

Abstract
Purpose
To conduct a first-in-human phase I study to determine the dose-limiting toxicities (DLT), characterize the pharmacokinetic profile, and document the antitumor activity of IPI-926, a new chemical entity that inhibits the Hedgehog pathway (HhP).
Experimental Design
Patients with solid tumors refractory to standard therapy were given IPI-926 once daily (QD) by mouth in 28-day cycles. The starting dose was 20 mg, and an accelerated titration schedule was used until standard 3 + 3 dose-escalation cohorts were implemented. Pharmacokinetics were evaluated on day −7 and day 22 of cycle 1.
Results
Ninety-four patients (32F, 62M; ages, 39–87) received doses ranging from 20 to 210 mg QD. Dose levels up to and including 160 mg administered QD were well tolerated. Toxicities consisted of reversible elevations in aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin, fatigue, nausea, alopecia, and muscle spasms. IPI-926 was not associated with hematologic toxicity. IPI-926 pharmacokinetics were characterized by a slow absorption (Tmax = 2–8 hours) and a terminal half-life (t1/2) between 20 and 40 hours, supporting QD dosing. Of those HhP inhibitor-naïve patients with basal cell carcinoma (BCC) who received more than one dose of IPI-926 and had a follow-up clinical or Response Evaluation Criteria in Solid Tumors (RECIST) assessment, nearly a third (8 of 28 patients) showed a response to IPI-926 at doses ≥130 mg.
Conclusions
IPI-926 was well tolerated up to 160 mg QD within 28-day cycles, which was established as the recommended phase II dose and schedule for this agent. Single-agent activity of IPI-926 was observed in HhP inhibitor–naïve patients with BCC.
Introduction
The hedgehog pathway (HhP) is critical for normal mammalian embryonic development and for adult tissue remodeling (1). The pathway is maintained in an inactive state by Patched (Ptch), a transmembrane receptor that represses the activity of Smoothened (Smo), another transmembrane receptor. When HhP ligands bind Ptch, repression of Smo is relieved and signals are transduced that activate the Gli proteins, a family of transcription factors that turn on the transcription of many genes involved in growth and development. Aberrant activation of the HhP is associated with many types of cancer, including basal cell carcinoma (BCC; refs. 2, 3), medulloblastoma (4), pancreatic adenocarcinoma (5), small cell lung cancer (SCLC; ref. 6), metastatic prostate cancer (7), glioma (8), and hematologic malignancies (9). High levels of HhP activation, either through mutation of pathway components or through constitutive expression of HhP genes, have been implicated in the initiation of cancer as well as cancer cell survival, growth, and metastasis.
Upregulation of HhP has been found in tumor-associated stroma and associated with vascularization (10). Excessive stromal expansion can limit intratumoral drug delivery. Administration of IPI-926, a potent and selective inhibitor of HhP, can promote enhanced intratumoral concentration of gemcitabine in transgenic pancreatic cancer models (11). Finally, there is a growing body of evidence on the relevance of the HhP in tumor-initiating cell (TIC) signaling (9, 12); HhP inhibition diminishes tumor metastasis, thought to be dependent on TIC (13), and has been shown to inhibit TIC proportion, number, and signaling (14).
IPI-926 is a derivative of the known HhP inhibitor cyclopamine, with enhanced affinity, specificity, and drug-like properties. IPI-926 selectively antagonizes the HhP by binding to the Smo receptor with an IC50 of 1.4 nmol/L. The cell-based EC50 for inhibition of the HhP by IPI-926 is between 5 and 7 nmol/L. Potent in vivo activity of IPI-926 has been shown in several tumor models, with the most extensive evaluation in the B837Tx medulloblastoma allograft model. In this model, oral administration of IPI-926 results in dose-dependent inhibition of HhP activity [as assessed by downregulation of Gli1 (glioma-associated oncogene homolog 1) mRNA], antitumor activity, and significant prolongation of survival of mice bearing orthotopically implanted tumors (15). In a pancreatic tumor model with elevated Shh (sonic hedgehog) expression, IPI-926 inhibited Shh signaling in surrounding stromal tissue (Gli1 mRNA) and slows tumor growth. IPI-926 also delays regrowth of SCLC tumors following chemotherapy in preclinical xenograft models (16).
The objectives of this first-in-human study were to determine the maximum tolerated dose (MTD), safety, pharmacodynamics, pharmacokinetics, and antitumor activity of IPI-926 when administered to patients with advanced cancers. A food effect substudy evaluated the pharmacokinetic profile of IPI-926 taken with food compared with administration in a fasting state. Finally, an expanded cohort studied the efficacy of IPI-926 at the MTD in patients with advanced or metastatic BCCs.
