-303 | 1-30 | pCDSSara-L23 | Essential |
| L24 | rplX | spc | All | None | pCDSSara-L24 | Nonessential |
| L27 | rpmA | L21 | All | None | pCDSSara-L27 | Nonessential |
| L28 | rpmB | L28 | 31-237 | 1-30 | pCDSSara-L28 | Essentialf |
| L29 | rpmC | S10 | All | None | pCDSSara-L29 | Nonessential |
| L30 | rpmD | spc | 1-171 | [172-180] | pCDSSara-L30 | Nonessential |
| L34 | rpmH | L34 | All | None | pCDSSara-L34 | Nonessential |
| S1 | rpsA | S1 | All | None | pCDSSara-S1 | Essential |
| S2 | rpsB | S2 | 31-726 | 1-30 | pCDSSara-S2 | Essential |
| S3 | rpsC | S10 | 31-702 | 1-30 | pCDSSara-S3 | Essential |
| S4 | rpsD | alpha | 1-528 | 529-621 | pCDSSara-S4 | Essential |
| S5 | rpsE | spc | 1-492 | [493-504] | pCDSSara-S5 | Essential |
| S7 | rpsG | str | All | None | pCDSSara-S7 | Essential |
| S8 | rpsH | spc | All | None | pCDSSara-S8 | Essentialf |
| S9 | rpsI | L13 | All | None | pCDSSara-S9 | Nonessential |
| S10 | rpsJ | S10 | 1-192 | 193-312 | pCDSSara-S10 | Essential |
| S11 | rpsK | alpha | All | None | pCDSSara-S11 | Essential |
| S12 | rpsL | str | All | None | pCDSSara-S12 | Essential |
| S13 | rpsM | alpha | All | None | pCDSSara-S13 | Essentialf |
| S14 | rpsN | spc | All | None | pCDSSara-S14 | Essential |
| S16 | rpsP | S16 | All | None | pCDSSara-S16 | Essential |
| S17 | rpsQ | S10 | All | None | pCDSSara-S17 | Nonessential |
| S18 | rpsR | S6 | All | None | pCDSStrc-S18 | Essential |
| S19 | rpsS | S10 | All | None | pCDSSara-S19 | Essential |
Deletion strains of rplA (L1), rplI (L9), rplK (L11), rplY (L25), rpmE (L31), rpmF (L32), rpmG (L33), rpmI (L35), rpmJ (L36), rpsF (S6), rpsO (S15), rpsT (S20), and rpsU (S21) are available from Keio collection 25.
The indicated region were replaced by the chloramphenicol acetyltransferase (CAT) gene as shown in Fig. S1. All strains exhibited chloramphenicol resistance at ≥7 μg/ml. The base-pair number in the gene is indicated.
The regions in brackets indicate non-coding sequences (Fig. S2), and the others indicate open reading frames (Fig. S2c and S2d).
The complementing plasmid carrying the ribosomal protein gene is listed. Gene expression is controlled by either arabinose (pCDSSara) or IPTG (pCDSStrc).
Essentiality of the plasmid gene copy was analyzed by both growth dependence on inducer and dispensability of the plasmid on LB media at 30 °C and 37 °C.
Essentiality of ribosomal protein genes
In our deletion mutants, the full-length, wild-type protein is conditionally expressed from an inducible copy of the targeted gene encoded within the complementing plasmid. If the gene is essential for survival of the cell under a given condition, the absence of arabinose or IPTG to induce gene expression should result in cell death. If the gene is nonessential, cells would survive in the absence of an inducer. It was found that 5 mutants (with chromosomal deletion of the gene encoding L15, L21, L29, L30, or S9) showed obvious growth in the absence of an inducer (Fig. 2a). To test the essentiality of the genes more thoroughly, all mutants were grown in liquid media in the absence of an inducer at either 30 °C or 37 °C, and then plated on media containing 5% sucrose to select for cells that have lost the complementing plasmid. Surviving colonies were analyzed for the absence of the target gene and for the presence of the desired chromosomal recombination event (Fig. S4).
Fig. 2. Growth analysis of deletion strains.

(a) Growth dependence of the deletion mutants on the complementing genes. All strains contained a genomic deletion (Δ) and a plasmid (p) copy of the ribosomal protein gene indicated. Overnight cultures of the mutants grown in the presence of 0.2% arabinose (for all except ΔrplL pL12 and ΔrpsR pS18) or 1 mM IPTG (for ΔrplL pL12 and ΔrpsR pS18) were diluted 100-fold, spotted on LB agar supplemented with antibiotics and either the inducer (+) or 0.2% glucose (−), and incubated for 15-36 h at 37 °C. (b) Temperature dependence of knockout strains lacking the complementing plasmid. pCDSSara plasmids containing nonessential ribosomal protein genes were removed by sacB-based counterselection, and overnight cultures of the resulting cells were diluted 100-fold, spotted, and grown on LB agar for 48 h at various temperatures.
