

Summary
Despite significant heritability of autism spectrum disorders (ASDs), their extreme genetic heterogeneity has proven challenging for gene discovery. Studies of primarily simplex families have implicated de novo copy number changes and point mutations, but are not optimally designed to identify inherited risk alleles. We apply whole exome sequencing (WES) to ASD families enriched for inherited causes due to consanguinity and find familial ASD associated with biallelic mutations in disease genes (AMT, PEX7, SYNE1, VPS13B, PAH, POMGNT1), some implicated for the first time in ASD. At least some of these genes show biallelic mutations in nonconsanguineous families as well. These mutations are often only partially disabling or present atypically, with patients lacking diagnostic features of the Mendelian disorders with which these genes are classically associated. Our study shows the utility of WES for identifying specific genetic conditions not clinically suspected and the importance of partial loss of gene function in ASDs.
Introduction
Despite studies suggesting that autism spectrum disorders (ASDs) are significantly heritable, the basis of this heritability remains largely unexplained (Devlin and Scherer, 2012). Autism is characterized by the triad of communication deficits, abnormal social interests, and restricted and repetitive behaviors. Genome-wide association studies (GWAS) have so far detected no strong contribution of common alleles (State, 2010), motivating renewed interest in rare variants (Malhotra and Sebat, 2012). Transmitted, rare copy number variants (CNVs), such as 16p11.2 microdeletion/duplication and 15q11.2-q13 duplication have been found to contribute, although the total number of cases accounted for by these conditions is small (Levy et al., 2011; Pinto et al., 2010; Weiss et al., 2008). Significant roles have also been demonstrated for diverse, de novo CNVs (Levy et al., 2011; Sanders et al., 2011; Sebat et al., 2007) and more recently, de novo, protein-altering point mutations (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2011; O’Roak et al., 2012; Sanders et al., 2012). In the cohorts examined, de novo events may be projected to account for up to 15–20% of ASD cases. Despite the high total rate of de novo point mutations, estimates of the number of contributing loci to autism susceptibility are in the several hundreds, so that validating specific causative genes is a significant challenge, since recurrent mutation in any given gene is so uncommon. Nonetheless, these studies have been successful at elucidating gene dosage-sensitive ASD molecular pathways, since the typical mutations observed are loss/disruption, or sometimes gain, of one functional copy of a gene or contiguous genes, rather than biallelic mutations of both copies of a gene. However, despite the importance of de novo mutations, much of the heritability of ASDs remains unaccounted for (Devlin and Scherer, 2012).
We hypothesized that at least some cases of autism reflect rare, inherited point mutations that existing study designs, often involving families with one or two affected individuals, are not designed to capture. Consanguineous and multiplex pedigrees have been extremely useful for identifying inherited mutations responsible for rare heritable conditions in the setting of extreme genetic heterogeneity, because single families can provide substantial genetic linkage evidence (Lander and Botstein, 1987; Woods et al., 2006). Applying high-throughput sequencing to such families has been extremely useful in identifying recessive causes of intellectual disability (Najmabadi et al., 2011). The potential role of biallelic mutations in ASDs is strongly supported by a number of syndromic recessive conditions that have already been associated with autistic symptoms (Betancur, 2011). Additional evidence supporting a role of biallelic mutations comes from studies that have implicated homozygous CNVs (Levy et al., 2011; Morrow et al., 2008) and long homozygous intervals as significantly associated with ASDs (Casey et al., 2012). Finally, a recent whole exome sequencing (WES) study has suggested a role for biallelic point mutations in a subset of patients with ASDs that show long runs of homozygosity (Chahrour et al., 2012).
In this study we apply WES to a cohort of consanguineous and/or multiplex families with ASD that also show shared ancestry between the parents, typically as cousins. We find several families where mapping and sequence analysis allow the identification of specific causative mutations, and show that many of these mutations represent partial loss of function in genes where null mutations cause distinctive Mendelian disorders. These hypomorphic mutations confirm the complex and heterogeneous nature of ASDs, but also highlight the importance of WES in identifying specific genetic causes underlying this heterogeneity.
Results
Identifying inherited mutations in three ASD families
We studied an ASD cohort recruited by the Homozygosity Mapping Collaborative for Autism (HMCA), an international, multicenter effort to identify genetically informative ASD families with consanguinity and/or multiple affected individuals (Morrow et al., 2008). We first performed genome-wide linkage analysis on the most informative families, using high-resolution single nucleotide polymorphism (SNP) arrays, reasoning that some families would show homozygous, biallelic mutations embedded within larger blocks of homozygosity inherited from the ancestor common to both parents. Families were prescreened to exclude those harboring autism-associated CNVs or other known diagnoses (Supplemental Experimental Procedures). Three families provided particularly strong genetic power to localize potential disease loci.
The first family had three children affected with ASD and two unaffected children, born to parents who were first cousins (Figure 1A; Table 1B; Supplemental Text). Mapping under a single locus, biallelic model (i.e., allowing for both homozygous and compound heterozygous mutations) excluded 99.3% of the genome and revealed a single linkage peak centered at 3p21.31, in a large homozygous interval, reaching the maximum LOD score obtainable in the pedigree, 2.96 (Figure 1B), suggesting a >900:1 likelihood that the responsible mutation was contained within this homozygous interval. WES of a single affected child was performed. The linked interval contained only a single rare, nonsynonymous change that was absent from known databases and population-matched controls: a homozygous single base substitution in the aminomethyltransferase (AMT) gene, encoding an enzyme essential for the degradation of glycine. The mutation resulted in p.I308F, altering an Ile residue that is highly conserved in all AMT orthologs (Figure 1C), and is packed tightly into a hydrophobic pocket (Figure 1D)(Okamura-Ikeda et al., 2005). Sanger validation confirmed that the mutation was heterozygous in both parents, homozygous in all affected children, and absent or heterozygous in the two unaffected children.
Figure 1.

Identification of mutations in AMT in a family with ASD. (A) AU-1700, a Saudi family with three children affected by autism. Shaded symbols indicate affected individuals. The triangle represents a miscarriage. WES was performed on samples from individuals indicated with a star. Genotyping by Sanger sequencing in additional family members was performed where indicated (+: reference allele, −: alternate allele). (B) Mapping to a locus on chromosome 3. Genome-wide linkage plot (top) and maximum obtainable LOD score in the family across the interval (bottom). (C) Ile308 residue is highly conserved across species. (D) Mapping of the I308F missense mutation onto the human AMT crystal structure (PDB accession 1WSV). Left, overview showing I308 in domain 3 of AMT. Right, detail illustrating the hydrophobic pocket in which I308 resides. Neighboring hydrophobic residues are shown in white. The white brackets indicate the different domains: AMT domain 1 (folding); AMT domain 2 (catalytic); AMT domain 3 (capping). (E) I308F results in protein misfolding and aggregation. C-terminal 6xHis-tagged human AMT, AMT I308F, and AMT I308A were expressed in E. coli. Left panel, when overexpressed at 30°C by induction with 25 μM IPTG, wildtype AMT was fully soluble, but I308F and I308A segregated to inclusion bodies despite overall similar expression levels. W: whole cell extract, S: supernatant, P: pellet. Right panel, slower induction at 22°C for 44 hours resulted in partially soluble mutants. Near-wildtype solubility could be achieved by co-expression with GroEL and GroES. (F) AMT I308F results in partial loss-of-function of glycine cleavage and synthesis activity. Wildtype and mutant 6xHis-human AMT were expressed in E. coli, purified, and assayed for glycine cleavage and glycine synthesis activity. Relative to wildtype (blue traces), AMT I308F (red traces) demonstrates significant reduction of activity in glycine cleavage (left) and glycine synthesis (right) assays. R320H, N145I, and G269D are previously reported NKH-associated alleles (Okamura-Ikeda et al., 2005).