Patients and Methods
Patient eligibility
Eligible patients had histologically confirmed malignancy (with basal cell cancer histology for BCC cohorts) for which standard curative or palliative measures did not exist, age ≥ 18 years, ECOG performance status ≤ 2, life expectancy ≥ 12 weeks, adequate bone marrow, hepatic, and renal function [absolute neutrophil count (ANC) ≥ 1,500/μL, platelet ≥ 100,000/μL, bilirubin <upper limit of normal (ULN), aspartate aminotransferase (AST) or alanine aminotransferase (ALT) <1.5 × ULN, and creatinine < 1.5 × ULN]. Patients could have received any prior therapy ending ≥28 days prior as long as there was no residual toxicity. Measurable disease was not a requirement for study entry. Patients with a history of active hepatitis B or C, brain metastases, or any severe, clinically significant, and/or uncontrolled medical condition were ineligible for the study. The Institutional Review Board of all participating centers granted approval and written informed consent was mandatory.
Treatment plan
IPI-926 was supplied by Infinity Pharmaceuticals, Inc. Treatment consisted of IPI-926 administered orally as a single dose 7 days before the initiation of the first treatment cycle to evaluate the single-dose pharmacokinetic profile, followed by IPI-926 given daily (QD) in 28-day cycles, in an outpatient setting. The IPI-926 starting dose was 20 mg QD, which is approximately one tenth of the severely toxic dose in 10% of animals (STD10) in the mouse following daily administration for 28 days. This is based on the U.S, Food and Drug Administration (FDA) decision tree guideline for selection of the relevant safety assessment species for oncologic products.
Treatment was administered until disease progression, intercurrent illness, unacceptable adverse event(s), or with-drawal of consent; treatment may have been temporary withheld due to adverse events. Retreatment required adequate laboratory parameters and resolution of nonhematologic toxicities (except alopecia and fatigue) to baseline or grade 1. Patients with treatment delays >14 days were removed from the study.
Assessments, follow-up, and monitoring
Before study entry, all patients had a clinical history and physical examination, performance status assessment, complete blood and platelet count, chemistries, urinalysis, pregnancy test (if applicable), EKG, and disease assessment by computed tomographic (CT) scan and/or skin exam for patients with BCCs. Hematology and chemistries were repeated weekly during the first 2 cycles and then every 2 weeks thereafter. CT scan of disease sites was conducted every 2 cycles. Adverse events were classified/graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events CTCAE v3.0. Adverse events were monitored from the initiation of treatment and followed until resolution or they returned to baseline. Response was evaluated by Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 (17). Patients were considered evaluable for toxicity once therapy started and were evaluable for efficacy if at least 1 complete cycle was administered.
Definition of dose-limiting toxicity, MTD, and dose-escalation
A dose-limiting toxicity (DLT) was defined as any drug-related grade ≥4 hematologic toxicity plus grade 3 febrile neutropenia and/or grade 3 thrombocytopenia with grade ≥2 hemorrhage; and any grade ≥3 nonhematologic toxicity (grade 3 or higher diarrhea, nausea, and vomiting were considered dose-limiting only if persistent despite optimal treatment). An accelerated titration design was used initially for dose escalation, where one patient was treated at each dose level until the occurrence of either a grade ≥2 toxicity or a predefined maximum steady state plasma concentration of IPI-926 was reached. Afterward, a modified Fibonacci scheme was implemented, in which 3 patients were treated per dose level. If no DLTs were encountered in any of the 3 patients in cycle 1, dose-escalation was allowed. If 1 of the 3 patients experienced a DLT, 3 more patients were to be enrolled at the same dose level, and if none of these 3 additional patients experienced a DLT, dose-escalation was allowed. The MTD was defined as the highest dose tested with at least 6 patients evaluable for toxicity of which fewer than 33% experienced a DLT attributable to the study drug(s).
Expanded phase
Several expanded cohorts were accrued: a main expansion cohort (patients with any advanced and/or metastatic solid tumor malignancies), 2 separate BCC cohorts, and a tumor biopsy cohort (patients who consented to pre- and postdose tumor biopsies). The objectives of these cohorts were to gain more experience in both a broad solid tumor and a BCC patient population, and learn about the drug pharmacodynamic effects. In addition, a food-effect sub-study was conducted in a subset of patients enrolled in the expanded phase.