Mutants that had lost their complementing plasmid included a more extensive set of non-essential genes, namely those encoding L15, L21, L24, L27, L29, L30, L34, S9, and S17 (Fig. 2b). Growth of the mutant lacking L21 was inhibited at lower temperature (30 °C), while growth of the mutants lacking L24 or L27 was inhibited at higher temperature (42 °C) (Fig. 2b). To our knowledge, mutants lacking L21 and L34 have not been reported. The absence of L21 was further confirmed by mass spectroscopy analysis of 14N-labeled ribosomes isolated from our L21 deletion strain, spiked with 15N-labeled ribosomes isolated from the parent DH10B strain 27. The results displayed a complete absence of protein L21 from intact 50S and 70S subunits isolated from our L21 deletion strain, and confirmed the stoichiometric presence of all other essential protein components in these ribosomes (Fig. 3). Here, “protein level” refers to the concentration (or abundance) of individual ribosomal proteins present in 14N labeled ribosomes isolated from the ΔL21 line, normalized to that of 15N labeled ribosomes isolated from the parent DH10B strain. The L35 signal was also missing from the data, but L35 was not detected in the wild-type sample either. In general, peptides from the shorter ribosomal proteins L31-L36 are more difficult to resolve by this technique 27. A similar analysis was performed for the L34 deletion strain, but for the same reason, no L34 peptides were identified for either the 14N-ribosomes from the deletion strain or the 15N-labeled wild-type ribosomes (data not shown). Mutants lacking L15, L24, L27, L29, L30, S9, or S17 have been isolated previously by various groups in diverse contexts 28; 29; 30; 31; 32. Mutants lacking L28 or S13 30; 31; 33 as well as an insertion mutant of the S8 gene 34 have also been isolated previously, but we were unable to remove the complementing plasmid from the corresponding strains we created, possibly due to differences in nature of mutation and strain background.
Fig. 3. Protein Level of 70S subunit from the ΔL21 mutant ribosomes.
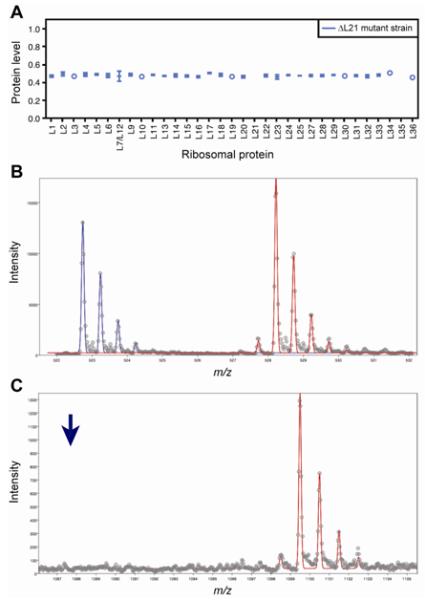
(a) Protein level for a single fraction from the 70S subunit peak. Error bars represent the standard deviation from multiple measurements from different peptides/ions in the same protein. In the case were a single measurement was obtained, an open circle is used. (b) A peptide from L5 (MWEFFER) is identified as being present in both the L21 mutant strain (blue) and the 15N ribosomal standard (red). (c) A peptide from L21 (MYAVFQSGGK) is identified as being present in the 15N ribosomal standard (red) but absent in the L21 mutant strain. The position of where the 14N peptide would appear is shown with a blue arrow.
Probing importance of protein-RNA contacts in the ribosomal 30S A and 50S P sites
Crystal structures of ribosomal complexes have revealed intricate interactions between ribosomal proteins and RNAs, including rRNA, tRNA, and mRNA, within the ribosome 4; 35; 36. The importance of these interactions is poorly understood, in part due to the lack of a facile experimental system to create and analyze mutations in ribosomal proteins. By using the mutant strains obtained in this study, we were able to create a series of truncations in ribosomal proteins L5, L27, and S19, which are positioned in proximity to functional ribosomal sites, to evaluate the significance of interactions formed by these proteins (Fig. 4a). Mutant versions of the genes were placed under control of a trc promoter in a pTrc99A-derived plasmid compatible with pCDSSara carrying the wild-type copy, and introduced into the corresponding deletion mutants. Because of the absence of the genomic allele, a homogeneous population of mutant or wild-type protein would be expressed within the cell in the presence of either IPTG or arabinose, respectively; in the presence of sucrose, surviving cells will have lost the sacB-containing vector, resulting in the production of only mutant protein by the cell.