Table 1.
Inherited mutations identified in ASDs. Severe (nonsense, frameshift) (A) and hypomorphic (missense) (B) mutations in known disease genes were identified in 11 ASD families. M: male, F: female, ID: intellectual disability.
| (A) Severe mutations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation | Known disease association | Family | Structure | Consanguinity | # Affected | # Unaffected | Linkage | Primary phenotype | Additional phenotypes |
| MECP2 p.E483X | Rett syndrome, ASD | AU-5400 | Multiplex | No | 2 (2M) | - | Yes | Autism | - |
| NLGN4X p.Q329X | Non-syndromic X-linked ID and/or ASD | AU-5700 | Simplex | Yes | 1 (M) | 1 | Yes | Autism | - |
| PAH p.198_205del | Phenylketonuria | AU-13100 | Simplex | Yes | 1 (M) | 2 | Yes | Autism | ID |
| PAH p.Q235X | Phenylketonuria | AU-4100 | Multiplex | Yes | 2 (2F) | - | Yes | Autism | - |
| VPS13B p.A3943fs | Cohen syndrome | AU-21100 | Simplex | Yes | 1 (M) | 3 | Yes | Autism | ID, dysmorphic features, hyperextensible joints |
| (B) Hypomorphic mutations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation | Known disease association | Family | Structure | Consanguinity | #Affected | # Unaffected | Linkage | Primary phenotype | Additional phenotypes |
| AMT p.I308F | Nonketotic hyperglycinemia | AU-1700 | Multiplex | Yes | 3 (2M, 1F) | 2 | Yes | Autism | ID, seizures |
| AMT p.D198G | Nonketotic hyperglycinemia | AU-11800 | Simplex | Yes | 1 (M) | 1 | Yes | Autism | ID, seizures |
| PEX7 p.W75C | Rhizomelic chondrodysplasia punctata | AU-3500 | Multiplex | Yes | 3 (2M, 1F) | 3 | Yes | PDD-NOS | ID, seizures, cataracts |
| POMGNT1 p.R367H | Muscle–eye–brain disease | AU-13300 | Simplex | Yes | 1 (M) | 1 | Yes | Autism | ID |
| SYNE1 p.L3206M | Autosomal Recessive Cerebellar Ataxia Type 1, Arthrogryposis Congenita, ASD, Bipolar Disease | AU-1600 | Multiplex | Yes | 4 (1M, 3F) | 1 | Yes | Autism | ID |
| VPS13B p.S824A | Cohen syndrome | AU-17800 | Simplex | Yes | 1 (M) | 1 | Yes | Autism | ID, dysmorphic features, hyperextensible joints |
Mutations in AMT classically cause nonketotic hyperglycinemia (NKH) (Applegarth and Toone, 2004), a neonatal syndrome leading to progressive lethargy, hypotonia, severe seizures, and death within the first year of life (Hamosh A, 2001). Patients with neonatal NKH have impaired activity of the glycine cleavage system, leading to abnormal elevation of glycine levels in serum or CSF. Rarer, atypical forms of NKH have been described in association with hypomorphic, missense AMT mutations (Applegarth and Toone, 2001; Dinopoulos et al., 2005), manifesting as later age of clinical onset, delays in expressive language, behavioral problems, and variable or absent seizures. Clinical and biochemical evidence suggests that the p.I308F mutation is hypomorphic. The three affected children in this family had a range of symptoms that would have suggested NKH had they all occurred in the same individual (Supplemental Text). The eldest child was twelve years old and had, in addition to a diagnosis of ASD, a history of severe epilepsy, with first seizures presenting by age 10 months, very consistent with NKH. The second child was nine years old and also suffered from a combination of autism and epilepsy, though her seizures were milder. The third child was two years old, suffered from language and motor delays, and carried a presumptive PDD diagnosis. He had had only a single febrile seizure. Though the two older children had had plasma amino acid screening that disclosed no abnormalities, milder forms of NKH typically have no abnormalities on serum biochemical analyses (Applegarth and Toone, 2001; Dinopoulos et al., 2005).
Direct biochemical analysis of the p.I308F mutation confirms that it has reduced activity. While wildtype AMT is fully soluble at 30°C when expressed in bacteria, mutant AMT p.I308F was poorly soluble (Figure 1E), indicating a protein folding defect, similar to that observed with NKH-associated AMT mutations (Figure S1; Table S1). This defect could be rescued by coexpressing GroES and GroEL heat shock proteins at 22°C (Figure 1E). AMT p.I308F, even after solubilization, retained only 45% (SD 4.1%) and 1.8% (SD 0.5%) of wildtype glycine cleavage and glycine synthesis specific activity, respectively, when assayed enzymatically (Figure 1F, Figure S2). When compared to classical NKH-associated alleles, glycine cleavage activity of AMT p.I308F is at the mild end of the range of previously reported values (Figure 1F, Figure S2), further suggesting that the affected autistic children in this family suffer from undiagnosed, atypical NKH presenting as ASD with seizures.
A second consanguineous family had three children diagnosed with ASD and three unaffected children, born to unaffected parents who were first cousins (Figure 2A; Table 1B; Supplemental Text). Parametric mapping excluded 97.2% of the genome and established linkage to a homozygous interval on 6p23 (Figure 2B) (LOD 2.78, the maximum obtainable in the pedigree, under a recessive model, indicating a >600:1 likelihood that this interval contained the disease-causing mutation). WES identified only one variant, absent from known databases and population-matched controls, in this region that was predicted to be pathogenic: PEX7 p.W75C. This change was homozygous and altered a highly conserved Trp residue within a WD-40 repeat of the predicted protein (Figures 2C and 2D). Sanger validation confirmed that this mutation was heterozygous in both parents, and heterozygous or wildtype in unaffected children.
Figure 2.

Identification of mutations in PEX7 in a family with ASD. (A) AU-3500, a family with three children affected with ASD. Shaded symbols indicate affected individuals. WES was performed on samples from individuals indicated with a star. Genotyping by Sanger sequencing in additional family members was performed where indicated (+: reference allele, −: alternate allele). (B) Mapping to a locus on chromosome 6. Genome-wide linkage plot (top) and maximum obtainable LOD score in the family across the interval (bottom). A homozygous missense mutation in the AU-3500 family disrupts a conserved tryptophan (p.W75C) (C) in the first WD40 repeat of PEX7, the receptor responsible for import of PTS2-containing proteins into the peroxisome (D), and an established cause of rhizomelic chondrodysplasia punctata. (E) PEX7 W75C and S25F, a previously characterized mutation from a patient with mild RCDP and ASD, result in partial loss-of-function in a peroxisomal import assay. In fibroblasts from patients with RCDP, PTS2-tagged mCherry accumulates in the cytoplasm due to lack of PEX7 transport activity (top). Transfection of wildtype PEX7 cDNA causes PTS2-mCherry to redistribute to small puncta, indicative of restoration of peroxisomal import (bottom). Transfection of mutant PEX7 W75C or S25F does not restore complete import, with a minority of cells showing partial import (middle). Quantification of defects in peroxisomal import mediated by ASD-associated PEX7 alleles, scored by visual assay. Error bars represent standard error (right). (F) Immunoblotting of whole cell lysates illustrates deficient maturation of PTS2-targeted proteins in a RCDP cell line transfected with ASD-associated PEX7 alleles. AGPS, PhyH, and thiolase are imported into the peroxisome and undergo cleavage into a smaller, mature peptide. p: preprotein, m: mature protein, Beta-tubulin: loading control. Quantification of processing defects by densitometry of transfected cell lysates (right).