Pharmacokinetic sampling and bioanalytical assay
Blood samples were collected with sodium heparin anti-coagulant for plasma isolation at the following time points following a single dose on day −7 and repeat daily dosing on day 22 during the first cycle: pretreatment, and 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 24, and 48 (day −7 only) hours following IPI-926 administration. In addition, pretreatment samples were obtained on days 1, 8, and 15 during cycles 1 and 2, day 22 of cycle 2, and every day 1 on subsequent cycles. In the food-effect substudy, samples were collected after single-dose administration under fasting and fed conditions to assess the impact of a high-fat meal on the pharmacokinetics of IPI-926. Plasma concentrations of IPI-926 and IPI-541 (N-hydroxy metabolite) were determined using a validated liquid chromatography-tandem mass spectrometric (LC-MS/MS) method.
IPI-926 and IPI-541 PK parameters were estimated using a noncompartmental approach (NCA) from the individual plasma concentration profiles using WinNonlin Enterprise version 5.3 (Pharsight Corporation). Actual sample collection times relative to the time of IPI-926 dose administration and the actual dose of IPI-926 administered were used to calculate PK parameters. Maximum plasma concentration (Cmax), time to maximum concentration (Tmax), area under the plasma concentration time curve (AUC), and elimination phase half-life (t1/2) were determined for both analytes. Apparent volume of distribution (Vz/F) and oral clearance (CL/F) were also determined for IPI-926 only.
Pharmacodynamic sampling and analysis
A biopsy of normal skin was conducted pretreatment and again 3 weeks into cycle 1. In the tumor biopsy expanded cohort, patients underwent sequential tumor biopsies at the same time points from accessible sites, according to each center’s policies. IPI-926 concentrations in normal skin biopsies were determined using an LC-MS/MS assay. RNA was isolated from the human skin samples and the level of Gli-1 mRNA expression was measured by reverse transcription polymerase chain reaction (RT-PCR).
Results
Patient characteristics
Between October 2009 and June 2011, 94 patients (32 female, 62 male; ages, 39–87 years) with advanced solid tumors received any amount of IPI-926. All 94 patients were evaluable for toxicity and response was evaluable in 88 patients. Of these 94 patients, 39 had a diagnosis of BCC. Demographics and other baseline characteristics are summarized in Table 1. Patients received IPI-926 for a median duration of 2.3 months (range, 0.03–23.5 months).
Table 1.
Patient characteristics
| BCC | Non-BCC | |
|---|---|---|
| Parameter | (n = 39) | (n = 55) |
| Age, y | ||
| Median (range) | 67 (50–87) | 60 (40–79) |
| Sex, n (%) | ||
| Male | 33 (85) | 29 (53) |
| Female | 6 (15) | 26 (47) |
| Cancer diagnosis, n (%) | ||
| BCC | 39 (100) | 0 (0) |
| Colorectal | 0 (0) | 7 (13) |
| Pancreatic | 0 (0) | 7 (13) |
| Chondrosarcoma | 0 (0) | 5 (9) |
| NSCLC | 0 (0) | 5 (9) |
| Ovarian | 0 (0) | 5 (9) |
| SCLC | 0 (0) | 3 (5) |
| Other | 0 (0) | 23 (42) |
| Years from cancer diagnosis to first dose | ||
| Median (range) | 13 (0.2–61) | 3 (0.5–22) |
| Stage at screening (BCC only), n (%) | ||
| II | 10 (26) | NA |
| III | 10 (26) | NA |
| IV | 19 (48) | NA |
| History of Gorlin syndrome, n (%) |
5 (13) | NA |
| Previous therapies, n (%) | ||
| Surgery | 37 (95) | 48 (87) |
| Systemic | 15 (38) | 49 (89) |
| Prior vismodegib | 9 (23) | 0 (0) |
| Radiotherapy | 18 (46) | 27 (49) |
Dose escalation
The starting dose of IPI-926 was 20 mg (Table 2). Five dose levels (20, 30, 40, 60, and 90 mg) were explored with 1-patient cohorts, and no ≥grade 2 toxicities were documented. At the 130-mg dose level, the first patient experienced grade 2 toxicity. Dosing at 130-mg dose level and escalation then proceeded per protocol in 3-patient cohorts following a modified Fibonacci schema. The next escalation was by 50% and went from 130 to 200 mg QD. At 200 mg, 2 patients experienced adverse events that met DLT criteria. The first patient, with a history of hepatitis C and alcohol use, experienced asymptomatic grade 3 AST elevation and grade 2 ALT elevation after receiving IPI-926 for 25 days. A second patient experienced an asymptomatic grade 3 ALT elevation after receiving IPI-926 for 21 days, which resolved when study drug was held; this patient resumed dosing at 130 mg QD.
Table 2.
