

Abstract
Activation of human natural killer (NK) cells is associated with the cleavage of CD16 from the cell surface, a process mediated by matrix metalloproteinases (MMPs). In this report, we examined whether inhibition of MMPs would lead to improved NK cell antibody-dependent cell-mediated cytotoxicity (ADCC) function. Using an in-vitro ADCC assay, we tested the anti-tumour function of NK cells with three different therapeutic monoclonal antibodies (mAbs) in the presence of MMPs inhibitor GM6001 or its control. Loss of CD16 was observed when NK cells were co-cultured with tumour targets in the presence of specific anti-tumour antibodies, and was found particularly on the majority of degranulating NK responding cells. Treatment with MMPs inhibitors not only prevented CD16 down-regulation, but improved the quality of the responding cells significantly, as shown by an increase in the percentage of polyfunctional NK cells that are capable of both producing cytokines and degranulation. Furthermore, MMPs inhibition resulted in augmented and sustained CD16-mediated signalling, as shown by increased tyrosine phosphorylation of CD3ζ and other downstream signalling intermediates, which may account for the improved NK cell function. Collectively, our results provide a foundation for combining MMPs inhibitors and therapeutic mAbs in new clinical trials for cancer treatment.
Keywords: monoclonal antibody, NK cell, polyfunctionality, tumour immunotherapy
Introduction
Natural killer (NK) cells play a vital role in antibody-based anti-tumour immunotherapies via antibody-dependent cell-mediated cytotoxicity (ADCC) mechanism, whereby the Fcγ receptor III (FcγRIII or CD16) expressed on NK cells binds to the Fc portion of an antibody that recognizes tumour antigens on the surface of cancer cells. During the process of ADCC, NK cells release cytokines such as interferon (IFN)-γ and tumour necrosis factor (TNF)-α, and cytotoxic granules containing perforin and granzymes that penetrate the tumour cell and promote its death 1–4. Human NK cells are divided into two distinct subsets according to their surface expression of CD56 and their different effector functions. The CD56dim subset of NK cells are the primary mediators of ADCC, because of their high expression of CD16 and cytotoxic granules 5.
Two isoforms of CD16, CD16A and CD16B, were identified in humans. CD16B is a glycosyl-phosphatidyl inositol (GPI)-anchored molecule that is found on neutrophils and eosinophils, and CD16A is expressed on monocytes and NK cells 6. CD16A contains a transmembrane domain that enables its association with the immunoreceptor tyrosine-based activation motif (ITAM) containing adaptors CD3ζ and the FcRγ chain 7,8. Immunoglobulin (Ig)G interacts with CD16 via its second, membrane-proximal Ig domain, which can be recognized by the monoclonal antibody (mAb) 3G8 9, and promotes CD3ζ phosphorylation that could lead to cytolytic signals and cytokine induction 8. Recent studies have shown that CD16 is not only essential for ADCC function of NK cells, but also important for NK cell spontaneous cytotoxicity through its association with the CD2 co-activating receptor 10. Upon activation NK cells enter a refractory phase, in which CD16 molecules are rapidly cleaved off from the surface in a process mediated by MMPs 11–13. This could lead to impaired NK cell function and impede the efficacy of antibody-based therapies that utilize ADCC as a mechanism of action 13.
MMPs are zinc-dependent endopeptidases that degrade a large variety of extracellular matrix proteins 14, and are responsible for the cleavage of several cell surface receptors such as CD16 and CD95L 12,15,16. Uncontrolled MMP activity will lead to various physiological and pathological abnormalities. Recent studies have also shown a strong correlation between elevated MMP levels within the tumour stroma and tumour cell invasion or metastasis 17–19. The activity of MMPs can be blocked by inhibitors such as GM6001, which targets a subset of MMPs. Several MMPs inhibitors have been tested in clinical trials for inflammation and cancer therapies 17.
In this report, we examined the NK cell-mediated ADCC activity of three therapeutic mAbs, including trastuzumab [anti-human epidermal growth factor receptor 2 (HER2)], rituximab (anti-CD20) and cetuximab [anti-epidermal growth factor receptor (EGFR)]. In all cases, cell surface levels of CD16 on NK cells were greatly down-regulated in response to mAb-coated tumour cells. Treatment with a MMPs inhibitor preserved the expression of CD16. More importantly, inhibition of MMPs activity resulted in a significant increase in the percentage of polyfunctional NK cells, i.e. cells that can perform more than one effector function. This effect was the result of increased and persistent CD16-mediated signalling, characterized by higher levels of phosphorylated CD3ζ and other signalling intermediates. Our results suggest that the use of MMPs inhibitors may increase the efficacy of therapeutic mAbs that utilize ADCC as a mechanism of action for the treatment of cancer.