PEX7 encodes a receptor required for import of PTS2(peroxisome targeting signal 2)-containing proteins into the peroxisome (Braverman et al., 1997). Null mutations in PEX7 cause rhizomelic chondrodysplasia punctata (RCDP), an inborn metabolic syndrome of abnormal facies, cataracts, skeletal dysplasia, epilepsy, and severe psychomotor defects, with most cases not surviving beyond two years of age (Braverman et al., 2002; Braverman et al., 1997; Motley et al., 1997). The affected children in this family however ranged in age from 18 to 31 years. They were not dysmorphic and did not exhibit skeletal dysplasia, though two had cataracts, and two had epilepsy (Supplemental Text). The cataracts and seizures in particular suggested partial loss of PEX7 function, since rare, atypical RCDP cases associated with hypomorphic compound heterozygous or homozygous mutations have been described that have some but not all of the features of the classical syndrome, lacking dysmorphic features, and showing only intellectual disability with variable cataracts (Braverman et al., 2002).
To evaluate whether the p.W75C missense change in this family could be pathogenic, we assayed its ability to rescue peroxisomal import in cultured fibroblasts from a RCDP patient. In RCDP fibroblasts, fluorescent mCherry fused to the PTS2 peroxisomal targeting sequence fails to be imported into peroxisomes and remains cytosolic (Figure 2E). Co-transfection of wildtype PEX7 fully restores peroxisomal import (Figure 2E). In contrast, transfection with PEX7 p.W75C failed to rescue (Figure 2E): the majority of cells showed cytosolic PTS2-mCherry, although a fraction showed partial rescue. To characterize this effect, we utilized a semi-quantitative assay of peroxisomal import. The PTS2 proteins thiolase, phytanoyl-CoA hydroxylase (PhyH), and alkylglycerone phosphate synthase (AGPS) are imported into the peroxisome and proteolytically processed into smaller, mature forms (Figure 2F). Peroxisomal uptake is thus reflected in the ratio of the mature protein to the preprotein. In RCDP cells, these three proteins all remain in the preprotein state, reflecting failure of peroxisomal import. Transfection of wildtype PEX7 fully restores processing, whereas transfection of PEX7 p.W75C produced only partial processing (Figure 2F). These results demonstrate that this allele is pathogenic, but partial loss-of-function, consistent with these individuals not exhibiting full features of the RCDP syndrome.
To our knowledge, a link between mild RCDP and ASDs has not been described previously. However, two previously reported patients with biochemical evidence of RCDP and cataracts, but lacking the dysmorphic features of RCDP, were found to be compound heterozygous for partial loss-of-function PEX7 mutations (Braverman et al., 2002); one was originally described as intellectually disabled and the second as neurotypical. We re-contacted these patients. A review of clinical records and reexamination of the first child revealed that she had subsequently been diagnosed with ASD, and the second child was diagnosed with severe ADHD, providing additional examples of the range of clinical expressivity of mild mutations in PEX7. Partial loss of function for one of the alleles in these patients, S25F, was verified in the fibroblast assays (Fig. 2E and F).
Analysis of a third large family pointed to a novel candidate autism gene potentially implicated in synaptic plasticity, SYNE1. In this family, five children were born to parents who were double first cousins. Four were affected with autism and the fifth child was unaffected (Figure 3A; Supplemental Text). The family showed linkage to two loci on chromosome 6q25 and 7q33 (LOD 2.83, maximal obtainable in the pedigree, indicating a >670:1 chance that the disease-causing gene lies in one of these intervals) (Figure 3B). WES was performed for the entire nuclear family. No rare, protein-altering variants were found in the 7q33 linkage interval, whereas 6q25 harbored only one protein-altering variant, absent from known databases and population-matched controls, that segregated with disease: a homozygous missense change in SYNE1 (p.L3206M) (Table 1). SYNE1 has previously been implicated as an ASD gene candidate by the presence of a de novo single nucleotide variant in a patient with ASD (O’Roak et al., 2011), and has been implicated in bipolar disorder in a GWAS study (Sklar et al., 2011) (Figure 3C). Truncating, presumably null, mutations in SYNE1 cause cerebellar ataxia (Gros-Louis et al., 2007) and a recessive form of arthrogryposis multiplex congenita (Attali et al., 2009) (Figure 3C), again suggesting that the ASD-associated allele may be hypomorphic, since the phenotype is milder. SYNE1 p.L3206M alters a highly conserved residue that lies within a spectrin repeat (Figure 3D; SIFT score 0.01).
Figure 3.

Identification of mutations in SYNE1 in a family with ASD. (A) AU-1600, a family with four children affected with ASD. Shaded symbols indicate affected individuals. WES was performed on samples from individuals indicated with a star. Genotyping by Sanger sequencing in additional family members was performed where indicated (+: reference allele, −: alternate allele). (B) Mapping rules out the majority of the genome and points out to only two candidate loci, one on chromosome 6 and the other on chromosome 7. Genome-wide linkage plot (top) and maximum obtainable LOD score in the family across the interval (bottom). (C) SYNE1 is a complex locus implicated in neuropsychiatric disease. SYNE1 encompasses multiple alternative transcripts (Known Genes, UCSC Genome Browser), and mutations have been associated with a wide range of neurodevelopmental phenotypes. The location of the recessively inherited missense change in this study is denoted by the green star. Late truncation (open triangle) causes autosomal recessive arthrogyroposis multiplex congenita (Attali et al., 2009), while truncating mutations further upstream (closed triangles) cause autosomal recessive cerebellar ataxia (Gros-Louis et al., 2007). An intronic SNP (red square) is a putative bipolar disorder susceptibility locus (Sklar et al., 2011). A de novo missense change was previously reported in ASD (blue star) (O’Roak et al., 2011). (D) The homozygous missense mutation p.L3206M disrupts a highly conserved residue. (E) Human neuronal ChIPseq and RNAseq data demonstrate transcriptional complexity and activity-dependent regulation of the SYNE1 locus. ChIPseq with antibodies to H3K4Me3 (active promoters) and H3K27Ac (enhancers) demonstrates at least four active promoters (P1–P4) for SYNE1. P3 lies immediately upstream of the residue mutated in AU-1600 (p.L3206M) and corresponds to the active promoter site for BC039121. RNAseq of cultured human neurons was performed before (0h; 0 hours) and after (6h; 6 hours) depolarization with KCl. Depolarization induces upregulation of full-length SYNE1 and the BC039121 isoform compared to untreated neurons (also see Figure S3). The experiment was performed on five biological replicates. Representative tracks are shown. The red square represents a magnification of the region around BC039121. Sense (+; pink) and antisense (−; blue) transcripts are indicated.
Full-length SYNE1 encodes a large 8,797 amino acid protein with two N-terminal actin-binding domains, multiple spectrin repeats, a transmembrane domain, and a C-terminal KASH domain. The SYNE1 mutation identified here is predicted to map to exon 61 of the full-length transcript (RefSeq NM_182961), although the SYNE1 locus is complex, with many predicted alternative splice forms (Simpson and Roberts, 2008).
To identify what human transcript(s) might be affected by the p.L3206M mutation, we mapped transcriptional start sites in human neurons using ChIPseq (Figure 3E). ChIPseq using antibodies to H3K4Me3, a mark associated with active promoter sites (Ernst et al., 2011), and to H3K27Ac, a mark associated with enhancer elements (Heintzman et al., 2009), demonstrated mapped read peaks corresponding to at least four major transcriptional start sites within the SYNE1 locus (P1–P4), one of which (P3) lies immediately upstream of the p.L3206M mutation (Figure 3E). 5′ and 3′ RACE (data not shown) confirmed the existence of at least one polyadenylated transcript emanating from this promoter, corresponding to the GenBank deposited mRNA clone BC039121, encompassing exons 57–63 of the predicted full-length SYNE1 mRNA. This is the minimal confirmed transcript that overlaps the p.L3206M mutation, although contributions of additional or even full-length transcripts cannot be excluded.