Dose-escalation scheme and DLTs
| IPI-926 dose, mg |
DLTsa per total patients treated, n |
DLT description |
|---|---|---|
| 20 | 0/1 | NA |
| 30 | 0/1 | NA |
| 40 | 0/1 | NA |
| 60 | 0/1 | NA |
| 90 | 0/1 | NA |
| 130 | 0/56 | NA |
| 160b | 1/18 | Grade 3 AST and ALT elevation |
| 180c | 2/5 | Grade 3 fatigue and anorexia |
| Grade 3 AST and ALT elevation |
||
| 200c | 2/3 | Grade 3 AST elevation |
| Grade 3 ALT elevation |
||
| 210c | 2/7 | Grade 3 AST and ALT elevation |
| Grade 2 ALT elevation and hyperbilirubinemia |
Number of patients with a DLT.
Dose selected for phase II.
Intolerable dose level.
On the basis of DLT observations in the 200-mg dose cohort, 5 patients were enrolled in an intermediate dose cohort, receiving 160 mg QD. In this cohort, 1 of 4 patients had asymptomatic grade 3 AST and ALT elevations during cycle 1, which met the DLT criteria. Two additional patients did develop grade 2 AST and ALT abnormalities (not meeting DLT criteria), which were reversible when study drug was held. An expansion cohort at 130 mg QD added further confirmation of the observed safety and tolerability at or below 160 mg QD.
Given the asymptomatic and reversible nature of the liver enzyme elevations observed, doses above 130 mg QD were further explored under modified DLT criteria related to abnormal liver functions These modifications allowed for transaminase elevations up to 10× ULN in the absence of other signs or symptoms of compromised liver function. In the setting of hyperbilirubinemia or prolonged prothrombin time (i.e., elevated INR), the transaminase limit for DLT consideration was 3× ULN.
Escalation continued first to 160 mg QD without incidence and then to 210 mg QD. At the 210-mg dose level, 2 patients experienced DLTs, one patient an asymptomatic grade 3 AST and ALT elevation and the other patient a grade 2 ALT elevation and bilirubin elevation. Other liver function parameters were not affected, and the alterations improved after study drug were discontinued. An intermediate dose level of 180 mg was then explored, but DLTs occurred in 2 more patients, one patient with grade 3 fatigue and anorexia and the other with grade 3 AST and ALT elevation. Altogether 18 patients were treated at the 160 mg QD dose level, with one DLT of reversible ALT and AST elevation. In addition, there were 3 patients with BCCs originally dosed at 130 mg who had their doses increased to 160 mg without experiencing a DLT. Therefore, the recommended dose for further study of once daily IPI-926 was 160 mg.
Toxicity
Overall treatment was generally well tolerated. The treatment-emergent adverse events are listed in Table 3. Overall, the most common clinical adverse events were fatigue, nausea, and alopecia, with the vast majority of these ≤grade 2. Overall ≥grade 3 AST and ALT elevations occurred in 11% and 28% of patients, respectively, treated at 160 mg QD, the MTD. Elevations were generally reversible and asymptomatic, which did not indicate the potential for an association between IPI-926 administration and severe drug-induced liver injury. Class-specific toxicities such as muscle spasms, alopecia, and dysgeusia were reported by 33%, 22%, and 22% of patients, respectively, at the MTD. No significant hematologic toxicities were documented. No patients died while on study, and 6 patients (6%) were discontinued from the study due to toxicity. Ten patients (11%) died due to disease progression within 30 days of study discontinuation.
Table 3.