Fig. 4. Mutational analysis of L27, L5, and S19 function.

(a) A schematic view of protein-tRNA interactions and the catalytic centers of the ribosome. The N-terminal 16 residues of L27, residues 70-83 of L5, and C-terminal 16 residues of S19 are shown in red. tRNA nucleotides comprising sites of potential interaction with the proteins are shown by stick representation (Protein Data Bank entries 318H and 318I). (b) Growth analysis of mutants containing homogeneous populations of various truncated ribosomal proteins. Overnight cultures of mutants carrying both pCDSSara and pTrc plasmids containing a full-length or truncated version of the protein, respectively, were diluted 100-fold and spotted on LB agar supplemented with antibiotics and the inducer indicated. pTrc plasmids containing the wild-type gene or a green fluorescent protein (GFP) gene were also used as controls.
With this system, we found that when the C-terminal tail of S19, which protrudes toward the decoding center in the 30S A site, is truncated by as many as 7 amino acids (S19CΔ7), cell growth is substantially perturbed (Fig. 4b). Furthermore, removal of 3 consecutive Lys residues from the C terminus (S19CΔ3), which were unresolved in the crystal structures 4, did not significantly affect growth. In the 50S P site, removal of the L5 loop interacting with tRNA (L5Δ70-83) caused a severe growth defect, consistent with a recent study suggesting that the yeast counterpart of this loop (“P-site loop”) is important for cell viability and tRNA binding 37. Finally, truncation of 9 amino acids from the N-terminal tail of L27 (L27NΔ9), which should remove all interactions with A-site and P-site tRNAs, minimally affected cell growth when over-expressed in this system. This is not surprising due to the fact that the presence of L27 was found to be non-essential for cell viability.
Mutated or truncated ribosomal protein variants may have altered affinities for the ribosome. Indeed, a high level of truncated L27 (L27ΔN16) expression was required to restore growth of the L27 deletion (Fig. 4b), which may imply a reduced interaction with the ribosome. To assess ribosomal incorporation of the truncated protein variants used in this study (L5Δ70-83, L27NΔ16, L27NΔ20, and S19CΔ16; we were not able to grow ΔS19 cells expressing the S19CΔ23 truncation, suggesting this truncation may be lethal) we again used quantitative mass spectrometry. 14N-labeled proteins expressed by the deletion lines harboring the various truncations were assessed in comparison with 15N-labeled wild-type ribosomes. We found that all truncated variants analyzed were retained on the ribosome. In ΔL27 mutants expressing the L27NΔ16 or L27NΔ20 truncations, we observed elevated levels of truncated L27 protein in 14N-labeled samples as compared to the wild-type 15N-labeled samples. We observed retention of the S19CΔ16 protein in ΔS19 cells, however we were not able to detect any changes to the relative abundance of truncated S19, or any other ribosomal proteins in ΔS19 cells expressing S19CΔ16. Ribosomes from ΔL5 cells expressing the L5 truncation mutant (L5Δ70-83) were found to contain slightly elevated levels of L5 protein as compared wild-type ribosomes, but all other proteins detected in these ribosomes were in wild-type abundance. Additionally, with the exception of ribosomes isolated from ΔS19 cells expressing S19CΔ16, ribosomes containing these truncated variants (L5Δ70-83, L27NΔ16, and L27NΔ20) also had slightly elevated levels of ribosomal protein L31 (Fig. S5).
Discussion
The deletion lines generated here should accelerate further studies of the various activities of the ribosome and individual ribosomal proteins, and shed light on the mechanism of translation and assembly of ribonucleoprotein complexes 38; 39; 40. For example, studies of ribosome assembly pathways have been limited by the small number of available deletion mutants and difficulties associated with in vitro reconstitution of ribosome assembly 41. Analysis of in vivo assembly intermediates upon depletion of various ribosomal proteins should provide insights regarding the involvement of these proteins in the assembly of prokaryotic ribosomes. Moreover, the deletion collection generated here allows for extensive analysis of point or deletion mutations of specific ribosomal protein genes. Our data suggest that E. coli DH10B can survive deletion of any one of 9 ribosomal protein genes among those targeted, including 2 genes (encoding L21 and L34) that are considered to be essential. L21 is encoded by the rplU gene, which is co-transcribed with its downstream gene, rpmA, encoding L27. Previous attempts to generate viable L21 deletion lines may have failed due to unintentional alteration of L27 expression. L21 is assembled into the ribosome near the 5′ end of 23S rRNA and forms extensive interactions with L20. A previous in vitro study showed that omitting L21 affects binding of L30 to assembly intermediates 42. However, we were unable to detect substantial alteration in the relative abundance of any ribosomal proteins in the large subunit by mass spectroscopy when L21 was absent in vivo. These results suggest that in vivo ribosome assembly pathways may differ in some aspects from those in vitro 43.