Materials and methods
Antibodies and reagents
Trastuzumab was provided by Dr Wen Jin Wu's laboratory (Food and Drug Administration, Silver Spring, MD, USA). Rituximab and cetuximab were purchased from the National Institutes of Health (NIH, Bethesda, MD, USA) pharmacy. MMPs inhibitor GM6001 and its control were obtained from Calbiochem (EMD Biosciences, Inc., La Jolla, CA, USA). Antibodies used for flow cytometric analysis were obtained from the following vendors: allophycocyanin (APC) anti-CD16 (clone CB16), phycoerythrin (PE) anti-CD56 (clone CMSSB), AlexaFluor 700 anti-CD3 (clone UCHT1), peridinin chlorophyll-cyanin 5·5 (PerCP-Cy5·5) anti-CD3 (clone SK7), fluorescein isothiocyanate (FITC) anti-CD107a (clone ebioH4A3), FITC anti-CD107b (clone ebioH4A4) and PE-Cy7 anti-TNF-α (clone Mab11), were from eBioscience (San Diego, CA, USA); PerCP-Cy5.5 anti-IFN-γ (clone B27) is from BD Biosciences, San Jose, CA, USA; Aqua Live/Dead® fixable dead cell stain kit is from Invitrogen (Carlsbad, CA, USA).
Cell lines
SKBr3 cells, derived from mammary adenocarcinoma, were kindly provided by Dr Wen Jin Wu's laboratory. Raji cells derived from a Burkitt's lymphoma and A431 cells derived from a vulva epidermoid carcinoma were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). SKBr3 and A431 cell were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Raji cells were cultured in RPMI-1640 medium containing 10% FBS.
Degranulation and cytokine production assays
Blood samples from healthy donors were collected under an institutional review board-approved protocol at the NIH with appropriate written consent. NK cells were isolated with human NK cell enrichment kit from Stem Cell Technology (> 90% purity of NK cells was achieved), using a Robosep (Stem Cell Technology, Vancouver, BC, Canada). Freshly isolated NK cells were incubated with SKBr3, Raji or A431 cells in the presence of the MMPs inhibitor GM6001 or control at 50 μM in Iscove's modified Dulbecco's medium (IMDM) medium (Invitrogen) containing 10% human AB serum (Valley Biomedical, Winchester, VA, USA). Trastuzumab, rituximab or cetuximab were added at a final concentration of 10 μg/ml. For degranulation assays, cells were incubated for 4 h at 37°C. FITC anti-CD107a and anti-CD107b were added to all conditions. For the measurement of cytokine production, cells were incubated for 6 h in the presence of GolgiStop (monensin) (BD Biosciences), according to the manufacturer's manual. After harvest, cells were labelled with appropriate fluorochrome-labelled antibodies. Flow cytometric analysis was performed and data were analysed using the FlowJo software (Treestar, Ashland, OR, USA).
Western blots
Freshly isolated NK cells were incubated at 4°C with purified anti-CD16, 20 μg/ml final concentration (clone 3G8) (BD Biosciences) for 30 min. After several washes to remove the unbound anti-CD16 mAb, NK cells were treated with either GM6001 (50 μM) or its control in the presence of purified goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA) at 20 μg/ml final concentration, for different time-points at 37°C. Next, cells were lysed for 30 min at 4°C with lysis buffer [50 mM Tris, 150 mM NaCl, 0·1% Triton X-100, 0·5% sodium deoxicholate, 2 mM ethylenediamine teraacetic acid (EDTA) and 0·1% sodium dodecyl sulphide (SDS)] containing cocktail protease inhibitors (Amersham Bioscience, Pittsburgh, PA, USA) and phosphatase inhibitors (Thermo Scientific, Logan, UT, USA). Next, lysates were spun down at 4°C for 10 min at 14 500 g to remove cell debris. For immunoblotting, total cell lysates were resolved on a 4−20% gradient polyacrylamide gel (Biorad, Hercules, CA, USA) and transferred to PVDF membranes (Biorad). Membranes were then blocked with 5% (w/v) non-fat milk in Tris-buffered saline-Tween 20 (TBS)-T (20 mM Tris-HCl pH 8, 150 mM NaCl and 0·05% Tween 20) for 1 h. Next, the membranes were probed with the appropriate anti-phosphotyrosine mAb overnight at 4°C. After extensive washing in TBS-T, the membranes were incubated with horseradish peroxidase (HRP)-labelled goat anti-mouse or goat anti-rabbit Ig antibody, and immunoreactivity was visualized by using the enhanced chemiluminescence (ECL) system (GE Healthcare, Pittsburgh, PA, USA). For loading control, additional gels were ran in parallel and membranes were probed with antibody against the corresponding total protein.