SYNE1 has been shown to have roles in cellular nuclear migration in C. elegans and Drosophila (Starr and Han, 2002; Zhang et al., 2002), anchoring of synaptic nuclei with postsynaptic membranes at the vertebrate neuromuscular junction (Grady et al., 2005), although based upon patients with SYNE1-associated cerebellar ataxia, it has been suggested that vertebrates may have compensatory mechanisms for these two processes, and that SYNE1 may have adapted to perform a specialized function in the brain (Gros-Louis et al., 2007). In rodents, a spectrin-rich splice form of SYNE1 called CPG2 has been shown to control dendritic spine shape and glutamate receptor turnover in response to neuronal activity (Cottrell et al., 2004). To test whether SYNE1 might be responsive to neuronal activity, we performed RNAseq on cultured human primary neurons, before and after depolarization. Transcription of full-length SYNE1 was induced 1.27-fold (n=5, S.E. 0.06, p=0.0203, t test, one-tailed) by neuronal activity, and transcription of BC039121 was induced by 1.50-fold (n=5, S.E. 0.11, p=0.0225, t test, one-tailed) across five biological replicates (Figures 3E and S2). This suggests that both full-length SYNE1 and the shorter BC039121 isoform may have neuronal activity-dependent roles in regulating synaptic strength, like other synaptic genes implicated in autism.
WES for known disease genes
Our findings of inherited, biallelic, hypomorphic ASD mutations in larger families prompted us to ask whether additional cases of ASD might be explained by either unsuspected or atypical presentations of known diseases. Over 450 genes have been identified that, when mutated, have neurocognitive impact (van Bokhoven, 2011). To increase the specificity of our analysis, we chose to analyze a limited subset of 70 of these genes, each associated with a monogenic, autosomal recessive or X-linked neurodevelopmental syndrome in which autistic features have been previously described (Table S2) (Betancur, 2011). We also screened for additional alleles of AMT, PEX7, and SYNE1. We used WES to screen for mutations in these genes in a total of 163 consanguineous and/or multiplex families using established heuristic filtering for rare, high penetrance disease (Bamshad et al., 2011; Stitziel et al., 2011) to identify homozygous, compound heterozygous, or hemizygous variants with allele frequencies of less than 1% (dbSNP132, 1000 Genomes Project, NHLBI Exome Sequencing Project, and population-matched controls consisting of 831 exomes from the Middle Eastern population; see Experimental Procedures for details) and which were predicted to be protein-altering (missense, nonsense, splice site, or frameshift). Candidate mutations were confirmed by Sanger sequencing in the entire family, and were required to segregate with disease status within the family (i.e. homozygous or hemizygous in the affected individuals, inherited in the heterozygous state from parents, and heterozygous or absent from unaffected siblings). An overview of the analytic strategy is shown in Figure 4.
Figure 4.

Schematic of the analytical pipeline. Tiered strategy for examining the cohort of ASD families for inherited mutations in known disease genes. Whole exome sequencing data was used to systematically survey 70 genes (listed in Table S2) known to be associated with autosomal recessive or X-linked neurodevelopmental illness and autistic features. Variants were filtered based on rarity (MAF<1%) in known databases and our internal, population-matched dataset (see Experimental Procedures for details), and predicted functional effect on the protein. All candidate variants were validated by Sanger sequencing and were required to segregate within the family. XL: X-linked, AR: autosomal recessive, MAF: minor allele frequency, 1000G: 1000 Genomes Project, NHLBI EVS5000: NHLBI Exome Sequencing Project.
In five families (Tables 1A and S4), our screen revealed novel molecular genetic diagnoses due to severe loss of function (nonsense or frameshift) hemizygous or homozygous mutations in known genes. One of these was a nonsense mutation in NLGN4X (p.Q329X), found in a single affected male child. NLGN4X is an X-linked gene encoding a neuronal synaptic adhesion molecule, and mutations in NLGN4X have been described in individuals with autism, Asperger syndrome, and intellectual disability (Jamain et al., 2003). This mutation was inherited from an unaffected mother, consistent with prior observations that carrier females may be asymptomatic (Südhof, 2008) (Table 1A; Figure S4).
In another family, two male children affected with autism carried a nonsense mutation in the X-linked gene MECP2 (p.E483X), the gene responsible for Rett syndrome (Table 1A). Their mutation was also inherited from the unaffected mother, who was heterozygous (Figure S4). The finding of MECP2 nonsense mutations in this family was unusual since these are typically lethal in males (Chahrour and Zoghbi, 2007), suggesting that this allele is likely hypomorphic. Consistent with this idea, p.E483X is a late truncation predicted to remove only the last four amino acids of the full-length protein.
Two consanguineous families had homozygous nonsense or frameshift mutations in PAH (Table 1A; Figure S4), the cause of phenylketonuria and one of the earliest neurometabolic syndromes described as a cause of ASD (Zecavati and Spence, 2009). These families were subsequently confirmed to have phenylketonuria by clinical laboratory testing.
An additional ASD family implicated a syndrome associated with dysmorphic features and microcephaly. We found a homozygous frameshift alteration in VPS13B/COH1 in the proband in a consanguineous family who had ASD and mild dysmorphic features (Figure 5A; Table 1A). The mutation (p.A3943fs) causes truncation of the C-terminal 54 amino acids of VPS13B/COH1. Recessive mutations in VPS13B/COH1 cause Cohen syndrome, a constellation of intellectual disability, facial dysmorphism, retinal dystrophy, truncal obesity, joint laxity, intermittent neutropenia, and postnatal microcephaly (Hennies et al., 2004) that has previously been associated with autistic symptoms in some cases (Douzgou and Petersen, 2011). However, significant variability in the features associated with Cohen syndrome makes clinical diagnosis challenging (Mochida et al., 2004; Seifert et al., 2006). The affected child in our cohort had several features that suggest a diagnosis of Cohen syndrome, including microcephaly (head circumference 49 cm at age 9, ≪3rd percentile) and the characteristic facial dysmorphisms typically seen in Cohen syndrome (Figures 5B and 5C; Supplemental Text).
Figure 5.
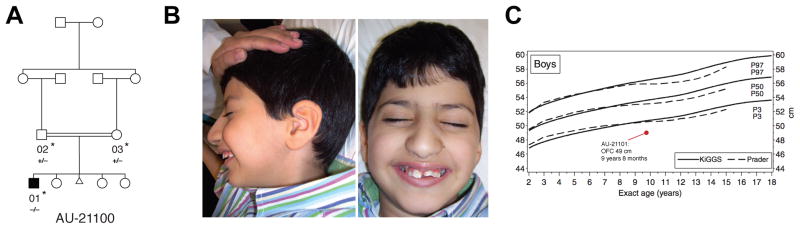
Identification of a mutation in VPS13B, the Cohen syndrome gene in a patient with ASD. (A) AU-21100, a family with one child affected with ASD. Shaded symbol indicates affected individual. The triangle represents a miscarriage. WES was performed on samples from individuals indicated with a star. Genotyping by Sanger sequencing in additional family members was performed where indicated (+: reference allele, −: alternate allele). (B) Photographs of proband, demonstrating mild facial dysmorphisms consistent with Cohen syndrome: prominent nasal tip, protruding maxilla, and short philtrum. For additional clinical details, see Supplemental Text. (C) Head circumference for proband compared to published population normative data (Schienkiewitz et al., 2011). KiGGS: norms from German Health Interview and Examination Survey for Children and Adolescents, 2003–2006. Prader: previously published Swiss growth curves from 1989.
In addition to severe loss-of-function mutations, a significant proportion of rare missense variants are also expected to be significantly deleterious (Kryukov et al., 2007), as underscored by our AMT and PEX7 findings. Eleven families were found to have rare, segregating, homozygous or hemizygous missense changes in known genes (Tables 1B, S3, and S4). While some of these may be expected to be functionally silent, we found clinical and/or biochemical evidence supporting their pathogenicity in at least four instances (Table 1B).