To further illustrate the utility of this strain collection, we carried out a preliminary study by generating truncation mutants of S19, L27, and L5. Each nucleotide triplet of mRNA is decoded by a specific aminoacyl-tRNA molecule, and this is contingent upon specific base pairing at the decoding center in the ribosomal 30S A site. The incoming aminoacyl-tRNA forms interactions at the anticodon stem loop with two proteins, S12 and S19. S12 has been extensively investigated because mutations in the gene affect both translational fidelity and antibiotic resistance (e.g., “restrictive” mutations causing hyperaccuracy) 14. However, S19 has been poorly studied until recent crystal structures revealed its C-terminal interactions with tRNA and possible involvement in decoding 4. The terminal 3 amino acids (4 amino acids in Thermus thermophilus) are unresolved in the crystal structures, and therefore the nature of their interactions with tRNA remains unclear. We observed nearly wild-type growth when the C-terminal tail of S19 was truncated by only 3 residues, suggesting minimal participation of these residues during protein translation. However, the 4 residues preceding them seem to play an important role in S19 function, as our truncation analysis revealed very poor growth when the C-terminal 7-13 residues of this protein were deleted. Although these data indicate the importance of S19 within the 30S A site, further studies are necessary to elucidate the precise role of S19 in translation.
The peptidyltransferase center is the catalytic center for peptide bond formation on the large subunit and is mainly comprised of rRNA, which supports the hypothesis that peptide bond formation was once catalyzed solely by RNA in the prebiotic environment 44; 45. However, in several crystal structures of bacterial ribosomes, the N-terminal tail of L27 protrudes into the peptidyltransferase center and contacts the backbone of P-site and A-site tRNAs 4; 36. Although L27 is non-essential, complete depletion of the protein strongly perturbs growth as well as several ribosomal functions (Fig. 2) 10; 28. It was previously shown that truncation of as few as 3 amino acids from the N-terminus of L27 decreases peptidyltransferase activity 10, though catalysis of peptide bond formation may not directly involve L27 46. We show here that increased expression levels of truncated L27 (as observed when expressing N-terminal deletions of 3-16 residues, but not observed when expressing an N-terminal deletion of 20 residues) can alleviate deleterious effects on growth, suggesting that the L27 N-terminal tail may also play a role in assembly of the 50S subunit.
L5 lies at the interface of the 30S and 50S subunits, generating inter-subunit bridge B1b, and makes extensive contacts with 5S rRNA. L5 also forms interactions with tRNA within the 50S P site, specifically at the elbow region of tRNA. We postulated that removal of the tRNA-interacting loop from L5 might perturb the precise positioning of the P-site tRNA at the peptidyltransferase center and thus affect the catalytic efficiency of peptide bond formation. Indeed, cells expressing an L5 mutant without this loop (L5Δ70-83) showed severely impaired growth. Although it is also plausible that the removal of the loop caused unfolding or a substantial decrease in affinity of L5 for the ribosome, yeast L11 (eukaryotic homolog of L5) with a similar truncation was retained on the ribosome and affected tRNA binding 37. Further investigation of the precise role of the L5-tRNA interaction in the P site remains to be performed in the future. Mutated or truncated ribosomal protein variants may have altered affinities for the ribosome. The methods described here would allow investigators to determine the ribosomal retention and relative abundance of such proteins in sucrose gradient-isolated ribosomes.
Engineering of the cellular translational machinery has allowed the biosynthesis of proteins containing non-native amino acids with diverse chemical properties 47. These amino acids can be genetically incorporated into cellular proteins by orthogonal aminoacyl-tRNA synthetase (AARS)/tRNA pairs in response to either nonsense codons or frame-shift (4-base) codons 48; 49. Notably, it was recently shown that the 16S rRNA can be engineered to enhance decoding of these frame-shift codons by a non-natural aminoacyl-tRNA in E. coli 50; 51. Just as with rRNA, ribosomal proteins are involved in conformational changes of the ribosome and interactions with other translation components. It is likely that engineering of functionally critical ribosomal proteins, by directed evolution, may also improve the translational efficiency with which non-native amino acids can be incorporated in response to nonsense or frame-shift condons. Fine-tuning of ribosomal protein function may also provide differential modulation of ribosomal activities, including decoding, peptidyltransfer, and intermolecular interactions. Since ribosomal proteins can be considered as later additions to the primitive all-RNA ribosome core 52, it is also of interest to examine whether any of the ribosomal protein components can be functionally replaced in their entirety by RNA, either by cis-acting insertion libraries placed near the site of protein deletion, or by trans-acting in vivo RNA aptamers.