Killing assay
SKBr3 cells were labelled with bis(acetoxymethyl)2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (BADTA) according to the manufacturer's manual (PerkinElmer™, Waltham, MA, USA). Freshly isolated NK cells were cultured with labelled SKBr3 cells at different E/T ratios in the absence or presence of trastuzumab (10 μg/ml) for 2·5 h at 37°C. SKBr3 cells alone served as spontaneous release (SR) and SKBr3 cells lysed with 1% Triton X-100 served as total release (TR). After the incubation, 25 μl of supernatant from each culture condition was transferred to a 96-well plate with 200 μl europium solution prepared according to the manufacturer's manual. Europium signals were measured with a PerkinElmer multi-label counter. Killing efficiency was then calculated according to the following formula: % of specific killing = [(experimental release − SR)/(TR-SR)] × 100%.
Statistical analysis
Graphs and statistical analysis were made with GraphPad Prism version 5.0 software. Student's t-test unpaired comparison was used to determine significant differences between each group. P-values < 0·05 were considered to be significant.
Results
CD16 cell surface down-regulation during ADCC with therapeutic mAbs
mAbs represent a promising strategy for cancer therapies due to high specificity and relatively low toxicity when compared to other anti-cancer drugs. Many therapeutic mAbs for cancer treatment are of the IgG1 isotype, with intact or engineered Fc regions that are capable of harnessing the immune system against cancer through the mechanism of ADCC 2,4. On human NK cells, CD16 is the receptor responsible for ADCC, and its cell surface expression is associated with the magnitude of NK cell-mediated ADCC, as was shown in the settings of HIV infection and cancer 13,20. More importantly, engagement of Fcγ receptors on effector cells is required for the anti-tumour actions of therapeutic mAbs such as rituximab and trastuzumab in xenograft models 21. It has been shown that cross-linking of CD16 on NK cells by either IgG or the anti-CD16 mAb clone 3G8 led to release of CD16 from the cell surface in a process mediated by MMPs 9. We decided to test antibody-mediated CD16 down-regulation on resting freshly isolated NK cells in a clinically relevant setting for cancer therapy, by using the therapeutic mAbs trastuzumab, rituximab and cetuximab. First, and as expected, we found a profound cell surface CD16 down-regulation only when NK cells were incubated with target cells in the presence of the specific therapeutic mAbs (Fig. 1a,b and Fig. S1). CD16 was not down-regulated when NK cells were incubated with only target cells or therapeutic mAbs, indicating that in these experimental conditions CD16 down-regulation requires the cross-linking of the receptor. Similar results were observed when IL-2-activated and -expanded NK cells were used instead of freshly isolated resting NK cells, indicating that CD16 down-regulation was independent of the activation status of NK cells (data not shown). Very importantly, CD16 down-regulation was associated with NK cell degranulation, as shown by the expression of CD107a/b mainly on CD16 low NK cells (Fig. 1c, upper panel). Surface expression of CD107a/b is a marker of lytic granule exocytosis 22. NK cells did not degranulate when cultured with only target cells or therapeutic mAbs (data not shown). In addition to target cell killing, NK cells also respond, producing cytokines when activated 23. As described for CD16 down-regulation we found that NK cells produce IFN-γ and TNF-α only when they were incubated with target cells in the presence of therapeutic mAbs (Fig. 2a, upper panel). Together, these effector functions resulted in the killing of target cells (Fig. S2).
Figure 1.

Matrix metalloproteinases (MMPs) inhibition preserved CD16 cell surface expression on natural killer (NK)-responding cells. Freshly isolated NK cells were incubated at a 1:1 ratio with the tumour cell lines SKBr3, Raji or A431, in the absence or presence of therapeutic monoclonal antibodies (mAbs) trastuzumab (anti-HER2), rituximab (anti-CD20) or cetuximab [anti-epidermal growth factor receptor (EGFR)], at 10 μg/ml for 4–6 h in the presence of the MMPs inhibitor GM6001 or control (50 μM). CD16 and CD107a/b expression were measured by flow cytometric analysis. Cells were gated electronically on CD3−CD56dim NK cells. (a) Representative dot-plots of CD16 expression on NK cells cultured with SKBr3 tumour cells in the presence or absence of trastuzumab and treated with GM6001 or its control (CTRL) are shown. (b) Bar graphs showing the percentage of NK cells expressing CD16 are shown. Bars represent the average ± standard error of the mean. Results are from four independent experiments with NK cells from four donors. (c) Representative dot-plots of CD16 and CD107a/b expression on NK cells cultured with tumour cell lines in the presence of therapeutic mAbs and treated with GM6001 or its control are shown. *P < 0·05 compared to CTRL.
Figure 2.