In one consanguineous ASD family, we identified a linked homozygous missense change in AMT (p.D198G) in a single affected child with ASD and intellectual disability (Table 1B). This variant was heterozygous in both parents and an unaffected sibling, and disrupts a highly conserved residue of AMT (SIFT score 0.01). Functional assays of AMT p.D198G demonstrated that, like p.I308F and other NKH-associated mutations, p.D198G is poorly soluble (Figure S5). AMT p.D198G also exhibited a temperature-sensitive protein stability defect (Figure S5). Enzyme specific activity was preserved (Figure S5), suggesting that pathogenicity may be due to protein misfolding/stability and not catalytic dysfunction, similar to what is observed for p.G47R, a known NKH-associated AMT mutation (Figure S1; Table S1). These findings suggest that this child may have also suffered from undiagnosed, atypical NKH.
A child affected with ASD and moderate intellectual disability was found to have a homozygous missense change (p.R367H) in POMGNT1 (Table 1B; Figure S4). POMGNT1 is responsible for an inherited dystroglycanopathy characterized by brain malformation, intellectual disability, developmental delay, hypotonia, and myopia; interestingly, rare patients have been reported with severe autistic features (Haliloglu et al., 2004; Hehr et al., 2007). The p.R367H missense variant in this patient disrupts a highly conserved residue, and this exact allele has been reported as causative in a patient with relatively mild clinical disease, as a compound heterozygous mutation in combination with a splice site mutation (Godfrey et al., 2007).
Finally, in another consanguineous family, the single affected child was homozygous for a rare missense variant (p.S824A) in VPS13B (Table 1B; Figure S4). The proband had in retrospect some but not all features of Cohen syndrome (autism with mild facial dysmorphism and joint laxity), consistent with mild versions reported previously (Hennies et al., 2004).
To begin to explore how these results might extend to nonconsanguineous families, we screened for mutations in genes implicated from our cohort (AMT, PEX7, SYNE1, VPS13B, PAH, POMGNT1) in 612 families from the Simons Simplex Collection (193 trios with parents and affected child, plus 419 quartets with parents, affected child, and unaffected sibling). An analysis of publicly released whole exome sequence data (Iossifov et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) showed a modest trend towards an excess of biallelic, inherited, rare (MAF<1%), protein-altering variants in cases (8/612 ) compared to control siblings (2/419) (p=0.21, Fisher’s exact test, two-tailed) in at least one of the genes we screened (Table S5). As expected for a nonconsanguineous cohort, all but one were found in the compound heterozygous state. Although functional validation of all of these mutations is not available, in at least two cases, phenotype data are supportive of the mutations’ pathogenicity. One affected male child was compound heterozygous for two different mutations in VPS13B (p.W963X/p.G2704R). Gly2704 is a highly conserved residue, while p.W963X leads to early truncation of the protein and has been previously reported in Cohen syndrome (Kolehmainen et al., 2004). Review of the clinical phenotype of this individual confirmed that he manifested, in addition to autism, features of Cohen syndrome including prominent microcephaly (<3 S.D.) and somatic dysmorphisms (Supplemental Text), making the diagnosis of a Cohen syndrome mutation highly likely. A second, unrelated male child affected with autism was compound heterozygous for two rare point mutations in VPS13B, p.S3303R and p.A3691T, both altering highly conserved residues. In addition to being autistic, this child also manifested dysmorphisms of the face and extremities as well as an abnormal hair growth pattern, known to characterize Cohen syndrome. These data confirm that biallelic mutations are also found in nonconsanguineous autism cohorts, but analysis of much larger numbers of genes and patients will be needed to quantify their prevalence.
Discussion
Our data combine WES with segregation analysis to demonstrate that biallelic, hypomorphic mutations underlie at least a subset of ASDs (Chahrour et al., 2012; Morrow et al., 2008). Although the extent to which such mutations contribute to ASD in general remains to be quantified, we demonstrate the utility of our approach in identifying three new ASD genes from a relatively small sample enriched for recessive inheritance. We present three families that simplify the identification of causative genes by narrowing genetic loci to 1–3% of the genome, and allow identification of single mutations that are rare and likely to be functional. Our analyses permit dissection of an otherwise highly heterogeneous disorder. We present additional evidence that biallelic mutations occur in other smaller families, as well as in European-American families from the Simons Simplex Collection. As high-quality whole exome sequencing data from additional cohorts becomes available, it will be valuable to quantify the prevalence of these biallelic mutations in ASD in general. A common theme of many of these mutations is hypomorphic mutations that partially impair gene function, though one or two null mutations are also identified.
The finding of partially disabling mutations in AMT and PEX7 suggests that mild forms of neurometabolic conditions may present predominantly with autistic symptoms, although such very mild mutations may be quite rare, especially in nonconsanguineous populations. Although several neurometabolic disorders have been associated with autistic symptoms (Zecavati and Spence, 2009), milder forms of other metabolic conditions may also be potentially missed by current newborn or biochemical screening tests, which have limits to their sensitivity (Watson et al., 2006). In analogous fashion, the rare biallelic variants we identified in other syndromic neurodevelopmental genes (VPS13B/COH1, SYNE1, MECP2, POMGNT1) also seem to represent mutations in genes in which complete knockout causes more severe syndromes, but which present with milder ASD phenotypes when partially inactivated. Exome sequencing will likely improve the ability to recognize difficult-to-diagnose cases. Metabolic conditions are especially critical to identify since for some neurometabolic conditions, interventions may be available.
In this study we focused on identifying rare, deleterious, penetrant variants that are causative of ASD in the families in question. Our data does not rule out contributions of common variation to ASD in other cases. While common variants are under less selective pressure than rare variants, and are more likely to be benign, the functional impact of most common variants is poorly understood. Some might be expected to lie in autism gene pathways, impact biochemical function, and modify disease risk. For example, a common deletion in TMLHE, encoding the first enzyme in carnitine biosynthesis, has been recently implicated as a risk factor for autism (Celestino-Soper et al., 2012).
Genes implicated in this study include ones known to regulate or be regulated by synaptic activity (MECP2, SYNE1) but also genes not traditionally thought of as having synaptic roles (AMT, PEX7, VPS13B/COH1). This could reflect an important role for nonsynaptic genes and suggest the involvement of hitherto unexpected pathways in ASDs. Alternatively, given the strength of genetic evidence implicating genes of the synapse as causative in ASDs, AMT, PEX7, and VPS13/COH1 may have involvement in synaptic pathways that have yet to be characterized. AMT for example, regulates turnover of glycine, a crucial inhibitory neurotransmitter (Baer, 2009; Keck and White, 2009). PEX7 regulates peroxisomal protein import, and peroxisomes are abundant in dendrites (Kou et al., 2011). Finally, VPS13/COH1 has essential roles in vesicle trafficking through the Golgi apparatus (Seifert et al., 2011). Hence, while null mutations in these genes have effects in many tissues, hypomorphic mutations may cause subtler defects primarily limited to neurons.
Our data support the interpretation that biallelic mutations in the proper setting can cause a spectrum of clinical phenotypes, which at one extreme cause a Mendelian disorder, but at the other extreme represent risk alleles for ASDs. In our multiplex pedigrees, siblings who share homozygosity for the identical biallelic mutation still can show a variety of phenotypes, ranging from ASD to intellectual disability, and including epilepsy and/or other features. This variable expressivity has parallels in known associations of ASD with Mendelian genes like FMR1 or TSC2, which are near fully penetrant for syndromic features of Fragile X or Tuberous Sclerosis, respectively, but only partially penetrant for ASD (Hagerman et al., 2010; Wiznitzer, 2004). Variability of phenotype is also characteristic of recurrent ASD-associated CNVs, such as 16p11.2, which has been linked not only to ASD but also to schizophrenia, epilepsy, ADHD, and obesity (Mccarthy et al., 2009; Shinawi et al., 2010; Walters et al., 2010; Weiss et al., 2008). The common theme of variability of phenotype despite underlying shared genetic susceptibility increasingly suggests that highly penetrant mutations associated strictly with ASD, and never with other conditions, may be extremely rare or nonexistent.