Materials and Methods
Plasmid design
pCDSSara and pCDSStrc were constructed by combining PCR-amplified fragments containing the pCloDF13 origin and the spectinomycin/streptomycin-resistance marker derived from pCDF-1b (Novagen), the sacB gene from B. subtilis, and the araBAD or trc promoter from pBAD (Invitrogen) or pTrc99A (GenBank: A13038.1), respectively. The ribosomal protein genes were PCR amplified from genomic DNA of E. coli DH10B (F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 endA1 recA1 deoR Δ(ara,leu)7697 araD139 galU galK nupG rpsL(StrR) λ−) with alteration of the native stop codon to TAA (if not already) and cloned into the XhoI and SpeI sites of pCDSSara or pCDSStrc (only for rplL and rpsR).
Knockout experiments
These plasmids (pCDSSara or pCDSStrc for rplL and rpsR) were transformed into DH10B carrying pKD46, the expression plasmid for λ-Red recombinase 26. These cells were grown at 30 °C in 2YT media containing 50 μg/ml spectinomycin and 100 μg/ml ampicillin. 0.2% arabinose (pCDSSara) or both 0.2% arabinose and 1 mM IPTG (pCDSStrc) were added to the media when the optical density at 600 nm (OD600) reached 0.15-0.2; cells were harvested when OD600 was approximately 0.5; and electrocompetent cells were prepared by washing with 10% glycerol 3 times at 4 °C. Freshly-prepared electrocompetent cells were transformed with 300-1000 ng of DNA containing a CAT gene sequence PCR-amplified from pEVOL 53. Primers containing 40-70 nt 3′ overhangs homologous to the target genomic regions were used to amplify the CAT gene. A spacer sequence containing a Shine-Dalgarno sequence was included in the template if necessary (Fig. S2). For the rplL, rpsB, and rpsR deletions, DNA fragments containing a CAT gene and ~100-bp extension with homology to the flanking genomic regions were constructed by overlap extension PCR to increase propensity for recombination. Transformants were recovered in 2YT containing 0.2% arabinose (pCDSSara) or 1 mM IPTG (pCDSStrc), plated on LB agar containing 50 μg/ml spectinomycin, 7-15 μg/ml chloramphenicol, and arabinose or IPTG, and incubated at 37 °C for 2-4 days. Surviving colonies were individually analyzed by genomic PCR using taq polymerase (Invitrogen) and primers annealing to the CAT gene (5′-CAT Fwd, 5′-AATTAACTGCAGTTACTGCCACTCATCGCAGTACTGTTG-3′; 3′-CAT Rev, 5′-AATTAACATATGGAGAAAAAAATCACTGGATATACC-3′) and/or to the flanking genomic regions ~100-bp upstream or downstream of the target gene (Fig. S3). Loss of pKD46 from all strains was confirmed by the loss of ampicillin resistance (data not shown).
Vector removal/exchange
To remove pCDSSara or pCDSStrc plasmids from the deletion mutants, cells were grown overnight on LB media containing 7 μg/ml chloramphenicol. 100 μl of the culture was spread on LB agar containing 7 μg/ml chloramphenicol and 5% sucrose and incubated at 30 °C or 37 °C for 2-4 days. Loss of the plasmid was confirmed on LB agar containing 50 μg/ml spectinomycin and the appropriate inducer. To test the facility for vector exchange, the IPTG-inducible pTrc-S16 plasmid was used for electro-transformation of our S16 deletion strain. These cells were recovered overnight (at least 8 h) in 2YT + IPTG at 37 °C, then plated on LB agar plates containing 100ug/ml ampicillin with 1mM IPTG and 5% sucrose. Single colonies surviving this sucrose counter-selection were picked and streaked on LB agar plates containing 100ug/ml ampicillin + 1mM IPTG, 100ug/ml ampicillin, or 50ug/ml spectinomycin + 0.2% arabinose (Fig. S1).
Truncation analysis
For expression of truncated ribosomal proteins, the pTrc99A vector was PCR amplified using primers SpeI-pTrc-Fwd 5′-AATTAAACTAGTGGCGGATGAGAGAAGATTTTCAGCC-3′ and XhoI-pTrc-Rev 5′-AATTAACTCGAGTTCCTCCTGTGTGAAATTGTTATCCGCTCACAATTCC-3′ and digested by XhoI and SpeI. The truncated L5 Δ70-83 gene was amplified by overlap extension PCR, and the truncated L27 and S19 genes were amplified by PCR. These fragments were digested and cloned with the XhoI and SpeI sites of the pTrc99A vector created above. The resulting plasmids contain genes under control of the trc promoter. The ΔrplE pL5 ΔrpmA pL27, and ΔrpsS pS19 strains were transformed with these plasmids, and transformants were recovered in 2YT containing 0.2% arabinose for 2 h and selected on LB agar supplemented with 50 μg/ml spectinomycin, 100 μg/ml ampicillin, and 0.2% arabinose overnight at 37 °C.