Matrix metalloproteinases (MMPs) inhibition improved natural killer (NK) cell polyfunctionality. (a) Representative dot plots of interferon (IFN)-γ- and tumour necrosis factor (TNF)-α-producing NK cells cultured with SKBr3 tumour cells in the absence or presence of trastuzumab, and treated with the MMPs inhibitor GM6001 or its control (CTRL) are shown. (b) Bar graph representation of the percentage of IFN-γ single-producing, TNF-α single-producing and IFN-γ and TNF-α double-producing NK cells cultured with SKBr3 tumour cells in the absence or presence of trastuzumab and with GM6001 or its control. Bars represent the average ± standard error of the mean (s.e.m.). Results are from four independent experiments with NK cells from four donors. (c) Bar graph representation of the mean fluorescence intensity (MFI) of IFN-γ and TNF-α expression by NK cells cultured with SKBr3 tumour cells in the presence of trastuzumab with GM6001 or its control. Data were normalized according to the controls. Results are from three independent experiments with NK cells from three donors. (d) Bar graph representation of the possible combinations of three effector functions (degranulation as shown by the expression of CD107a/b, IFN-γ and TNF-α production) on the x-axis and the percentage of distinct functional populations within the CD56dim NK cells on the y-axis is shown. Bars represent the average ± s.e.m. Results are from three independent experiments with NK cells from three donors. *P < 0·05 compared to CTRL.
MMPs inhibition preserved CD16 expression and improved NK cell function
We reasoned that inhibiting CD16 down-regulation would result in enhanced NK cell-mediated ADCC activity, and consequently improve the efficacy of therapeutic mAbs. To prove this hypothesis, we examined the role of the MMPs inhibitor GM6001 in NK cell-mediated ADCC. As expected, GM6001 was able to preserve CD16 expression on the majority of NK cells during ADCC (Fig. 1a,b and Fig. S1), and significantly on the degranulating CD107a/b+ NK effector cells (Fig. 1c). We did not find a significant change in the percentage of degranulating CD107a/b+ NK cells when ADCC assay was performed in the presence of the MMPs inhibitor GM6001 (data not shown). However, and very importantly, the inhibition of CD16 down-regulation resulted in a significant increase in the percentage of cytokine-producing NK cells (Fig. 2a,b and Fig. S3). We then analysed the level of cytokine production by the cells by measuring the median fluorescence intensity (MFI) of cytokine staining, a value known to be correlated with the amount of cytokine produced by an NK cell. We observed that the MFI of IFN-γ and TNF-α expression was increased when the MMPs inhibitor GM6001 was present during ADCC (Fig. 2c), indicating that not only the number of IFN-γ- and TNF-α-producing NK cells was increased by inhibiting trastuzumab-mediated CD16 down-regulation, but also on a per cell basis, NK cells tend to produce more IFN-γ and TNF-α when MMPs are inhibited. These results confirmed our expectation that preserving CD16 cell surface expression during ADCC led to an increase, at least in part, in the NK cell effector functions.
Studies have shown that there are differences in the quality of effector cells based on whether they have the ability to perform more than one effector function, i.e. degranulation and/or production of two or more cytokines 24. In particular, it has been demonstrated that the number of polyfunctional NK cells is correlated strongly with the outcome of certain infectious diseases 24. Given that during ADCC more NK cells produced cytokines when CD16 was not down-regulated, we wanted to analyse the polyfunctionality of NK cells during ADCC in the presence of the MMPs inhibitor GM6001. We found that the percentage of bifunctional and trifunctional NK cells increased significantly when CD16 cleavage was inhibited by GM6001 (Fig. 2d). Altogether, our results indicate that preservation of CD16 on the cell surface, by means of MMPs inhibition, results in an increase in the number of polyfunctional NK cells during ADCC.