Summary of highest treatment-emergent nonhematologic and hematologic toxicity (all subjects treated)
| Treatment-emergent adverse events (frequency ≥ 20%) |
<130 mg (n = 5) | 130 mg (n = 56) | 160 mg (n = 18) | >160 mg (n = 15) | ||||
|---|---|---|---|---|---|---|---|---|
| Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Hematologic (lab) | ||||||||
| Hemoglobin | 3 (60) | 0 (0) | 39 (64) | 2 (4) | 12 (66) | 1 (6) | 12 (80) | 1 (7) |
| Thrombocytopenia | 0 (0) | 0 (0) | 12 (21) | 3 (5) | 4 (22) | 0 (0) | 1 (7) | 0 (0) |
| Nonhematologic | ||||||||
| ALT elevation (lab) | 1 (20) | 0 (0) | 40 (71) | 4 (7) | 10 (55) | 5 (28) | 11 (73) | 4 (27) |
| AST elevation (lab) | 1 (20) | 0 (0) | 41 (73) | 2 (4) | 14 (78) | 2 (11) | 12 (80) | 3 (20) |
| Fatigue | 2 (40) | 0 (0) | 29 (52) | 3 (5) | 8 (44) | 3 (17) | 6 (40) | 2 (13) |
| Nausea | 1 (20) | 0 (0) | 23 (41) | 0 (0) | 6 (33) | 0 (0) | 7 (47) | 0 (0) |
| Alopecia | 1 (20) | 0 (0) | 17 (30) | 0 (0) | 4 (22) | 0 (0) | 2 (13) | 0 (0) |
| Muscle spasms | 2 (40) | 0 (0) | 11 (20) | 1 (2) | 6 (33) | 0 (0) | 4 (27) | 0 (0) |
| Decreased appetite | 0 (0) | 0 (0) | 12 (21) | 0 (0) | 5 (28) | 0 (0) | 4 (27) | 1 (7) |
| Vomiting | 0 (0) | 0 (0) | 11 (20) | 0 (0) | 6 (33) | 0 (0) | 5 (33) | 0 (0) |
| Diarrhea | 0 (0) | 0 (0) | 15 (27) | 0 (0) | 2 (11) | 1 (6) | 2 (13) | 0 (0) |
| Dysgeusia | 1 (20) | 0 (0) | 6 (11) | 1 (2) | 4 (22) | 1 (6) | 1 (7) | 0 (0) |
Efficacy
Overall 88 patients were evaluable for response. Of these, 55 patients had non-BBC solid tumors and 33 patients had BCCs. Of the 55 evaluable patients with non-BCC solid tumors, the best response was stable disease in 29 patients (53%), of which 9 patients (16%) maintained for at least 3 months and 5 patients for at least 6 months (9%). Of the patients who maintained stable disease for at least 6 months, 2 patients had chondrosarcoma, 1 neuroendocrine tumor, 1 ovarian cancer, and 1 adenocystic carcinoma.
Of the 39 total patients with BCCs treated in this study, 10 were stage II, 10 were stage III, and 19 were stage IV. Of these 28 were considered evaluable (i.e., having received more than one dose of IPI-926 and a post-baseline clinical or RECIST assessment) and were vismodegib-naïve. In this population, IPI-926 showed substantial antitumor activity (Fig. 3A). Eight patients achieved an objective response [2 complete response (CR) and 6 partial response (PR); Fig. 3B and C]. All the responses occurred in patients with locally advanced disease. In many instances, improvement of clinical symptoms such as pain or itching occurred within a week of initiating daily dosing. Interestingly, several patients with basal cell nevus syndrome reported improvement in noncancer features such as palmar inflammation and pitting.
Figure 3.

Antitumor efficacy in patients with BCCs. A, time on study and best response in evaluable vismodegib-naïve patients with BCC. PD, progressive disease; SD, stable disease. B, selected paired clinical photographs taken at baseline (left) and at the end of cycle 3 (right), examples of clinical response. C, selected paired scans of a patient with a supraclavicular lesion taken at baseline (left) and at the end of cycle 3 (right), examples of response per RECIST 1.0.
Nine of the 39 patients with BCCs experienced progressive disease during or after receiving vismodegib; of these, one patient received IPI-926 for 50 weeks and another for 18 weeks. There were no objective responses among the patients previously treated with vismodegib. Among the full cohort of patients with BCCs treated (N = 39), there was no apparent difference in efficacy between the 130- and 160-mg dose levels, although the small number of patients treated is insufficient to conduct a formal comparison of efficacy. As of (April 13, 2012), 9 patients with BCCs remained on treatment with IPI-926 in a rollover protocol.
Pharmacokinetics
In the dose-escalation phase of the study, following single- and multiple-dose administration of IPI-926, the mean total (AUC0–24) and peak (Cmax) exposures of IPI-926 generally increased with increasing dose (Tables 4 and 5 and Fig. 1).
Table 4.