Sample preparation for mass spectrometry
Cells were grown at 37 °C in minimal media (per liter: 12.08 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 2 g glucose, 20.3 mg MgCl2, 24.6 mg MgSO4, 30 mg Na2EDTA, 7.5 mg CaCl2, 25.05 mg FeCl3, 0.27 mg ZnSO4, 0.27 mg CuSO4, 0.27 mg CoCl2, 0.18 mg MnSO4, 20 mg biotin, 20 mg folic acid, 50 mg thiamine, 50 mg niacinamide, 50 mg riboflavin, 50 mg Ca-pantothenate, and 0.001 mg vitamin B12, with either 1 g (14NH4)2SO4 or 1 g (15NH4)2SO4 for 14N- or 15N-labeling of mutant or DH10B ribosomal proteins, respectively), harvested when OD600 was approximately 0.5 by centrifugation at 7,000 × g for 10 min, and stored at −80 °C for subsequent analysis. Frozen cell pellets for the L21 and L34 mutants and the parent DH10B strain were thawed and re-suspended in high magnesium lysis buffer (20 mM Tris HCl pH 7.5, 100 mM NH4Cl, 10mM MgCl2, 0.5 mM EDTA, 6 mM β-mercaptoethanol) and then lysed in a bead-beater (BioSpec Products, Inc, Bartesville, OK) using 0.1 mm zirconia/silica beads. Insoluble debris was removed by two centrifugation steps: an initial spin at 13000 × g for 10 min followed by a second spin at 13000 × g for 90 min. A 1mL aliquot of the supernatant was layered on a 35 mL 10-40% sucrose gradient (50 mM Tris HCl pH 7.8, 10 mM MgCl2, 100 mM NH4Cl) in a SW 32 rotor (Beckman Coulter, Fullerton, CA) and centrifuged at 26,000 rpm at 4 °C for 16 h in order to separate out subunits. The gradients were fractionated with a Brandel fractionator. Subunit concentration in the fractions was estimated by measuring the optical density at 260 nm. The quantitative mass spectrometry experiments were performed on two fractions from the sucrose gradient peak corresponding to the 50S subunit and two fractions from the 70S subunit for both the ΔL21 and ΔL34 mutants. Eights fractions were run in total. For all fractions, 50 pmol of 14N sample was used and 50 pmol of 15N 70S was added as a standard. Samples were then prepared for electrospray ionization time-of flight (ESI-TOF) analysis as described previously 43.
ESI-TOF mass spectrometry
The peptides were analyzed on an Agilent 1100 Series high performance liquid chromatography (HPLC) instrument coupled to an Agilent ESI-TOF instrument with capillary flow electrospray. Peptides were separated on an ACN gradient in 0.1% formic acid at a flow rate of 7 μL/min. The steps of the gradient were 5-15% ACN over 10 min, 15-50% ACN over 70 min and 50-95% ACN over 4 min. Data were collected over the m/z range of 100-1300.
Quantitative analysis of protein levels using mass spectrometry
Briefly, a feature list is generated using software from Agilent (Mass Hunter and Mass Profiler), where a feature is the entire isotopic envelope from a single ion and contains the monoisotopic peak and isotopomers. Features are defined by the m/z value of the monoisotopic peak, the charge of the ion and the retention time on the column. Feature lists are compared to the theoretical digest of all ribosomal proteins from the E. coli 70S ribosome. Experimental features were assigned possible identities when the theoretical ion’s charge was identical and its mass was within 50 ppm of the experimental feature’s mass. 14N/15N pairs were extracted from the possible identities when the features to which they matched had a retention time within 0.1 min of each other. For the cases where multiple identities were found for the same feature or the same peptide was matched the multiple proteins, these features were discarded. For the remaining feature pairs, segments of the complete mass spectrum are extracted and theoretical isotope distributions were fit to extracted spectra using the program Isodist 54. Two distributions were fit, one unlabeled (natural abundance deletion strains) and one fully labeled (15N fixed at 99.3%, corresponding to the external standard, in this case wild-type ribosomes). The fits for all extracted spectra were evaluated visually, and the spectra with poor fitting distributions were rejected. The amplitudes given by Isodist yield relative amounts of unlabeled and 15N labeled peptide in the sample and can be used to calculate the abundance of each protein 44. In addition all samples were also run searching for and extracting only the 15N standard peaks. By searching for the presence of 15N and not 14N we could verify that the absent L21 protein in the L21 deletion strain was truly absent in the deletion strain and not the standard (Fig. 3b and 3c).