Our data extend the observation that hypomorphic alleles can commonly cause conditions that may be dramatically different from null mutations in the same gene (Walsh and Engle, 2010). Hypomorphic, biallelic mutations, combined with CNVs and heterozygous stop mutations (Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) which completely inactivate one of two alleles, suggest that a common theme for ASD mutations in general might be partial loss of gene function and/or dosage sensitivity. In other words, ASD, and potentially other neuropsychiatric conditions, may be united by incomplete loss of function of specific synaptic genes. Such incomplete loss might provide a general model for the complex genetic architecture, and genetic heterogeneity, of ASDs. In this respect, neuropsychiatric conditions may increasingly come to be understood as much by their allelic architecture as by the specific causative genes.
Experimental Procedures
Human studies
All human studies were reviewed and approved by the institutional review board of the Boston Children’s Hospital, Beth Israel Deaconess Medical Center, and local institutions.
Patient recruitment
The families presented were collected by the Homozygosity Mapping Collaborative for Autism (HMCA) (Morrow et al., 2008), with referral centers in Turkey, the Kingdom of Saudi Arabia, Kuwait, United Arab Emirates, Oman, Jordan, and Pakistan. Inclusion criteria included a diagnosis of autism or ASD by a neurologist, child psychiatrist, or psychologist, and families with multiple affected children and/or suspected consanguinity. See Supplemental Experimental Procedures for further phenotyping details. Additional clinical information on families described here is provided in Supplemental Text.
Genome-wide linkage and homozygosity scans
Genome-wide SNP screens were performed at the Broad Institute and Dana Farber Cancer Institute. Families were genotyped using Affymetrix 500K (NspI/Sty) or Affymetrix 6.0 microarrays. Linkage disequilibrium-based SNP pruning was performed with PLINK, followed by filtering of loci homozygous in all samples and those with Mendelian inheritance errors. Multipoint LOD scores were calculated using MERLIN, assuming a recessive mode of disease inheritance, full penetrance, and a disease allele frequency of 0.0001. Runs of homozygosity were calculated using custom Perl scripts, allowing for no more than 2 consecutive heterozygous SNPs in a run and 3 heterozygous calls in every 10 consecutive SNPs. Intervals homozygous for the same haplotype and shared by all affected individuals were used to narrow the locus in each family. See Supplemental Experimental Procedures for details.
Whole exome sequencing and data analysis
DNA samples were sequenced at the Broad Institute. Whole blood DNA was subject to exome capture (SureSelect v2, Agilent Technologies) and whole exome sequence (Illumina HiSeq) was obtained on a total of 277 affected children and 409 parents, with a mean target coverage of 85.6% at >=20X and a mean read depth of 158X. For this study, families harboring known autism-associated CNVs were excluded (Supplemental Experimental Procedures). Reads were aligned to NCBI human genome build v37 and variants were called and annotated using GATK. ANNOVAR (Wang et al., 2010), and custom pipelines. All reported variants were confirmed by Sanger sequencing. See Supplemental Experimental Procedures for additional details.
SSC exome reanalysis
Exome data from 612 families from the Simons Simplex Collection were obtained from dbGAP and NDAR. Raw sequence reads were aligned with BWA and variants were called with Samtools and annotated as previously described (Sanders et al., 2012).
Sanger resequencing
See Supplemental Experimental Procedures for details.
Data visualization
See Supplemental Experimental Procedures for details.
AMT expression and enzymatic assays
Wildtype and mutant human AMT proteins with a C-terminal His6-tag were expressed and purified as previously described (Okamura-Ikeda et al., 2005). I308F, I308A, D198G, or D198A substitutions were introduced using site-directed mutagenesis, and enzymatic activities were determined as previously described (Okamura-Ikeda et al., 2010).
For heat stability studies, wildtype and mutant AMTs (about 0.5 mg/ml in 20 mM Tris-HCl PH 7.5, 1 mM DTT, 20 mM (p-amidinophenyl) methanesulfonyl fluoride, and 10% glycerol) were incubated for 1, 2, and 3 hours at 37°C and 42°C. After incubation, the solutions were centrifuged and the protein concentrations in the supernatants were determined using Coomassie Plus (Thermo Scientific, USA) with BSA as standard. The remaining protein concentrations in the supernatant were showed as a percent of the initial concentrations.
PEX7 peroxisomal import assays
A peroxisomal import marker was generated by fusing mCherry fluorescent protein to the PTS2 signal located in the first 26 amino acids of rat 3-ketoacyl-CoA thiolase [P07871.2] (Tsukamoto et al., 1994). N-terminal c-myc tagged variants of PEX7 (W75C and S25F) were engineered using PCR based site-directed mutagenesis of the N-myc-PEX7 cDNA [NM_000288] in pCDNA1. PEX7 and PTS2-mCherry were transiently transfected into an immortalized RCDP1 fibroblast line with a PEX7 null genotype (p.L292X];[S132X) and processed at 3 days for direct and indirect immunofluorescence as previously reported (Braverman et al., 2002). Recovery of peroxisomal import was assessed by blinded visual scoring of 100 cells each from 3 separate transfections. Peroxisomal import was confirmed by co-localization of the peroxisome membrane protein PEX14. Whole cell lysates from similarly transfected fibroblasts were used for immunoblotting with antibodies to the endogenous PTS2 proteins thiolase, PhyH, and AGPS (Zhang et al., 2010). PEX7 protein expression was confirmed with a c-myc antibody (SC789, Santa Cruz Biotechnology Inc., Santa Cruz, CA).
RNAseq of human neurons
Primary human neuronal cells were purchased from Sciencell (Carlsbad, CA). For RNAseq experiments, neuronal cultures from 3 biological replicates were grown for around two weeks (DIV13-16). At the final day in culture, neurons in the experimental group were stimulated for 6 hours with 55mM KCl and were harvested along with the unstimulated control neurons using Trizol (Invitrogen). Strand-specific and paired-end cDNA libraries were generated using the PE RNAseq library kit (Illumina). RNAseq was performed using HiSeq 2000 at the Broad Institute. 76-bp RNAseq reads were aligned to the human GRCh37/hg19 assembly using BWA. See Supplemental Experimental Procedures for details. For quantification of SYNE1 expression in response to depolarization, for each biological replicate, expression levels (normalized reads per bp) of individual exons were calculated, which allowed the calculation of the average expression level over any isoform of SYNE1 comprising subsets of these exons. Then fold-change ratios (6 hours/unstimulated) of these levels were calculated for each replicate and isoform, and finally each isoform’s mean and standard error over the replicates’ fold changes.
ChIPseq of human neurons
The mini-ChIP assays were performed as previously described (Adli and Bernstein, 2011) on human neuronal cells that had been cultured for around two weeks. Briefly, cells were cross-linked, lysed, and the fragmented chromatin was then immunoprecipated with H3K27Ac (abcam Cat# ab4728) and H3K27me3 (Millipore Cat# 074490) antibodies. The ChIP DNA was recovered and precipitated following standard procedures. The ChIP DNA libraries were then constructed using ChIP-Seq DNA Sample Prep Kit (Illumina) and subsequently sequenced using HiSeq 2000 (Illumina) in Biopolymers facility at Harvard Medical School. ChIPseq reads were aligned to the human genome (GRCh37/hg19 assembly). See Supplemental Experimental Procedures for details.