Summary of plasma IPI-926 PK parameters on day −7 of cycle 1 following oral IPI-926 single-dose administration in patients with advanced and/or metastatic solid tumor malignancies (dose-escalation phase)
| Arithmetic mean (CV%) |
||||
|---|---|---|---|---|
| Dose, mg | AUC0–24, ng h/mL | Cmax, ng/mL | tmax,a h | t1/2, h |
| 20 (n = 1) | 58.5 (NC) | 6.66 (NC) | 6.00 (NC) | NC |
| 30 (n = 1) | 68.3 (NC) | 5.44 (NC) | 6.08 (NC) | NC |
| 40 (n = 1) | 122 (NC) | 10.6 (NC) | 5.00 (NC) | NC |
| 60 (n = 1) | 367 (NC) | 28.2 (NC) | 4.00 (NC) | NC |
| 90 (n = 1) | 263 (NC) | 20.2 (NC) | 4.00 (NC) | NC |
| 130 (n = 8) | 823 (86.7) | 59.9 (95.0) | 6.44 (3.00–22.0) | 19.3 (NC)b |
| 160 (n = 10) | 1272 (30.6) | 87.4 (31.8) | 4.52 (2.00–10.0) | 17.3 (13.6)c |
| 180 (n = 4) | 1676 (52.8) | 106 (57.0) | 5.47 (3.00–8.00) | 16.1 (13.9)d |
| 200 (n = 3) | 1828 (41.2) | 123 (44.0) | 4.00 (3.00–6.00) | 12.7 (NC)b |
| 210 (n = 6) | 1915 (27.9) | 117 (29.9) | 6.00 (5.00–8.10) | NC |
Abbreviation: NC, not calculated.
Median (min – max).
n = 1.
n = 4.
n = 2.
Table 5.
Summary of plasma IPI-926 PK parameters on day 22 of cycle 1 following oral IPI-926 QD administration in patients with advanced and/or metastatic solid tumor malignancies (dose-escalation phase)
| Arithmetic mean (CV%) |
||||
|---|---|---|---|---|
| Dose, mg | AUC0–24, ng h/mL | Cmax, ng/mL | tmax,a h | CL/F, L/h |
| 20 (n = 1) | 180 (NC) | 13.1 (NC) | 2.13 (NC) | 111 (NC) |
| 30 (n = 1) | 508 (NC) | 27.2 (NC) | 5.05 (NC) | 59.0 (NC) |
| 40 (n = 1) | 800 (NC) | 39.7 (NC) | 5.00 (NC) | 50.0 (NC) |
| 60 (n = 1) | 2095 (NC) | 105 (NC) | 5.08 (NC) | 28.6 (NC) |
| 90 (n = 1) | NC | NC | NC | NC |
| 130 (n = 5) | 6147 (30.0)b | 336 (29.7) | 3.13 (2.98–8.08) | 23.0 (36.0)b |
| 160 (n = 4) | 8296 (32.7) | 450 (23.6) | 4.99 (3.93–6.02) | 21.2 (38.5) |
| 180 (n = 1) | 12829 (NC) | 644 (NC) | 5.00 (NC) | 14.0 (NC) |
| 200 (n = 1) | 15074 (NC) | 723 (NC) | 6.00 (NC) | 13.3 (NC) |
| 210 (n = 5) | 11276 (33.9) | 595 (25.2) | 5.08 (5.00–6.00) | 20.6 (35.5) |
Abbreviation: NC, not calculated.
Median (min – max).
n = 4.
Figure 1.

Representative IPI-926 plasma concentration–time profiles. Mean (+SD) plasma concentrations versus time profiles of IPI-926 on days −7 and 22 of cycle 1 following oral administration of IPI-926 (160 mg) in patients with advanced and/or metastatic solid tumor malignancies.
Consistent with the exposures observed in the doseescalation phase of the study, the mean total exposure (AUC0–24) and peak concentration (Cmax) of IPI-926 appeared to increase in a dose-proportional manner after single and multiple doses of 130- to 160-mg IPI-926 in the expanded phases. Mean oral clearance (CL/F) was 27.4 L/h for the 130-mg dose level and 20.1 L/h for 160-mg dose level. The mean total exposure (AUC0–24) of the N-hydroxy metabolite, IPI-541, was approximately 10% of that observed for the parent over the dose range evaluated. On the basis of the low exposure of IPI-541, and the fact that it binds Smo with lower affinity and inhibits the HhP with lesser potency than IPI-926, it is likely that the overall activity and toxicity profile observed in this study is predominantly due to IPI-926.
There was no apparent effect on the pharmacokinetic parameters of IPI-926, as shown by the comparison of the geometric mean total (AUClast) and peak (Cmax) exposures when 130-mg IPI-926 was administered with and without a high fat meal. Of note, the geometric mean total exposure (AUClast) of the metabolite IPI-541 appeared to be slightly larger and the median time to peak concentrations (tmax) longer (from ~4–8 hours) when IPI-926 was administered with food.
Pharmacodynamics
Normal skin biopsies in 48 of 65 patients (74%) showed a decrease in Gli-1 transcript level after 21 days of treatment with IPI-926 (data not shown). Moreover, there was an observed decrease in Gli1 by immunohistochemistry (IHC) and RT-PCR (Fig. 2A–C) in the stroma of a patient with rectal cancer following treatment with IPI-926, which to our knowledge is the first time this has been described in a non–HhP-driven tumor.