Supplementary Material
Acknowledgments
We thank Drs. Huiwang Ai, Tsotne Javahishvili, and Charles E. Melancon for helpful comments and discussions. We are also grateful to Lyn’Al L. Nosaka and Han Xiao for technical assistance. We thank Dr. Eli Chapman for providing us with the pTrc99A plasmid (supplied as pTrc99A-GroEL). This work was supported by the Uehara Memorial Foundation (fellowship to S.S.), and the National Institutes of Health (NIH) grants R37-GM-053757 (to J.R.W) and grant 5R01 GM062159 (to P.G.S.).
Abbreviations used
- CAT
chloramphenicol acetyltransferase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- RP
ribosomal protein
- SD
Shine-Dalgarno
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available.
Propensity for complementary vector removal/exchange, genomic deletion strategies, and verification of deletion strains by PCR can be found as supplementary material online.
References
- 1.Roberts E, Sethi A, Montoya J, Woese CR, Luthey-Schulten Z. Molecular signatures of ribosomal evolution. Proc. Natl. Acad. Sci. USA. 2008;105:13953–13958. doi: 10.1073/pnas.0804861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 3.Wimberly BT, Brodersen DE, Clemons WM, Jr., Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 4.Jenner L, Demeshkina N, Yusupova G, Yusupov M. Structural rearrangements of the ribosome at the tRNA proofreading step. Nat. Struct. Mol. Biol. 2010;17:1072–1078. doi: 10.1038/nsmb.1880. [DOI] [PubMed] [Google Scholar]
- 5.Hoang L, Fredrick K, Noller HF. Creating ribosomes with an all-RNA 30S subunit. Proc. Natl. Acad. Sci. USA. 2004;101:12439–12443. doi: 10.1073/pnas.0405227101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takyar S, Hickerson RP, Noller HF. mRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 7.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy F. V. t., Weir JR, Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 2009;326:694–699. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 10.Maguire BA, Beniaminov AD, Ramu H, Mankin AS, Zimmermann RA. A protein component at the heart of an RNA machine: the importance of protein L27 for the function of the bacterial ribosome. Mol. Cell. 2005;20:427–435. doi: 10.1016/j.molcel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Cukras AR, Southworth DR, Brunelle JL, Culver GM, Green R. Ribosomal proteins S12 and S13 function as control elements for translocation of the mRNA:tRNA complex. Mol. Cell. 2003;12:321–328. doi: 10.1016/s1097-2765(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 12.Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell. 2005;121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Fei J, Kosuri P, MacDougall DD, Gonzalez RL. Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol. Cell. 2008;30:348–359. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Zaher HS, Green R. Hyperaccurate and error-prone ribosomes exploit distinct mechanisms during tRNA selection. Mol. Cell. 2010;39:110–120. doi: 10.1016/j.molcel.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dabbs ER. Mutational alterations in 50 proteins of the Escherichia coli ribosome. Mol. Gen. Genet. 1978;165:73–78. doi: 10.1007/BF00270378. [DOI] [PubMed] [Google Scholar]
- 16.Isono S, Isono K. Mutations affecting the structural genes and the genes coding for modifying enzymes for ribosomal proteins in Escherichia coli. Mol. Gen. Genet. 1978;165:15–20. doi: 10.1007/BF00270371. [DOI] [PubMed] [Google Scholar]
- 17.Zengel JM, Young R, Dennis PP, Nomura M. Role of ribosomal protein S12 in peptide chain elongation: analysis of pleiotropic, streptomycin-resistant mutants of Escherichia coli. J. Bacteriol. 1977;129:1320–1329. doi: 10.1128/jb.129.3.1320-1329.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaman S, Fitzpatrick M, Lindahl L, Zengel J. Novel mutations in ribosomal proteins L4 and L22 that confer erythromycin resistance in Escherichia coli. Mol. Microbiol. 2007;66:1039–1050. doi: 10.1111/j.1365-2958.2007.05975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorini L, Kataja E. Phenotypic Repair by Streptomycin of Defective Genotypes in E. Coli. Proc. Natl. Acad. Sci. USA. 1964;51:487–493. doi: 10.1073/pnas.51.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozaki M, Mizushima S, Nomura M. Identification and functional characterization of the protein controlled by the streptomycin-resistant locus in E. coli. Nature. 1969;222:333–339. doi: 10.1038/222333a0. [DOI] [PubMed] [Google Scholar]
- 21.Birge EA, Kurland CG. Reversion of a streptomycin-dependent strain of Escherichia coli. Mol. Gen. Genet. 1970;109:356–369. doi: 10.1007/BF00267704. [DOI] [PubMed] [Google Scholar]
- 22.Brownstein BL, Lewandowski LJ. A mutation suppressing streptomycin dependence. I. An effect on ribosome function. J. Mol. Biol. 1967;25:99–109. doi: 10.1016/0022-2836(67)90281-1. [DOI] [PubMed] [Google Scholar]