Data access
Whole exome sequence data is available online (The National Database for Autism Research (NDAR) Collection ID: NDARCOL0001918).
Supplementary Material
Highlights.
Familial ASD traced to hypomorphic mutations in AMT
Familial ASD traced to hypomorphic mutation in PEX7
Familial ASD traced to hypomorphic mutation in SYNE1
Whole exome sequencing yields molecular diagnoses due to missed or atypical disease
Acknowledgments
We are grateful to Ed Gilmore, Chiara Manzini, Jenny Yang, and Mark Daly for stimulating discussions and helpful comments on the manuscript, and Thomas Lehner for support from the National Institute of Mental Health (NIMH). We are also grateful for the participation of the many families that enrolled in our studies as well as for the collaborative support of the Kuwait Center for Autism and the Dubai Autism Center. TWY was supported by a National Institute of Health (NIH) T32 grant (T32 NS007484-08), the Clinical Investigator Training Program (CITP) at Harvard-MIT Health Sciences and Technology and Beth Israel Deaconess Medical Center in collaboration with Pfizer, Inc. and Merck and Company, Inc., and the Nancy Lurie Marks Junior Faculty MeRIT Fellowship. MHC was supported by a NIH T32 grant (T32 NS007473-12). GHM was supported by the Young Investigator Award of NARSAD as a NARSAD Lieber Investigator. AP was supported by the National Institutes of Neurological Disease and Stroke (K23NS069784). ADG was supported by the National Institute of General Medical Sciences (T32GM07753). Research was supported by grants from the National Institute of Mental Health (RO1 MH083565; 1RC2MH089952) to CAW, the NIH to MEG (RO1 NS048276), the NIMH to EMM (1K23MH080954-01), the Dubai Harvard Foundation for Medical Research, the Nancy Lurie Marks Foundation, the Simons Foundation, the Autism Consortium, and the Manton Center for Orphan Disease Research. Sequencing at the Broad Institute was supported by the ARRA Grand Opportunities grant 1RC2MH089952. CAW is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions
TWY identified AMT, PEX7, and SYNE1 mutations, helped design AMT and PEX7 functional studies, designed and performed exome sequencing analyses for candidate genes, contributed to CNV analyses, and wrote the manuscript. MHC designed and performed exome sequencing analyses for candidate genes, analyzed Sanger validation data and SSC exome data, and wrote the manuscript. MEC helped analyze AU-1700 and AU-3500. SJ designed PEX7 functional experiments with NEB and SJ performed them. KOI designed and performed AMT functional studies and analyzed results. BA designed and analyzed RNAseq, ChIPseq, and qPCR experiments. DAH analyzed RNAseq and qPCR experiments. MA performed ChIPseq experiments, and ANM analyzed the data. ADG performed RACE experiments for SYNE1. KS and KM designed the CNV analysis, and KS compiled the CNV catalog and identified pathogenic CNVs. ETL and SJS helped analyze SSC whole exome data. GHM performed clinical phenotyping of Middle Eastern families as well as detailed molecular analyses of AU-8600. JNP organized clinical information and patient samples. CMS assisted with exome sequencing analyses and performed follow up Sanger validation. MF and JR performed follow up Sanger validation. RHN performed clinical phenotyping of AU-17800. JW performed clinical phenotyping of AU-17700 and AU-17800. RMJ performed clinical phenotyping of AU-1600, AU-10000, and AU-10200. RSH performed genome wide linkage studies and homozygosity analyses. BYK assisted with characterization of the AMT mutation. MAS organized clinical information and patient samples, and referred AU-17700 and AU-17800. NM referred and clinically characterized AU-4200, AU-4400, AU-4900, AU-5700, AU-6100, AU-6200, AU-6300, AU-8600 AU-11100, AU-11800, AU-11900, AU-12100, AU-12400, AU-14900, AU-15800, AU-16700, AU-20700, AU-22500, AU-23400, and AU-24300. AH referred and characterized AU-3500 and AU-3600. SB referred and characterized AU-1700. GG referred and characterized AU-1700, AU-3100, AU-4100, and AU-6000. FMH helped characterize AU-17700 and AU-17800. EL and AP performed clinical phenotyping of AU-1600, AU-10000, and AU-10200. OO referred and characterized AU-13100, AU-13400, AU-18000, AU-20300, and AU-22000. SA, SAA and LB referred and characterized AU-1600. SA referred and characterized AU-9600. TBO and AT referred and characterized AU-21100. LAG and VE referred and characterized AU-3200. CS organized and coordinated exome sequencing. LR evaluated the second compound heterozygous PEX7 family. SBG directed exome sequencing. KM designed the CNV analysis. MWS oversaw SSC exome analyses. MEG oversaw SYNE1 RNAseq and qPCR experiments. HT designed and performed AMT functional experiments. NEB designed PEX7 functional experiments, recruited the nonconsanguineous family with two sisters affected by PEX7 mutation, and contributed to interpretation of PEX7 sequencing data. EMM helped characterize AU-1700, performed linkage studies on AU-1600, AU-1700, and AU-3500, helped design the exome sequencing experiment, and contributed to finding the SYNE1 mutation. CAW directed the overall research and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 2011;6:1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegarth DA, Toone JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Molecular Genetics and Metabolism. 2001;74:139–146. doi: 10.1006/mgme.2001.3224. [DOI] [PubMed] [Google Scholar]
- Applegarth DA, Toone JR. Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. Journal of inherited metabolic disease. 2004;27:417–422. doi: 10.1023/b:boli.0000031222.38328.59. [DOI] [PubMed] [Google Scholar]
- Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, Glick B, Nevo Y, Ben-Neriah Z, Melki J. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Human Molecular Genetics. 2009;18:3462–3469. doi: 10.1093/hmg/ddp290. [DOI] [PubMed] [Google Scholar]
- Baer K. Localisation of glycine receptors in the human forebrain, brainstem, and cervical spinal cord: an immunohistochemical review. Frontiers in Molecular Neuroscience. 2009:2. doi: 10.3389/neuro.02.025.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Reviews Genetics. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- Braverman N, Chen L, Lin P, Obie C, Steel G, Douglas P, Chakraborty PK, Clarke JTR, Boneh A, Moser A, et al. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Human mutation. 2002;20:284–297. doi: 10.1002/humu.10124. [DOI] [PubMed] [Google Scholar]
- Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nature Genetics. 1997;15:369. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, Shields DC, Abrahams BS, Almeida J, Bacchelli E, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Human genetics. 2012;131:565–579. doi: 10.1007/s00439-011-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celestino-Soper PBS, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, Cai G, Sadikovic B, Lee K, Lo C, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, Stevens CR, Schubert CR, Collaboration AAS, Greenberg ME, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS genetics. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell JR, Borok E, Horvath TL, Nedivi E. CPG2A brain- and synapse-specific protein that regulates the endocytosis of glutamate receptors. Neuron. 2004;44:677–690. doi: 10.1016/j.neuron.2004.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Current Opinion in Genetics and Development. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Dinopoulos A, Matsubara Y, Kure S. Atypical variants of nonketotic hyperglycinemia. Molecular Genetics and Metabolism. 2005;86:61–69. doi: 10.1016/j.ymgme.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Douzgou S, Petersen M. Clinical variability of genetic isolates of Cohen syndrome. Clinical genetics. 2011;79:501–506. doi: 10.1111/j.1399-0004.2011.01669.x. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain: a journal of neurology. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- Grady RM, Starr DA, Ackerman GL, Sanes JR, Han M. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc Natl Acad Sci U S A. 2005;102:4359–4364. doi: 10.1073/pnas.0500711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nature Genetics. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- Hagerman R, Hoem G, Hagerman P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Molecular autism. 2010;1:12. doi: 10.1186/2040-2392-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haliloglu G, Gross C, Senbil N, Talim B, Hehr U, Uyanik G, Winkler J, Topaloglu H. Clinical spectrum of muscle-eye-brain disease: from the typical presentation to severe autistic features. Acta myologica: myopathies and cardiomyopathies: official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the study of striated muscle diseases. 2004;23:137–139. [PubMed] [Google Scholar]