Figure 2.
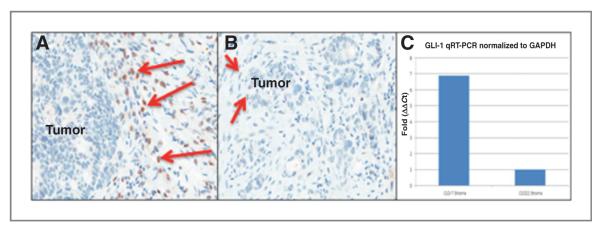
Pharmacodynamic studies. A, rectal adenocarcinoma analyzed by Gli-1 IHC, showing evidence of strong Gli-1 stromal staining (red arrows) of baseline biopsy (cycle 1, day −7), whereas tumor epithelial cell nuclei appear negative. B, posttreatment biopsy of rectal adenocarcinoma analyzed by Gli-1 IHC, showing reduced Gli-1 stromal staining (red arrows), following treatment with IPI-926 (cycle 1, day 22). C, laser capture microdissection (LCM) quantitative RT-PCR conducted on rectal adenocarcinoma patient biopsy, showing an about 7-fold Gli-1 transcript reduction in stroma, following treatment with IPI-926 (cycle 1, day 22).
Discussion
This report summarizes results from the first-in-human clinical and pharmacologic study of IPI-926, an HhP inhibitor. The accelerated titration escalation design proved to be a valuable strategy and facilitated a 10-fold dose escalation from the initial dose of 20 mg until nontolerability was established at 200 mg. On the basis of the DLTs observed at doses at and above 180 mg QD, the MTD of IPI-926 was determined to be 160 mg QD. Although one DLT consisting of grade 3 ALT and grade 3 AST elevation occurred in 1 of 6 patients dosed at 160 mg QD, this dose, bolstered by additional data from patients dosed at 130 mg, was considered to have a toxicity profile acceptable for phase II drug development. The modified Fibonacci schema that called for a 50% increase from 130 to 200 mg went from a level with grade 2 toxicity to a level that overestimated past the MTD level. Our experience highlights the importance of constant appraisal of phase I designs (18).
The clinical toxicity of IPI-926 was predominantly comprised of events of fatigue, nausea, muscle cramps, dysgeusia, and alopecia, which are consistent with what has been reported with other HhP inhibitors (19). Of note, the toxicity and efficacy profiles of IPI-926 substantially overlap with those from other inhibitors of the HhP (e.g., vismodegib and LDE-225), indicating homogeneous pathway inhibition with relatively consistent adverse effects. Of these side effects, the distribution of the alopecia was distinct from the typical chemotherapy-induced hair loss that predominantly affects the scalp. In patients receiving IPI-926, the alopecia distribution affected primarily the eyebrows, eyelashes, and extremities rather than the scalp. These observations are potentially relevant to the pathophysiology of BCCs, given that mounting evidence points toward the hair follicle as a site of initiation in BCC tumorigenesis (20, 21). Interestingly, HhP agonists have been postulated as potential therapies in treating conditions of decreased proliferation and aberrant follicular cycling in the scalp including androgenic alopecia (pattern hair loss; ref. 22).
IPI-926 pharmacokinetics were characterized by prolonged absorption, slow clearance relative to most small-molecule therapeutics, and an extended elimination half-life. Overall, the pharmacokinetic profile supports a once daily administration schedule projected to provide sustained pathway abrogation.
IPI-926 showed substantial antitumor activity in patients with vismodegib-naïve BCCs, confirming drug specificity. However, disease progression did ultimately develop, and in a time period similar to other tumors that are known to develop secondary mutations with other targeted agents (23, 24).
In our study, patients with BCCs who had progressed after vismodegib, an SMO inhibitor that has a chemical structure unrelated to cyclopamine, derived limited benefit from IPI-926. This seems to suggest that despite markedly different chemical configurations, the SMO inhibitory specificity of both agents is similar and resistance mechanisms could overlap.
In summary, IPI-926 showed single-agent activity in patients with BCCs and a manageable toxicity profile that was consistent with its class. The recommended single-agent dose of IPI-926 given QD is 160 mg.
Translational Relevance.
The hedgehog pathway (HhP) is dysregulated in a variety of solid tumors and is proposed to provide key growth and survival signals to tumor cells. Therefore, inhibitors of the HhP represent a promising class of therapeutic agents with several small-molecule HhP inhibitors in clinical development. This article reports data from a phase I, first-in-human study of IPI-926, an oral, selective inhibitor of the Smoothened receptor, a key transducer of ligand-dependent HhP activity. Results from this trial show that IPI-926 may be administered with a tolerable toxicity profile in patients with advanced solid tumors and has substantial efficacy in patients with basal cell carcinoma of the skin.