- 23.Siller E, DeZwaan DC, Anderson JF, Freeman BC, Barral JM. Slowing bacterial translation speed enhances eukaryotic protein folding efficiency. J Mol. Biol. 2010;396s:1310–1318. doi: 10.1016/j.jmb.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 24.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharpe Elles LM, Sykes MT, Williamson JR, Uhlenbeck OC. A dominant negative mutant of the E. coli RNA helicase DbpA blocks assembly of the 50S ribosomal subunit. Nucleic Acids Res. 2009;37:6503–6514. doi: 10.1093/nar/gkp711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wower IK, Wower J, Zimmermann RA. Ribosomal protein L27 participates in both 50 S subunit assembly and the peptidyl transferase reaction. J. Biol. Chem. 1998;273:19847–19852. doi: 10.1074/jbc.273.31.19847. [DOI] [PubMed] [Google Scholar]
- 29.Stoffler-Meilicke M, Dabbs ER, Ehrlich R, Stoffler G. A mutant from Escherichia coli which lacks ribosomal proteins S17 and L29 used to localize these two proteins on the ribosomal surface. Eur. J. Biochem. 1985;150:485–490. doi: 10.1111/j.1432-1033.1985.tb09048.x. [DOI] [PubMed] [Google Scholar]
- 30.Dabbs ER. Selection for Escherichia coli mutants with proteins missing from the ribosome. J. Bacteriol. 1979;140:734–737. doi: 10.1128/jb.140.2.734-737.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubunenko M, Baker T, Court DL. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J. Bacteriol. 2007;189:2844–2853. doi: 10.1128/JB.01713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franceschi FJ, Nierhaus KH. Ribosomal protein L20 can replace the assembly-initiator protein L24 at low temperatures. Biochemistry. 1988;27:7056–7059. doi: 10.1021/bi00418a058. [DOI] [PubMed] [Google Scholar]
- 33.Cukras AR, Green R. Multiple effects of S13 in modulating the strength of intersubunit interactions in the ribosome during translation. J. Mol. Biol. 2005;349:47–59. doi: 10.1016/j.jmb.2005.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang Y, Durfee T, Glasner JD, Qiu Y, Frisch D, Winterberg KM, Blattner FR. Systematic mutagenesis of the Escherichia coli genome. J. Bacteriol. 2004;186:4921–4930. doi: 10.1128/JB.186.15.4921-4930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 36.Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 37.Rhodin MH, Dinman JD. A flexible loop in yeast ribosomal protein L11 coordinates P-site tRNA binding. Nucleic Acids Res. 2010;38:8377–8389. doi: 10.1093/nar/gkq711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 39.Kaczanowska M, Ryden-Aulin M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. Rev. 2007;71:477–494. doi: 10.1128/MMBR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cech TR. Crawling out of the RNA world. Cell. 2009;136:599–602. doi: 10.1016/j.cell.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Sykes MT, Williamson JR. A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys. 2009;38:197–215. doi: 10.1146/annurev.biophys.050708.133615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herold M, Nierhaus KH. Incorporation of six additional proteins to complete the assembly map of the 50 S subunit from Escherichia coli ribosomes. J. Biol. Chem. 1987;262:8826–8833. [PubMed] [Google Scholar]
- 43.Sykes MT, Shajani Z, Sperling E, Beck AH, Williamson JR. Quantitative proteomic analysis of ribosome assembly and turnover in vivo. J. Mol. Biol. 2010;403:331–345. doi: 10.1016/j.jmb.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 45.Noller HF. Evolution of Protein Synthesis from an RNA World. Cold Spring Harb. Perspect Biol. 2010:a003681. doi: 10.1101/cshperspect.a003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodnina MV, Beringer M, Wintermeyer W. How ribosomes make peptide bonds. Trends Biochem. Sci. 2007;32:20–26. doi: 10.1016/j.tibs.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 47.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 48.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 49.Anderson JC, Wu N, Santoro SW, Lakshman V, King DS, Schultz PG. An expanded genetic code with a functional quadruplet codon. Proc. Natl. Acad. Sci. USA. 2004;101:7566–7571. doi: 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007;25:770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 51.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 52.Bokov K, Steinberg SV. A hierarchical model for evolution of 23S ribosomal RNA. Nature. 2009;457:977–980. doi: 10.1038/nature07749. [DOI] [PubMed] [Google Scholar]
- 53.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 54.Sperling E, Bunner AE, Sykes MT, Williamson JR. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal. Chem. 2008;80:4906–4917. doi: 10.1021/ac800080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.