MMPs inhibition resulted in sustained signalling through phosphorylation of CD3ζ
We hypothesized that sustained CD16-mediated signalling, as a result of prolonged engagement of CD16 on NK cells treated with the MMPs inhibitor GM6001, may contribute to the increased polyfunctionality of NK cells during ADCC. Phosphorylation of multiple molecular components is essential for NK cell effector functions 25. CD3ζ is an ITAM-containing adaptor molecule that associates with CD16 7,26, and is tyrosine phosphorylated after the receptor engagement, initiating the signalling pathway that governs antibody-dependent NK cell activation 8,27. We therefore decided to measure the levels of tyrosine phosphorylated CD3ζ and other downstream signalling intermediates that are triggered by extended ligation of CD16 on NK cells treated with the MMPs inhibitor GM6001. To exclude other potential activation signals from the tumour cells, such as those provided by adhesion molecules, we cross-linked CD16 with the anti-CD16 mAb clone 3G8, which mimics ADCC conditions 9. Similar to the results obtained with cell lines and therapeutic mAbs, cross-linking of CD16 with anti-CD16 mAbs induced the down-regulation of the receptor, which could be inhibited by incubating NK cells with the MMPs inhibitor GM6001 (Fig. 3a). In our experimental settings, activation through CD16 alone with anti-CD16 mAb was insufficient to trigger cytokine production (Fig. 3b). However, CD16 cross-linking in combination with GM6001 led to a significant cytokine production by NK cells (Fig. 3b). Therefore, we used this system and measured the phosphorylation status of several molecular targets in a time–course ranging from 0 to 240 min. Tyrosine phosphorylation of CD3ζ peaked 5 min after CD16 ligation and returned gradually to the basal levels at 240 min. However, when MMPs were inhibited with GM6001, increased and extended phosphorylation of CD3ζ was achieved in response to CD16 engagement (Fig. 3c,d). Next, we also measured the phosphorylation of two downstream signalling intermediates, i.e. extracellular-regulated kinase (ERK) and phospholipase C-gamma1 (PLC-γ1) and, as expected, augmented and prolonged phosphorylation was also detected when CD16 was engaged in NK cells treated with the MMPs inhibitor GM6001 (Fig. S4). These results confirm that increased and extended signalling is a consequence of prolonged engagement of CD16, and indicate that during ADCC the inhibition of MMPs led to the maintenance of the receptor on the cell surface and to a better quality in the responding NK cells, as shown by an increase in their polyfunctionality. Although we cannot exclude the participation of other NK receptors interacting with the respective ligands on tumour cells, CD16-mediated signalling is necessary and dominant, as there was no evidence of NK cell activation when cells were treated only with MMPs inhibitor (Fig. 2a and Fig. S3).
Figure 3.
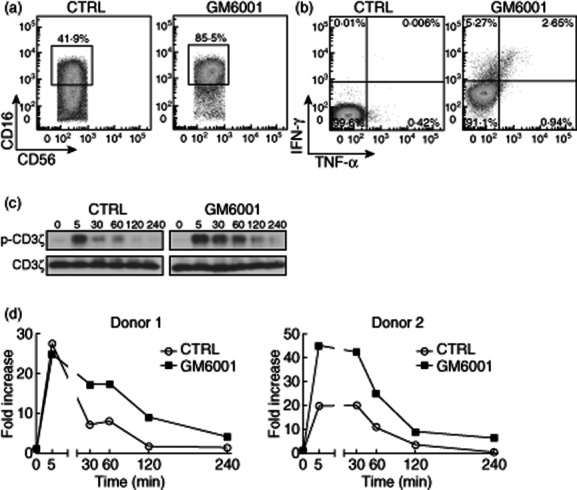
Cross-linking of CD16 in the presence of matrix metalloproteinases (MMPs) inhibition led to increased and sustained CD16 mediated signalling through phosphorylation of CD3ζ. (a) Freshly isolated natural killer (NK) cells were incubated with mouse anti-human CD16 (clone 3G8) at 20 μg/ml for 30 min on ice, washed with phosphate-buffered saline (PBS) to remove unbound antibodies and then followed by 4 h incubation at 37°C with goat anti-mouse secondary immunoglobulin (IgG) at 20 μg/ml in the presence of the MMPs inhibitor GM6001 or its control (CTRL). Then, flow cytometric analyses were performed. Cells were gated electronically on CD3−CD56dim NK cells and representative dot-plots of the CD16 expression from two experiments is shown with NK cells from two donors. (b) Cells were treated as in (a). Representative dot-plots of interferon (IFN)-γ- and tumour necrosis factor (TNF)-α-producing NK cells from two experiments are shown with NK cells from two donors. (c) Cells were stimulated as in (a) and (b), and harvested at different time-points (0, 5, 30, 60, 120 and 240 min) and lysed. Total cell lysates were resolved on 4–20% Mini-protean® precast gels, transferred to a polyvinylidene difluoride (PVDF) membrane, followed by immunoblotting with appropriate antibodies. A representative image of Western blot is shown. (d) Graph representations of the quantification of phospho-CD3ζ from two independent experiments with NK cells from two donors are shown. Data were analysed using ImageJ software and normalized according to total CD3ζ.
Discussion
In this report, we show for the first time an important role of MMPs in regulating the quality of human NK cell activity in response to three therapeutic mAbs used clinically for cancer treatment. After activation with tumour targets coated with therapeutic mAbs, such as trastuzumab, rituximab and cetuximab, CD16 expression on NK cells was down-regulated dramatically and this down-regulation was observed on the majority of degranulating CD107a/b+ NK-responding cells. Inhibition of MMPs with GM6001 preserved the CD16 expression during the process of ADCC and increased NK cell polyfunctionality. For the first time, we show further that sustained CD16-mediated signalling, as shown by an increased and prolonged tyrosine phosphorylation of CD3ζ and downstream intermediates, was achieved by MMP inhibition, therefore accounting for improved NK cell effector functions during therapeutic mAbs mediated ADCC.