- Hamosh A, JM . Non-ketotic hyperglycinemia. In: Scriver BACR, Sly WS, Valle D, editors. In The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 2065–2078. [Google Scholar]
- Hehr U, Uyanik G, Gross C, Walter MC, Bohring A, Cohen M, Oehl-Jaschkowitz B, Bird LM, Shamdeen GM, Bogdahn U, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle–eye–brain disease. neurogenetics. 2007;8:279–288. doi: 10.1007/s10048-007-0096-y. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennies HC, Rauch A, Seifert W, Schumi C, Moser E, Al-Taji E, Tariverdian G, Chrzanowska KH, Krajewska-Walasek M, Rajab A, et al. Allelic Heterogeneity in the COH1 Gene Explains Clinical Variabilityin Cohen Syndrome. The American Journal of Human Genetics. 2004;75:138–145. doi: 10.1086/422219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-h, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Bourgeron T, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature Genetics. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T, White J. Glycinergic inhibition in the hippocampus. Rev Neurosci. 2009;20:13–22. doi: 10.1515/revneuro.2009.20.1.13. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler AD. The Human Genome Browser at UCSC. Genome Research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolehmainen J, Wilkinson R, Lehesjoki AE, Chandler K, Kivitie-Kallio S, Clayton-Smith J, Träskelin AL, Waris L, Saarinen A, Khan J, et al. Delineation of Cohen Syndrome Following a Large-Scale Genotype-Phenotype Screen. The American Journal of Human Genetics. 2004;75:122–127. doi: 10.1086/422197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou J, Kovacs GG, Hoftberger R, Kulik W, Brodde A, Forss-Petter S, Honigschnabl S, Gleiss A, Brugger B, Wanders R, et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011;122:271–283. doi: 10.1007/s00401-011-0836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. American Journal of Human Genetics. 2007;80:727. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science (New York, NY) 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee Y-h, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. Rare De Novo and Transmitted Copy-Number Variation in Autistic Spectrum Disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics (Oxford, England) 2009 doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nature Genetics. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Rajab A, Eyaid W, Lu A, Al-Nouri D, Kosaki K, Noruzinia M, Sarda P, Ishihara J, Bodell A, et al. Broader geographical spectrum of Cohen syndrome due to COH1 mutations. Journal of medical genetics. 2004;41:e87. doi: 10.1136/jmg.2003.014779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science (New York, NY) 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley AM, Hettema EH, Hogenhout EM, Brites P, ten Asbroek AL, Wijburg FA, Baas F, Heijmans HS, Tabak HF, Wanders RJ, et al. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a nonfunctional PTS2 receptor. Nature Genetics. 1997;15:377–380. doi: 10.1038/ng0497-377. [DOI] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011 doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, MacKenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature Genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura-Ikeda K, Hosaka H, Maita N, Fujiwara K, Yoshizawa AC, Nakagawa A, Taniguchi H. Crystal structure of aminomethyltransferase in complex with dihydrolipoyl-H-protein of the glycine cleavage system: implications for recognition of lipoyl protein substrate, disease-related mutations, and reaction mechanism. The Journal of biological chemistry. 2010;285:18684–18692. doi: 10.1074/jbc.M110.110718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura-Ikeda K, Hosaka H, Yoshimura M, Yamashita E, Toma S, Nakagawa A, Fujiwara K, Motokawa Y, Taniguchi H. Crystal Structure of Human T-protein of Glycine Cleavage System at 2.0Å Resolution and its Implication for Understanding Non-ketotic Hyperglycinemia. Journal of Molecular Biology. 2005;351:1146–1159. doi: 10.1016/j.jmb.2005.06.056. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010 doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schienkiewitz A, Schaffrath Rosario A, Dortschy R, Ellert U, Neuhauser H. German head circumference references for infants, children and adolescents in comparison with currently used national and international references. Acta paediatrica (Oslo, Norway: 1992) 2011;100:e28–33. doi: 10.1111/j.1651-2227.2011.02173.x. [DOI] [PubMed] [Google Scholar]
- Sebat Lakshmi, Malhotra Troge, Lese-Martin Walsh, Yamrom Yoon, Krasnitz Kendall, et al. Strong Association of De Novo Copy Number Mutations with Autism. Science (New York, NY) 2007 doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert W, Holder-Espinasse M, Spranger S, Hoeltzenbein M, Rossier E, Dollfus H, Lacombe D, Verloes A, Chrzanowska KH, Maegawa GHB, et al. Mutational spectrum of COH1 and clinical heterogeneity in Cohen syndrome. Journal of medical genetics. 2006;43:e22. doi: 10.1136/jmg.2005.039867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert W, Kuhnisch J, Maritzen T, Horn D, Haucke V, Hennies HC. Cohen Syndrome-associated Protein, COH1, Is a Novel, Giant Golgi Matrix Protein Required for Golgi Integrity. Journal of Biological Chemistry. 2011;286:37665–37675. doi: 10.1074/jbc.M111.267971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SHL, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. Journal of medical genetics. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JG, Roberts RG. Patterns of evolutionary conservation in the nesprin genes highlight probable functionally important protein domains and isoforms. Biochemical Society transactions. 2008;36:1359–1367. doi: 10.1042/BST0361359. [DOI] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Nurnberger JI, Rietschel M, Blackwood D, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature Genetics. 2011 doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr DA, Han M. Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science. 2002;298:406–409. doi: 10.1126/science.1075119. [DOI] [PubMed] [Google Scholar]
- State MW. The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron. 2010;68:254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome biology. 2011;12:227. doi: 10.1186/gb-2011-12-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto T, Hata S, Yokota S, Miura S, Fujiki Y, Hijikata M, Miyazawa S, Hashimoto T, Osumi T. Characterization of the signal peptide at the amino terminus of the rat peroxisomal 3-ketoacyl-CoA thiolase precursor. The Journal of biological chemistry. 1994;269:6001–6010. [PubMed] [Google Scholar]
- van Bokhoven H. Genetic and Epigenetic Networks in Intellectual Disabilities. Annu Rev Genet. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- Walsh CA, Engle EC. Allelic diversity in human developmental neurogenetics: insights into biology and disease. Neuron. 2010;68:245–253. doi: 10.1016/j.neuron.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M, Mann M, Lloyd-Puryear M, Rinaldo P, Howell R ACoMGNSE Group. Newborn Screening: Toward a Uniform Screening Panel and System--Executive Summary. Pediatrics. 2006;117:S296. doi: 10.1542/peds.2005-2633I. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MAR, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England Journal of Medicine. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wiznitzer M. Autism and tuberous sclerosis. Journal of Child Neurology. 2004;19:675–679. doi: 10.1177/08830738040190090701. [DOI] [PubMed] [Google Scholar]
- Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, Stern R, Raymond FL, Sandford R, Malik Sharif S, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. American Journal of Human Genetics. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecavati N, Spence SJ. Neurometabolic disorders and dysfunction in autism spectrum disorders. Current neurology and neuroscience reports. 2009;9:129–136. doi: 10.1007/s11910-009-0021-x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG. The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics. 2002;80:473–481. [PubMed] [Google Scholar]
- Zhang R, Chen L, Jiralerspong S, Snowden A, Steinberg S, Braverman N. Recovery of PEX1-Gly843Asp peroxisome dysfunction by small-molecule compounds. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5569–5574. doi: 10.1073/pnas.0914960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.