Acknowledgments
The authors thank all the patients and their caregivers for their participation and generosity, as well as the clinical teams for enabling the successful conduct of the trial.
Grant Support This study was supported by Infinity Pharmaceuticals, Inc.
Footnotes
Disclosure of Potential Conflicts of Interest A. Jimeno has commercial research support from Inifinity. G.J. Weiss has honoraria from speakers’ bureau from Genentech, Eli Lilly, Pfizer, and Quintiles. A.L.S. Chang has commercial research support from Genentech and Novartis. K. Faia has ownership interest (including patents) in Infinity Pharmaceuticals. C.M. Rudin is a consultant/advisory board member of Eli Lilly.
No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions Conception and design: A. Jimeno, W.H. Miller, R.W. Ross, C.M. Rudin
Development of methodology: A. Jimeno, W.H. Miller, G. Skliris
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Jimeno, G.J. Weiss, W.H. Miller, S.N. Gettinger, B. Eigl, A.L.S. Chang, S. Devens, K. Faia, K.D. Lewis, R. Tibes, W.H. Sharfman, R.W. Ross, C.M. Rudin
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A. Jimeno, G.J. Weiss, W.H. Miller, A.L.S. Chang, J. Dunbar, K. Faia, J.L. Kutok, R. Tibes, R.W. Ross
Writing, review, and/or revision of the manuscript: A. Jimeno, G.J. Weiss, W.H. Miller, S.N. Gettinger, A.L.S. Chang, J. Dunbar, J.L. Kutok, K.D. Lewis, R. Tibes, W.H. Sharfman, R.W. Ross, C.M. Rudin
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): R. Tibes
Study supervision: A. Jimeno, W.H. Miller, B. Eigl, S. Devens, W.H. Sharfman, R.W. Ross
Prior presentations: Posters presented portions of the data at the National Cancer Institute (NCI) Molecular Therapeutics Meeting, Milan, Italy (Oct 8–10, 2010); the 2011 American Society of Clinical Oncology (ASCO) Annual Meeting, Chicago, IL (Jun 4–8, 2011).
References
- 1.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–54. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 4.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–61. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 5.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 6.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 7.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 8.Ehtesham M, Sarangi A, Valadez JG, Chanthaphaychith S, Becher MW, Abel TW, et al. Ligand-dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene. 2007;26:5752–61. doi: 10.1038/sj.onc.1210359. [DOI] [PubMed] [Google Scholar]
- 9.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048–53. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, et al. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol Cancer Res. 2012;10:1147–57. doi: 10.1158/1541-7786.MCR-12-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parisi MJ, Lin H. The role of the hedgehog/patched signaling pathway in epithelial stem cell proliferation: from fly to human. Cell Res. 1998;8:15–21. doi: 10.1038/cr.1998.2. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimeno A, Feldmann G, Suarez-Gauthier A, Rasheed Z, Solomon A, Zou GM, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8:310–4. doi: 10.1158/1535-7163.MCT-08-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremblay M, McGovern K. In: Cyclopamine and its derivatives for cancer therapeutics, in hedgehog signaling activation in human cancer and its clinical implications. Xie J, editor. Springer Publishing; New York: 2011. [Google Scholar]
- 16.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17:1504–8. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 18.Ivy SP, Siu LL, Garrett-Mayer E, Rubinstein L. Approaches to phase I clinical trial design focused on safety, efficiency, and selected patient populations: a report from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin Cancer Res. 2010;16:1726–36. doi: 10.1158/1078-0432.CCR-09-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–11. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang GY, Wang J, Mancianti ML, Epstein EH., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell. 2011;19:114–24. doi: 10.1016/j.ccr.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grachtchouk M, Pero J, Yang SH, Ermilov AN, Michael LE, Wang A, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J Clin Invest. 2011;121:1768–81. doi: 10.1172/JCI46307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paladini RD, Saleh J, Qian C, Xu GX, Rubin LL. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol. 2005;125:638–46. doi: 10.1111/j.0022-202X.2005.23867.x. [DOI] [PubMed] [Google Scholar]
- 23.Rosell R, Molina MA, Costa C, Simonetti S, Gimenez-Capitan A, Bertran-Alamillo J, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011;17:1160–8. doi: 10.1158/1078-0432.CCR-10-2158. [DOI] [PubMed] [Google Scholar]
- 24.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]