The involvement of MMPs in cancer development may start at an early stage, when the cross-talk between malignant and surrounding stromal cells is required for tumour cells to grow and metastasize 28–30. Additionally, the formation and alteration of the extracellular matrix of the tumour environment is also important for tumour progression 28. MMPs are expressed abundantly by a large variety of cell types including cancer cells 30,31. Use of specific or common inhibitors that target MMPs has been explored for decades, for example for the treatment of inflammatory and vascular diseases 17. Although many of them have shown promising effects in preclinical studies, the results from clinical trials with these agents as single therapy appear to be disappointing in cancer treatment 17. Conversely, the oral administration of a MMPs inhibitor, i.e. GI54902, has been shown to reduce the lipopolysaccaride (LPS)-induced release of soluble CD27 and CD16 in the circulation in healthy humans 32. The results of this study suggest that the combination therapies of MMP inhibitors, and therapeutic mAbs may offer greater probability to succeed in cancer treatment, as expression of CD16 is critical for the ADCC mechanism of mAbs.
Cytokines are released in response to a large array of cellular insults and stresses, including tumour, infection and inflammation. IFN-γ has been shown to have a direct effect on tumour cell survival and can sensitize some tumour cells for killing by effector cells 33–35. The direct anti-proliferative effect of IFN-γ requires the ligation of IFN-γ receptors (IFN-γR) and the signal transduction through the IFN-γRβ 36. IFN-γ can facilitate apoptosis of breast cancer cell lines by activation of the Fas/Fas ligand pathway through increasing caspase-8 expression, induction of cytochrome C and caspase-9 37. An additional role for IFN-γ in inhibiting cancer development is shown by the spontaneous development of mammary tumours in mice deficient in IFN-γ production 38. Moreover, IFN-γ and TNF-α are important cytokines that play a crucial role in modulating the anti-tumour immune response by activating effector cells such as T cells, macrophages, neutrophils and NK cells, which would abrogate tumour targets that are negative or have modified IFN-γR and TNF-α receptor expression to escape from the immune attack. Therefore, modulation of the production of such cytokines could have a significant impact on the outcomes of therapeutic mAbs-mediated cancer therapy. Our results demonstrate clearly a significant increase in the IFN-γ- and TNF-α-producing NK cells, and on a per cell basis, in response to mAbs-coated target cells when CD16 is maintained on the NK cell surface through inhibition of MMPs activity. The increased IFN-γ and TNF-α production could trigger a sequence of activation of both innate and adaptive immune components within the cytokine network and lead to amplified effector functions that may have the potential to eliminate tumour cells in vivo.
Several publications have shown that a higher number of polyfunctional T cells and NK cells correlates with better prognosis during infections and cancer 39–42. From our studies, we conclude that inhibiting the down-regulation of CD16 during therapeutic mAbs mediated ADCC with MMPs inhibitors will lead to a significant increase in the number of polyfunctional NK cells, and therefore to an increase in the therapeutic mAbs anti-tumour effect. In summary, in light of our results, we propose that an enhanced and sustained CD16-mediated signal during ADCC, by means of MMPs inhibition, may lead to an increase in the efficacy of therapeutic mAbs that use ADCC as a mechanism of action.
Acknowledgments
This work was supported by the intramural programme of the US Food and Drug Administration and by the NCI-FDA Interagency Oncology Task Force fellowship programme. The authors would like to thank Dr Wen Jin Wu and Dr Milos Dokmanovic for their generous gift of trastuzumab.
Disclosure
The authors declare no conflicting interests.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Matrix metalloproteinases (MMPs) inhibition preserved CD16 cell surface expression on natural killer (NK) responding cells. Freshly isolated NK cells were cultured at a 1:1 ratio with the tumour cell lines Raji or A431, in the absence or presence of therapeutic monoclonal antibodies (mAbs) rituximab or cetuximab, respectively, at 10 μg/ml for 4 h in the presence of the MMPs inhibitor GM6001 or its control (CTRL) (50 μM). CD16 expression was determined by flow cytometric analysis. Cells were gated electronically on CD3–CD56dim NK cells. Representative dot-plots of CD16 expression on NK cells from five independent experiments with NK cells from five donors are shown.
Fig. S2. Natural killer (NK) cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) activity led to killing of the tumour cells. SKBr3 cells were labelled with bis(acetoxymethyl)2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (BADTA), according to the manufacture's manual (PerkinElmer™). Freshly isolated NK cells were cultured with labelled SKBr3 cells at different effector/target cell (E/T) ratios in the presence or absence of trastuzumab (10 μg/ml) for 2·5 h at 37°C. SKBr3 cells alone served as spontaneous release (SR) and SKBr3 cells lysed with 1% Triton X-100 served as total release (TR). After the incubation, 25 μl of supernatant from each culture condition was transferred to a 96-well plate with 200 μl europium solution, prepared according to the manufacture's manual. Europium signals were measured with a PerkinElmer multi-label counter. Killing efficiency was then calculated according to the following formula: % of specific killing = [(experimental release − SR)/(TR-SR)] × 100%.
Fig. S3. Matrix metalloproteinases (MMPs) inhibition increased the percentage of cytokine-producing natural killer (NK) cells. Freshly isolated NK cells were cultured at a 1:1 ratio with the tumour cell lines Raji or A431, in the absence or presence of rituximab or cetuximab, respectively, at 10 μg/ml for 6 h in the presence of the MMPs inhibitor GM6001 or its control (CTRL) (50 μM). Monensin was added to the cultures according to the manufacturer's manual. Interferon (IFN)-γ and tumour necrosis factor (TNF)-α was measured by flow cytometric analysis. Cells were gated on electronically CD3–CD56dim NK cells. Representative dot-plots of IFN-γ- and TNF-α-producing NK cells from three independent experiments with NK cells from three donors are shown.
Fig. S4. Cross-linking of CD16 in the presence of matrix metalloproteinases (MMPs) inhibition led to increased and sustained phosphorylation of extracellular-regulated kinase (ERK) and phospholipase C-gamma1 (PLC-γ1). Freshly isolated natural killer (NK) cells were incubated with mouse anti-human CD16 (clone 3G8) at 20 μg/ml for 30 min on ice, washed with phosphate-buffered saline (PBS) to remove unbound antibodies and then followed by incubation at 37°C with goat anti-mouse secondary immunoglobulin (Ig)G at 20 μg/ml in the presence of the MMPs inhibitor GM6001 or its control (CTRL) (50 μM). Cells were harvested at different time-points (0, 5, 30, 60, 120 and 240 min) and lysed. Total cell lysates were resolved on 4–20% Mini-protean® precast gels, transferred to a polyvinylidene difluoride (PVDF) membrane, and followed by immunoblotting with appropriate antibodies. A graphic representation of the quantification of phospho-ERK and phospho-PLC-γ1 is shown. Data were analysed using ImageJ software and normalized according to total ERK and PLC-γ1.
References
- 1.Jiang XR, Song A, Bergelson S, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–111. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 2.Kohrt HE, Houot R, Marabelle A, et al. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4:511–527. doi: 10.2217/imt.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 4.Shuptrine CW, Surana R, Weiner LM. Monoclonal antibodies for the treatment of cancer. Semin Cancer Biol. 2012;22:3–13. doi: 10.1016/j.semcancer.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 6.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 7.Lanier LL, Yu G, Phillips JH. Analysis of Fc gamma RIII (CD16) membrane expression and association with CD3 zeta and Fc epsilon RI-gamma by site-directed mutation. J Immunol. 1991;146:1571–1576. [PubMed] [Google Scholar]
- 8.Vivier E, Morin P, O'Brien C, Druker B, Schlossman SF, Anderson P. Tyrosine phosphorylation of the Fc gamma RIII(CD16): zeta complex in human natural killer cells. Induction by antibody-dependent cytotoxicity but not by natural killing. J Immunol. 1991;146:206–210. [PubMed] [Google Scholar]
- 9.Mota G, Moldovan I, Calugaru A, et al. Interaction of human immunoglobulin G with CD16 on natural killer cells: ligand clearance, FcgammaRIIIA turnover and effects of metalloproteinases on FcgammaRIIIA-mediated binding, signal transduction and killing. Scand J Immunol. 2004;59:278–284. doi: 10.1111/j.0300-9475.2004.01398.x. [DOI] [PubMed] [Google Scholar]
- 10.Grier JT, Forbes LR, Monaco-Shawver L, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. 2012;122:3769–3780. doi: 10.1172/JCI64837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borrego F, Lopez-Beltran A, Pena J, Solana R. Downregulation of Fc gamma receptor IIIA alpha (CD16-II) on natural killer cells induced by anti-CD16 mAb is independent of protein tyrosine kinases and protein kinase C. Cell Immunol. 1994;158:208–217. doi: 10.1006/cimm.1994.1268. [DOI] [PubMed] [Google Scholar]
- 12.Grzywacz B, Kataria N, Verneris MR. CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia. 2007;21:356–359. doi: 10.1038/sj.leu.2404499. author reply 9. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q, Sun Y, Rihn S, et al. Matrix metalloprotease inhibitors restore impaired NK cell-mediated antibody-dependent cellular cytotoxicity in human immunodeficiency virus type 1 infection. J Virol. 2009;83:8705–8712. doi: 10.1128/JVI.02666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical–biological functions and (Q)SARs. Bioorg Med Chem. 2007;15:2223–2268. doi: 10.1016/j.bmc.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Harrison D, Phillips JH, Lanier LL. Involvement of a metalloprotease in spontaneous and phorbol ester-induced release of natural killer cell-associated Fc gamma RIII (CD16-II) J Immunol. 1991;147:3459–3465. [PubMed] [Google Scholar]
- 16.Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61:577–581. [PubMed] [Google Scholar]
- 17.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 18.Jackson C, Nguyen M, Arkell J, Sambrook P. Selective matrix metalloproteinase (MMP) inhibition in rheumatoid arthritis – targetting gelatinase A activation. Inflamm Res. 2001;50:183–186. doi: 10.1007/s000110050743. [DOI] [PubMed] [Google Scholar]
- 19.Rundhaug JE. Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res., 9: 00–00, 2003. Clin Cancer Res. 2003;9:551–554. [PubMed] [Google Scholar]
- 20.Hatjiharissi E, Xu L, Santos DD, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007;110:2561–2564. doi: 10.1182/blood-2007-01-070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 22.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Pullyblank AM, Guillou PJ, Monson JR. Interleukin 1 and tumour necrosis factor alpha may be responsible for the lytic mechanism during anti-tumour antibody-dependent cell-mediated cytotoxicity. Br J Cancer. 1995;72:601–606. doi: 10.1038/bjc.1995.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eller MA, Koehler RN, Kijak GH, et al. Human immunodeficiency virus type 1 infection is associated with increased NK cell polyfunctionality and higher levels of KIR3DL1+ NK cells in Ugandans carrying the HLA-B Bw4 motif. J Virol. 2011;85:4802–4811. doi: 10.1128/JVI.00111-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanier LL, Yu G, Phillips JH. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature. 1989;342:803–805. doi: 10.1038/342803a0. [DOI] [PubMed] [Google Scholar]
- 27.Lanier LL. Natural killer cell receptor signaling. Curr Opin Immunol. 2003;15:308–314. doi: 10.1016/s0952-7915(03)00039-6. [DOI] [PubMed] [Google Scholar]
- 28.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 29.Herszenyi L, Lakatos G, Hritz I, Varga MZ, Cierny G, Tulassay Z. The role of inflammation and proteinases in tumor progression. Dig Dis. 2012;30:249–254. doi: 10.1159/000336914. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann UB, Eggert AA, Blass K, Brocker EB, Becker JC. Expression of matrix metalloproteinases in the microenvironment of spontaneous and experimental melanoma metastases reflects the requirements for tumor formation. Cancer Res. 2003;63:8221–8225. [PubMed] [Google Scholar]
- 31.Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 32.Dekkers PE, ten Hove T, Lauw FN, et al. The metalloproteinase inhibitor GI5402 inhibits endotoxin-induced soluble CD27 and CD16 release in healthy humans. Infect Immun. 2000;68:3036–3039. doi: 10.1128/iai.68.5.3036-3039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109. doi: 10.1016/s1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 34.van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- 35.Wall L, Burke F, Barton C, Smyth J, Balkwill F. IFN-gamma induces apoptosis in ovarian cancer cells in vivo and in vitro. Clin Cancer Res. 2003;9:2487–2496. [PubMed] [Google Scholar]
- 36.Soh J, Donnelly RJ, Kotenko S, et al. Identification and sequence of an accessory factor required for activation of the human interferon gamma receptor. Cell. 1994;76:793–802. doi: 10.1016/0092-8674(94)90354-9. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz-Ruiz C, Munoz-Pinedo C, Lopez-Rivas A. Interferon-gamma treatment elevates caspase-8 expression and sensitizes human breast tumor cells to a death receptor-induced mitochondria-operated apoptotic program. Cancer Res. 2000;60:5673–5680. [PubMed] [Google Scholar]
- 38.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 39.Betts MR, Nason MC, West SM, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulet S, Song R, Kamya P, et al. HIV protective KIR3DL1 and HLA-B genotypes influence NK cell function following stimulation with HLA-devoid cells. J Immunol. 2010;184:2057–2064. doi: 10.4049/jimmunol.0902621. [DOI] [PubMed] [Google Scholar]
- 41.Ding ZC, Huang L, Blazar BR, et al. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–2239. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wren L, Parsons MS, Isitman G, et al. Influence of cytokines on HIV-specific antibody-dependent cellular cytotoxicity activation profile of natural killer cells. PLoS ONE. 2012;7:e38580. doi: 10.1371/journal.pone.0038580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.